Hepatitis C Virus Life Cycle and Lipid Metabolism

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
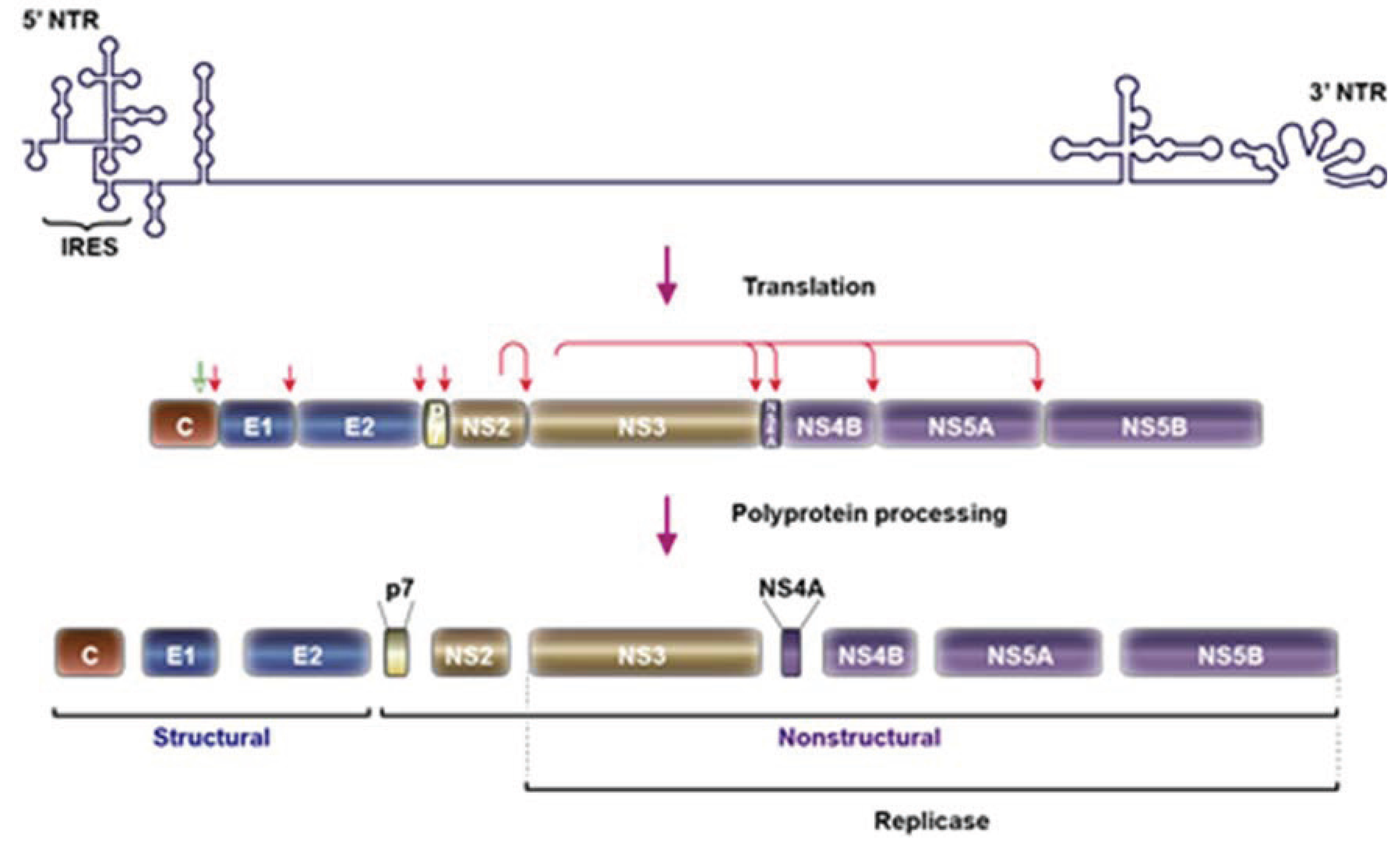
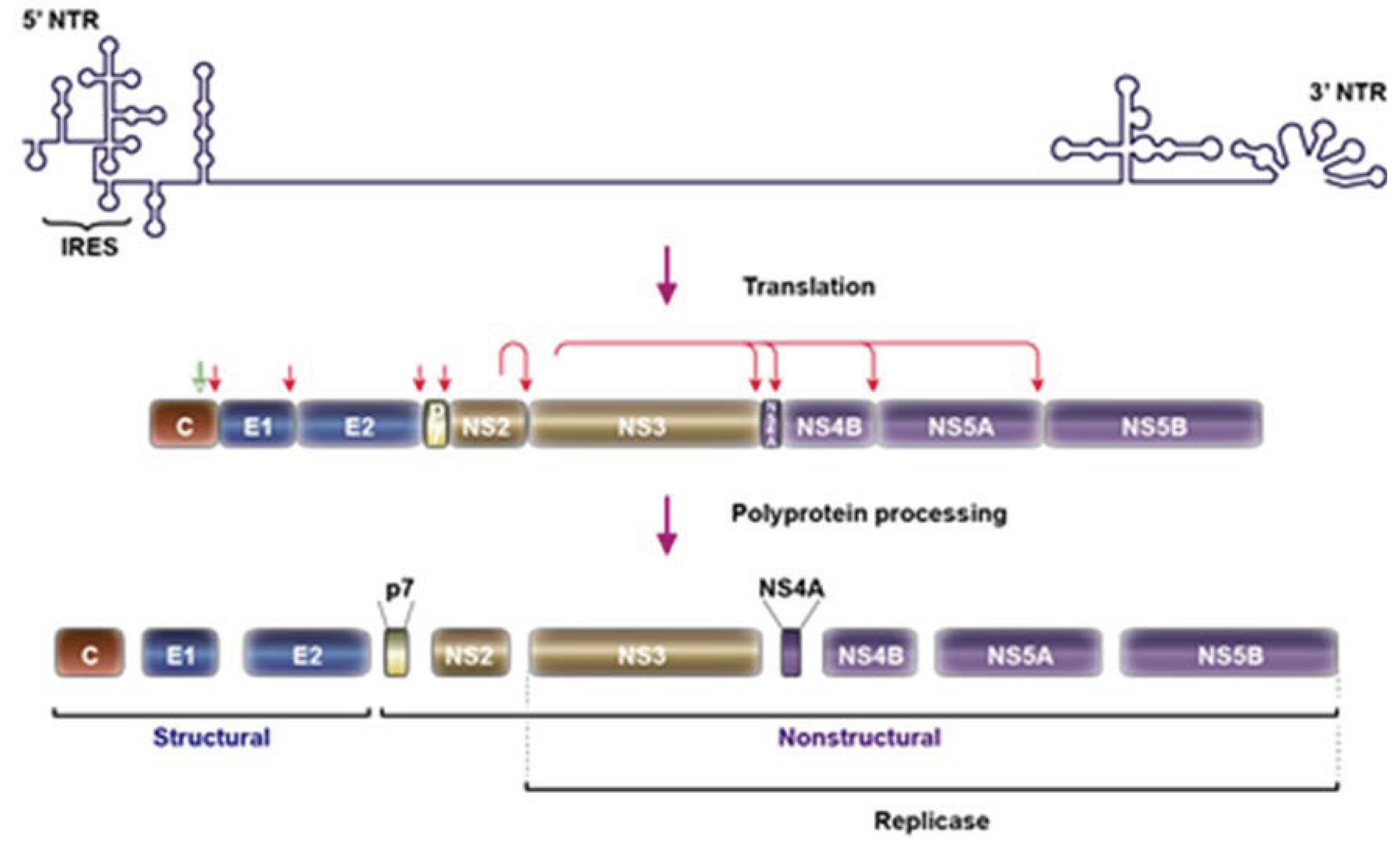
2. HCV Particle
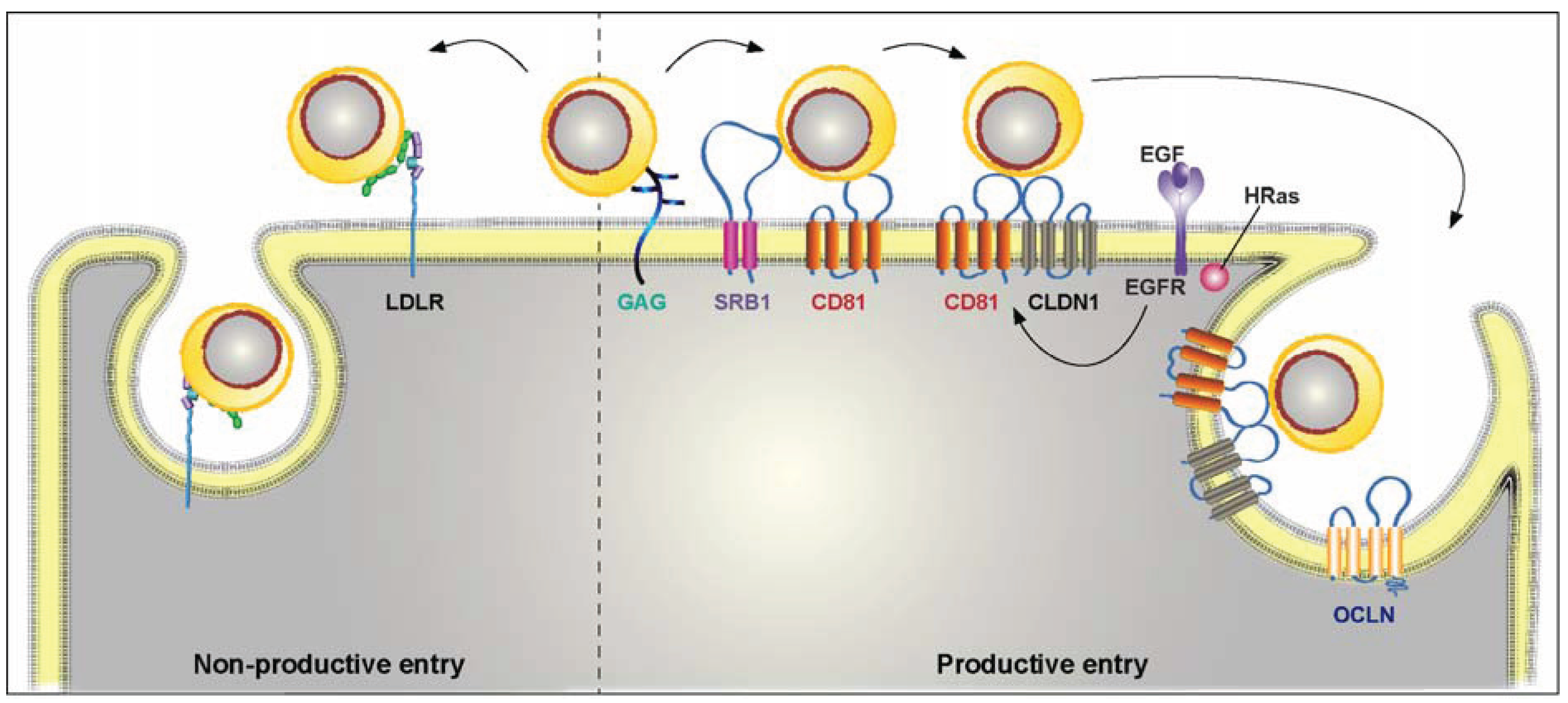
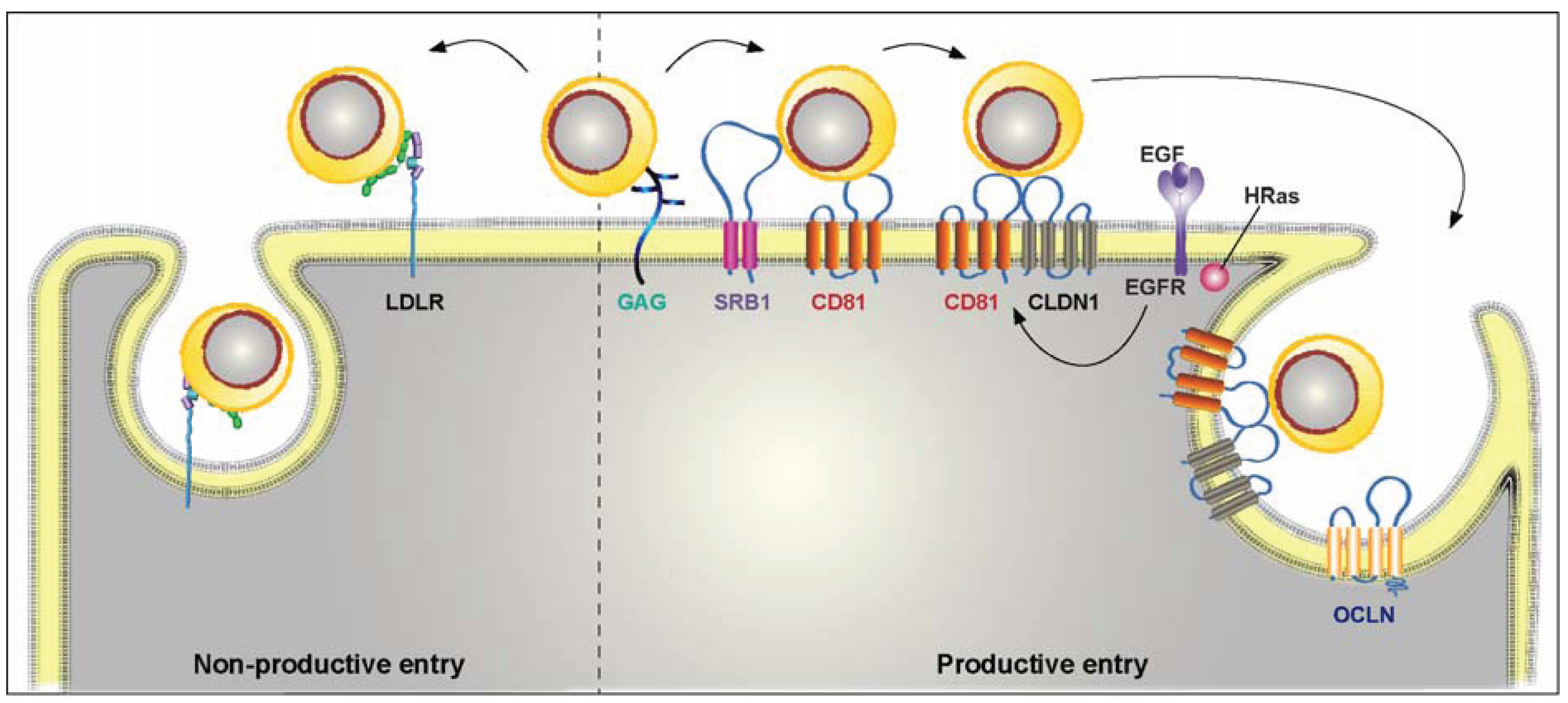
3. HCV Entry
4. HCV Replication
4.1. HCV-Induced Membrane Rearrangements
4.2. Role of Phosphatidylinositol-4 Phosphate in HCV Replication

4.3. Interplay between HCV Replication and Phospholipids and Fatty Acid Metabolism
4.4. Role of GBF1-Arf1-COP-I Pathway in HCV Replication
5. HCV Assembly
5.1. Lipid Droplets and HCV Assembly Modules
5.2. HCV Core Recruitment to LD
5.3. HCV Replication Complex Recruitment to LD
5.4. HCV NS2 Complex
5.5. HCV Virion Budding and Secretion
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Levrero, M. Viral hepatitis and liver cancer: The case of hepatitis C. Oncogene 2006, 25, 3834–3847. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.M. New hepatitis C therapies: The toolbox, strategies, and challenges. Gastroenterology 2014, 146, 1176–1192. [Google Scholar] [CrossRef] [PubMed]
- Dienes, H.P.; Popper, H.; Arnold, W.; Lobeck, H. Histologic observations in human hepatitis non-A, non-B. Hepatology 1982, 2, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, E.A.; Chung, R.T. HCV and host lipids: An intimate connection. Semin. Liver Dis. 2013, 33, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Goossens, N.; Negro, F. Is genotype 3 of the hepatitis C virus the new villain? Hepatology 2014, 59, 2403–2412. [Google Scholar] [CrossRef] [PubMed]
- Corey, K.E.; Kane, E.; Munroe, C.; Barlow, L.L.; Zheng, H.; Chung, R.T. Hepatitis C virus infection and its clearance alter circulating lipids: Implications for long-term follow-up. Hepatology 2009, 50, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Andre, P.; Komurian-Pradel, F.; Deforges, S.; Perret, M.; Berland, J.L.; Sodoyer, M.; Pol, S.; Brechot, C.; Paranhos-Baccala, G.; Lotteau, V. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J. Virol. 2002, 76, 6919–6928. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.T.; Murray, C.L.; Eastman, D.K.; Tassello, J.; Rice, C.M. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J. Virol. 2007, 81, 8374–8383. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of hepatitis C virus. Nat. Rev. MicroBiol. 2007, 5, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wolk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; et al. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Krausslich, H.G.; Mizokami, M.; et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar]
- Zhong, J.; Gastaminza, P.; Cheng, G.; Kapadia, S.; Kato, T.; Burton, D.R.; Wieland, S.F.; Uprichard, S.L.; Wakita, T.; Chisari, F.V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 2005, 102, 9294–9299. [Google Scholar] [CrossRef]
- Pumeechockchai, W.; Bevitt, D.; Agarwal, K.; Petropoulou, T.; Langer, B.C.; Belohradsky, B.; Bassendine, M.F.; Toms, G.L. Hepatitis C virus particles of different density in the blood of chronically infected immunocompetent and immunodeficient patients: Implications for virus clearance by antibody. J. Med. Virol. 2002, 68, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Bradley, D.; McCaustland, K.; Krawczynski, K.; Spelbring, J.; Humphrey, C.; Cook, E.H. Hepatitis C virus: Buoyant density of the factor VIII-derived isolate in sucrose. J. Med. Virol. 1991, 34, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Felmlee, D.J.; Sheridan, D.A.; Bridge, S.H.; Nielsen, S.U.; Milne, R.W.; Packard, C.J.; Caslake, M.J.; McLauchlan, J.; Toms, G.L.; Neely, R.D.; et al. Intravascular transfer contributes to postprandial increase in numbers of very-low-density hepatitis C virus particles. Gastroenterology 2010, 139. [Google Scholar] [CrossRef]
- Nielsen, S.U.; Bassendine, M.F.; Burt, A.D.; Martin, C.; Pumeechockchai, W.; Toms, G.L. Association between hepatitis C virus and very-low-density lipoprotein (VLDL)/LDL analyzed in iodixanol density gradients. J. Virol. 2006, 80, 2418–2428. [Google Scholar] [CrossRef] [PubMed]
- Thomssen, R.; Bonk, S.; Propfe, C.; Heermann, K.H.; Kochel, H.G.; Uy, A. Association of hepatitis C virus in human sera with beta-lipoprotein. Med. Microbiol. Immunol. 1992, 181, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Meuleman, P.; Ploss, A.; Vanwolleghem, T.; Syder, A.J.; McKeating, J.A.; Lanford, R.E.; Feinstone, S.M.; Major, M.E.; Leroux-Roels, G.; et al. Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc. Natl. Acad. Sci. USA 2006, 103, 3805–3809. [Google Scholar]
- Catanese, M.T.; Uryu, K.; Kopp, M.; Edwards, T.J.; Andrus, L.; Rice, W.J.; Silvestry, M.; Kuhn, R.J.; Rice, C.M. Ultrastructural analysis of hepatitis C virus particles. Proc. Natl. Acad. Sci. USA 2013, 110, 9505–9510. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.S.; Jiang, J.; Cai, Z.; Luo, G. Human apolipoprotein E is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 2007, 81, 13783–13793. [Google Scholar] [CrossRef] [PubMed]
- Gastaminza, P.; Cheng, G.; Wieland, S.; Zhong, J.; Liao, W.; Chisari, F.V. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol. 2008, 82, 2120–2129. [Google Scholar] [CrossRef] [PubMed]
- Meunier, J.C.; Russell, R.S.; Engle, R.E.; Faulk, K.N.; Purcell, R.H.; Emerson, S.U. Apolipoprotein C1 association with hepatitis C virus. J. Virol. 2008, 82, 9647–9656. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.Y.; Lin, C.C.; Lee, J.C.; Wang, S.W.; Cheng, P.N.; Wu, I.C.; Chang, T.T.; Lai, M.D.; Shieh, D.B.; Young, K.C. Very low-density lipoprotein/lipo-viro particles reverse lipoprotein lipase-mediated inhibition of hepatitis C virus infection via apolipoprotein C-III. Gut 2012, 62, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Merz, A.; Long, G.; Hiet, M.S.; Bruegger, B.; Chlanda, P.; Andre, P.; Wieland, F.; Krijnse-Locker, J.; Bartenschlager, R. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J. Biol. Chem. 2011, 286, 3018–3032. [Google Scholar] [CrossRef] [PubMed]
- Tomiyasu, K.; Walsh, B.W.; Ikewaki, K.; Judge, H.; Sacks, F.M. Differential metabolism of human VLDL according to content of apoE and apoC-III. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1494–1500. [Google Scholar] [CrossRef] [PubMed]
- Jammart, B.; Michelet, M.; Pecheur, E.I.; Parent, R.; Bartosch, B.; Zoulim, F.; Durantel, D. Very-low-density lipoprotein (VLDL)-producing and hepatitis C virus-replicating HepG2 cells secrete no more lipoviroparticles than VLDL-deficient Huh7.5 cells. J. Virol. 2013, 87, 5065–5080. [Google Scholar] [CrossRef]
- Vieyres, G.; Thomas, X.; Descamps, V.; Duverlie, G.; Patel, A.H.; Dubuisson, J. Characterization of the envelope glycoproteins associated with infectious hepatitis C virus. J. Virol. 2010, 84, 10159–10168. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D. Virion assembly and release. Curr. Top. Microbiol. Immunol. 2013, 369, 199–218. [Google Scholar] [PubMed]
- Bartenschlager, R.; Penin, F.; Lohmann, V.; Andre, P. Assembly of infectious hepatitis C virus particles. Trends MicroBiol. 2011, 19, 95–103. [Google Scholar] [PubMed]
- Icard, V.; Diaz, O.; Scholtes, C.; Perrin-Cocon, L.; Ramiere, C.; Bartenschlager, R.; Penin, F.; Lotteau, V.; Andre, P. Secretion of hepatitis C virus envelope glycoproteins depends on assembly of apolipoprotein B positive lipoproteins. PLOS ONE 2009, 4, e4233. [Google Scholar] [CrossRef] [PubMed]
- Boyer, A.; Dumans, A.; Beaumont, E.; Etienne, L.; Roingeard, P.; Meunier, J.C. The association of hepatitis C virus glycoproteins with apolipoproteins E and B early in assembly is conserved in lipoviral particles. J. Biol. Chem. 2014, 289, 18904–18913. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Acosta, E.G.; Stoeck, I.K.; Long, G.; Hiet, M.S.; Mueller, B.; Fackler, O.T.; Kallis, S.; Bartenschlager, R. Apolipoprotein E likely contributes to a maturation step of infectious hepatitis C virus particles and interacts with viral envelope glycoproteins. J. Virol. 2014, 88, 12422–12437. [Google Scholar] [CrossRef] [PubMed]
- Dao Thi, V.L.; Granier, C.; Zeisel, M.B.; Guerin, M.; Mancip, J.; Granio, O.; Penin, F.; Lavillette, D.; Bartenschlager, R.; Baumert, T.F.; et al. Characterization of hepatitis C virus particle subpopulations reveals multiple usage of the scavenger receptor BI for entry steps. J. Biol. Chem. 2012, 287, 31242–31257. [Google Scholar]
- Bartosch, B.; Dubuisson, J.; Cosset, F.L. Infectious hepatitis C pseudo-particles containing functional E1E2 envelope protein complexes. J. Exp. Med. 2003, 197, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Drummer, H.E.; Maerz, A.; Poumbourios, P. Cell surface expression of functional hepatitis C virus E1 and E2 glycoproteins. FEBS Lett. 2003, 546, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.; Zhang, J.; Flint, M.; Logvinoff, C.; Cheng-Mayer, C.; Rice, C.M.; McKeating, J.A. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. USA 2003, 100, 7271–7276. [Google Scholar] [CrossRef] [PubMed]
- Koutsoudakis, G.; Kaul, A.; Steinmann, E.; Kallis, S.; Lohmann, V.; Pietschmann, T.; Bartenschlager, R. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J. Virol. 2006, 80, 5308–5320. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Jiang, J.; Luo, G. Syndecan-1 serves as the major receptor for attachment of hepatitis C virus to the surfaces of hepatocytes. J. Virol. 2013, 87, 6866–6875. [Google Scholar] [CrossRef] [PubMed]
- Lefevre, M.; Felmlee, D.J.; Parnot, M.; Baumert, T.F.; Schuster, C. Syndecan 4 is involved in mediating HCV entry through interaction with lipoviral particle-associated apolipoprotein E. PLOS ONE 2014, 9, e95550. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.; Schafer, C.; Adah, M.I.; Zhang, F.; Linhardt, R.J.; Toyoda, H.; Kinoshita-Toyoda, A.; Toida, T.; Van Kuppevelt, T.H.; Depla, E.; et al. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J. Biol. Chem. 2003, 278, 41003–41012. [Google Scholar]
- Jiang, J.; Luo, G. Apolipoprotein E but not B is required for the formation of infectious hepatitis C virus particles. J. Virol. 2009, 83, 12680–12691. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Cun, W.; Wu, X.; Shi, Q.; Tang, H.; Luo, G. Hepatitis C virus attachment mediated by apolipoprotein E binding to cell surface heparan sulfate. J. Virol. 2012, 86, 7256–7267. [Google Scholar] [CrossRef] [PubMed]
- Agnello, V.; Abel, G.; Elfahal, M.; Knight, G.B.; Zhang, Q.-X. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 12766–12771. [Google Scholar] [CrossRef] [PubMed]
- Molina, S.; Castet, V.; Fournier-Wirth, C.; Pichard-Garcia, L.; Avner, R.; Harats, D.; Roitelman, J.; Barbaras, R.; Graber, P.; Ghersa, P.; et al. The low-density lipoprotein receptor plays a role in the infection of primary human hepatocytes by hepatitis C virus. J. Hepatol. 2007, 46, 411–419. [Google Scholar]
- Owen, D.M.; Huang, H.; Ye, J.; Gale, M., Jr. Apolipoprotein E on hepatitis C virion facilitates infection through interaction with low-density lipoprotein receptor. Virology 2009, 394, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Albecka, A.; Belouzard, S.; de Beeck, A.O.; Descamps, V.; Goueslain, L.; Bertrand-Michel, J.; Terce, F.; Duverlie, G.; Rouille, Y.; Dubuisson, J. Role of low-density lipoprotein receptor in the hepatitis C virus life cycle. Hepatology 2012, 55, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Andreo, U.; Maillard, P.; Kalinina, O.; Walic, M.; Meurs, E.; Martinot, M.; Marcellin, P.; Budkowska, A. Lipoprotein lipase mediates hepatitis C virus (HCV) cell entry and inhibits HCV infection. Cell. MicroBiol. 2007, 9, 2445–2456. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Hishiki, T.; Sugiyama, K.; Ogawa, K.; Funami, K.; Kato, A.; Ohsaki, Y.; Fujimoto, T.; Takaku, H.; Shimotohno, K. Lipoprotein lipase and hepatic triglyceride lipase reduce the infectivity of hepatitis C virus (HCV) through their catalytic activities on HCV-associated lipoproteins. Virology 2010, 407, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.J.; Hu, J.; Hu, Z.; Kraemer, F.B.; Azhar, S. Scavenger receptor class B type I (SR-BI): A versatile receptor with multiple functions and actions. Metab. Clin. Exp. 2014, 63, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002, 21, 5017–5025. [Google Scholar] [CrossRef] [PubMed]
- Maillard, P.; Huby, T.; Andreo, U.; Moreau, M.; Chapman, J.; Budkowska, A. The interaction of natural hepatitis C virus with human scavenger receptor SR-BI/Cla1 is mediated by apoB-containing lipoproteins. FASEB J. 2006, 20, 735–737. [Google Scholar] [PubMed]
- Dao Thi, V.L.; Dreux, M.; Cosset, F.L. Scavenger receptor class B type I and the hypervariable region-1 of hepatitis C virus in cell entry and neutralisation. Expert Rev. Mol. Med. 2011, 13, e13. [Google Scholar] [CrossRef] [PubMed]
- Zahid, M.N.; Turek, M.; Xiao, F.; Thi, V.L.; Guerin, M.; Fofana, I.; Bachellier, P.; Thompson, J.; Delang, L.; Neyts, J.; et al. The postbinding activity of scavenger receptor class B type I mediates initiation of hepatitis C virus infection and viral dissemination. Hepatology 2013, 57, 492–504. [Google Scholar]
- Bartosch, B.; Verney, G.; Dreux, M.; Donot, P.; Morice, Y.; Penin, F.; Pawlotsky, J.M.; Lavillette, D.; Cosset, F.L. An interplay between the hyper-variable region 1 of the HCV E2 glycoprotein, the scavenger receptor BI and HDL promotes both enhancement of infection and protection against neutralizing antibodies. J. Virol. 2005, 79, 8217–8229. [Google Scholar] [CrossRef] [PubMed]
- Meunier, J.C.; Engle, R.E.; Faulk, K.; Zhao, M.; Bartosch, B.; Alter, H.; Emerson, S.U.; Cosset, F.L.; Purcell, R.H.; Bukh, J. Evidence for cross-genotype neutralization of hepatitis C virus pseudo-particles and enhancement of infectivity by apolipoprotein C1. Proc. Natl. Acad. Sci. USA 2005, 102, 4560–4565. [Google Scholar] [CrossRef] [PubMed]
- Voisset, C.; Callens, N.; Blanchard, E.; op de Beeck, A.; Dubuisson, J.; Vu-Dac, N. High density lipoproteins facilitate hepatitis C virus entry through the scavenger receptor class B type I. J. Biol. Chem. 2005, 280, 7793–7799. [Google Scholar] [CrossRef] [PubMed]
- Von Hahn, T.; Lindenbach, B.D.; Boullier, A.; Quehenberger, O.; Paulson, M.; Rice, C.M.; McKeating, J.A. Oxidized low-density lipoprotein inhibits hepatitis C virus cell entry in human hepatoma cells. Hepatology 2006, 43, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Pileri, P.; Uematsu, Y.; Campagnoli, S.; Galli, G.; Falugi, F.; Petracca, R.; Weiner, A.J.; Houghton, M.; Rosa, D.; Grandi, G.; et al. Binding of hepatitis C virus to CD81. Science 1998, 282, 938–941. [Google Scholar]
- Bankwitz, D.; Steinmann, E.; Bitzegeio, J.; Ciesek, S.; Friesland, M.; Herrmann, E.; Zeisel, M.B.; Baumert, T.F.; Keck, Z.Y.; Foung, S.K.; et al. Hepatitis C virus hypervariable region 1 modulates receptor interactions, conceals the CD81 binding site, and protects conserved neutralizing epitopes. J. Virol. 2010, 84, 5751–5763. [Google Scholar]
- Prentoe, J.; Jensen, T.B.; Meuleman, P.; Serre, S.B.; Scheel, T.K.; Leroux-Roels, G.; Gottwein, J.M.; Bukh, J. Hypervariable region 1 differentially impacts viability of hepatitis C virus strains of genotypes 1 to 6 and impairs virus neutralization. J. Virol. 2011, 85, 2224–2234. [Google Scholar] [CrossRef] [PubMed]
- Feneant, L.; Levy, S.; Cocquerel, L. CD81 and hepatitis C virus (HCV) infection. Viruses 2014, 6, 535–572. [Google Scholar] [PubMed]
- Charrin, S.; le Naour, F.; Silvie, O.; Milhiet, P.E.; Boucheix, C.; Rubinstein, E. Lateral organization of membrane proteins: Tetraspanins spin their web. BioChem. J. 2009, 420, 133–154. [Google Scholar] [PubMed]
- Harris, H.J.; Clerte, C.; Farquhar, M.J.; Goodall, M.; Hu, K.; Rassam, P.; Dosset, P.; Wilson, G.K.; Balfe, P.; Ijzendoorn, S.C.; et al. Hepatoma polarization limits CD81 and hepatitis C virus dynamics. Cell. MicroBiol. 2013, 15, 430–445. [Google Scholar]
- Potel, J.; Rassam, P.; Montpellier, C.; Kaestner, L.; Werkmeister, E.; Tews, B.A.; Couturier, C.; Popescu, C.I.; Baumert, T.F.; Rubinstein, E.; et al. EWI-2wint promotes CD81 clustering that abrogates hepatitis C virus entry. Cell. MicroBiol. 2013, 15, 1234–1252. [Google Scholar]
- Kapadia, S.B.; Barth, H.; Baumert, T.; McKeating, J.A.; Chisari, F.V. Initiation of HCV infection is dependent on cholesterol and cooperativity between CD81 and scavenger receptor B type I. J. Virol. 2007, 81, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Voisset, C.; Lavie, M.; Helle, F.; op de Beeck, A.; Bilheu, A.; Bertrand-Michel, J.; Tercé, F.; Cocquerel, L.; Wychowski, C.; Vu-Dac, N.; et al. Ceramide enrichment of the plasma membrane induces CD81 internalization and inhibits hepatitis C virus entry. Cell. MicroBiol. 2008, 10, 606–617. [Google Scholar]
- Evans, M.J.; von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wolk, B.; Hatziioannou, T.; McKeating, J.A.; Bieniasz, P.D.; Rice, C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Ploss, A.; Evans, M.J.; Gaysinskaya, V.A.; Panis, M.; You, H.; de Jong, Y.P.; Rice, C.M. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 2009, 457, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.J.; Davis, C.; Mullins, J.G.; Hu, K.; Goodall, M.; Farquhar, M.J.; Mee, C.J.; McCaffrey, K.; Young, S.; Drummer, H.; et al. Claudin association with CD81 defines hepatitis C virus entry. J. Biol. Chem. 2010, 285, 21092–21102. [Google Scholar]
- Harris, H.J.; Farquhar, M.J.; Mee, C.J.; Davis, C.; Reynolds, G.M.; Jennings, A.; Hu, K.; Yuan, F.; Deng, H.; Hubscher, S.G.; et al. CD81 and claudin 1 coreceptor association: Role in hepatitis C virus entry. J. Virol. 2008, 82, 5007–5020. [Google Scholar]
- Farquhar, M.J.; Hu, K.; Harris, H.J.; Davis, C.; Brimacombe, C.L.; Fletcher, S.J.; Baumert, T.F.; Rappoport, J.Z.; Balfe, P.; McKeating, J.A. Hepatitis C virus induces CD81 and claudin-1 endocytosis. J. Virol. 2012, 86, 4305–4316. [Google Scholar] [CrossRef] [PubMed]
- Dubuisson, J.; Cosset, F.L. Virology and cell biology of hepatitis C virus life cycle–An update. J. Hepatol 2014, 61, S3–S13. [Google Scholar] [CrossRef] [PubMed]
- Lupberger, J.; Zeisel, M.B.; Xiao, F.; Thumann, C.; Fofana, I.; Zona, L.; Davis, C.; Mee, C.J.; Turek, M.; Gorke, S.; et al. EGFR and EPHA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 2011, 17, 589–595. [Google Scholar]
- Zona, L.; Lupberger, J.; Sidahmed-Adrar, N.; Thumann, C.; Harris, H.J.; Barnes, A.; Florentin, J.; Tawar, R.G.; Xiao, F.; Turek, M.; et al. HRas signal transduction promotes hepatitis C virus cell entry by triggering assembly of the host tetraspanin receptor complex. Cell. Host Microbe 2013, 13, 302–313. [Google Scholar]
- Sourisseau, M.; Michta, M.L.; Zony, C.; Israelow, B.; Hopcraft, S.E.; Narbus, C.M.; Parra Martin, A.; Evans, M.J. Temporal analysis of hepatitis C virus cell entry with occludin directed blocking antibodies. PLOS Pathog. 2013, 9, e1003244. [Google Scholar] [CrossRef] [PubMed]
- Benedicto, I.; Molina-Jimenez, F.; Bartosch, B.; Cosset, F.L.; Lavillette, D.; Prieto, J.; Moreno-Otero, R.; Valenzuela-Fernandez, A.; Aldabe, R.; Lopez-Cabrera, M.; et al. The tight junction-associated protein occludin is required for a postbinding step in hepatitis C virus entry and infection. J. Virol. 2009, 83, 8012–8020. [Google Scholar]
- Liu, S.; Yang, W.; Shen, L.; Turner, J.R.; Coyne, C.B.; Wang, T. Tight junction proteins claudin-1 and occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. J. Virol. 2009, 83, 2011–2014. [Google Scholar] [CrossRef] [PubMed]
- Sainz, B., Jr.; Barretto, N.; Martin, D.N.; Hiraga, N.; Imamura, M.; Hussain, S.; Marsh, K.A.; Yu, X.; Chayama, K.; Alrefai, W.A.; et al. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. 2012, 18, 281–285. [Google Scholar]
- Blanchard, E.; Belouzard, S.; Goueslain, L.; Wakita, T.; Dubuisson, J.; Wychowski, C.; Rouille, Y. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J. Virol. 2006, 80, 6964–6972. [Google Scholar] [CrossRef] [PubMed]
- Coller, K.E.; Berger, K.L.; Heaton, N.S.; Cooper, J.D.; Yoon, R.; Randall, G. RNA interference and single particle tracking analysis of hepatitis C virus endocytosis. PLOS Pathog. 2009, 5, e1000702. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Suzuki, R.; Kataoka, C.; Watashi, K.; Aizaki, H.; Kato, N.; Matsuura, Y.; Suzuki, T.; Wakita, T. Alternative endocytosis pathway for productive entry of hepatitis C virus. J. Gen. Virol. 2014, 95, 2658–2667. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.R.; Mateu, G.; Dreux, M.; Grakoui, A.; Cosset, F.L.; Melikyan, G.B. Hepatitis C virus is primed by CD81 protein for low pH-dependent fusion. J. Biol. Chem. 2011, 286, 30361–30376. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.G.; Whidby, J.; Miller, M.T.; Scarborough, H.; Zatorski, A.V.; Cygan, A.; Price, A.A.; Yost, S.A.; Bohannon, C.D.; Jacob, J.; et al. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature 2014, 509, 381–384. [Google Scholar]
- Kong, L.; Giang, E.; Nieusma, T.; Kadam, R.U.; Cogburn, K.E.; Hua, Y.; Dai, X.; Stanfield, R.L.; Burton, D.R.; Ward, A.B.; et al. Hepatitis C virus E2 envelope glycoprotein core structure. Science 2013, 342, 1090–1094. [Google Scholar]
- Krey, T.; d’Alayer, J.; Kikuti, C.M.; Saulnier, A.; Damier-Piolle, L.; Petitpas, I.; Johansson, D.X.; Tawar, R.G.; Baron, B.; Robert, B.; et al. The disulfide bonds in glycoprotein E2 of hepatitis C virus reveal the tertiary organization of the molecule. PLOS Pathog. 2010, 6. [Google Scholar] [CrossRef]
- Lavillette, D.; Pecheur, E.I.; Donot, P.; Fresquet, J.; Molle, J.; Corbau, R.; Dreux, M.; Penin, F.; Cosset, F.L. Characterization of fusion determinants points to the involvement of three discrete regions of both E1 and E2 glycoproteins in the membrane fusion process of hepatitis C virus. J. Virol. 2007, 81, 8752–8765. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, V.; Korner, F.; Koch, J.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Egger, D.; Wölk, B.; Gosert, R.; Bianchi, L.; Blum, H.E.; Moradpour, D.; Bienz, K. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 2002, 76, 5974–5984. [Google Scholar] [CrossRef] [PubMed]
- Gosert, R.; Egger, D.; Lohmann, V.; Bartenschlager, R.; Blum, H.E.; Bienz, K.; Moradpour, D. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 2003, 77, 5487–5492. [Google Scholar] [CrossRef] [PubMed]
- Rouille, Y.; Helle, F.; Delgrange, D.; Roingeard, P.; Voisset, C.; Blanchard, E.; Belouzard, S.; McKeating, J.; Patel, A.H.; Maertens, G.; et al. Subcellular localization of hepatitis C virus structural proteins in a cell culture system that efficiently replicates the virus. J. Virol. 2006, 80, 2832–2841. [Google Scholar]
- Wolk, B.; Buchele, B.; Moradpour, D.; Rice, C.M. A dynamic view of hepatitis C virus replication complexes. J. Virol. 2008, 82, 10519–10531. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, P.; Blanchard, E.; Roingeard, P. Ultrastructural and biochemical analyses of hepatitis C virus-associated host cell membranes. J. Gen. Virol. 2010, 91, 2230–2237. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, P.; Beaumont, E.; Uzbekov, R.; Brand, D.; Gaillard, J.; Blanchard, E.; Roingeard, P. Sequential biogenesis of host cell membrane rearrangements induced by hepatitis C virus infection. Cell. Mol. Life Sci. 2013, 70, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Romero-Brey, I.; Merz, A.; Chiramel, A.; Lee, J.Y.; Chlanda, P.; Haselman, U.; Santarella-Mellwig, R.; Habermann, A.; Hoppe, S.; Kallis, S.; et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLOS Pathog. 2012, 8. [Google Scholar] [CrossRef]
- Belov, G.A.; Nair, V.; Hansen, B.T.; Hoyt, F.H.; Fischer, E.R.; Ehrenfeld, E. Complex dynamic development of poliovirus membranous replication complexes. J. Virol. 2012, 86, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Limpens, R.W.; van der Schaar, H.M.; Kumar, D.; Koster, A.J.; Snijder, E.J.; van Kuppeveld, F.J.; Barcena, M. The transformation of enterovirus replication structures: A three-dimensional study of single- and double-membrane compartments. MBio 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Knoops, K.; Kikkert, M.; Worm, S.H.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLOS Biol. 2008, 6. [Google Scholar] [CrossRef] [PubMed]
- Ulasli, M.; Verheije, M.H.; de Haan, C.A.; Reggiori, F. Qualitative and quantitative ultrastructural analysis of the membrane rearrangements induced by coronavirus. Cell. MicroBiol. 2010, 12, 844–861. [Google Scholar] [CrossRef] [PubMed]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell. Host Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, L.K.; Hoenen, A.; Morgan, G.; Mackenzie, J.M. The endoplasmic reticulum provides the membrane platform for biogenesis of the flavivirus replication complex. J. Virol. 2010, 84, 10438–10447. [Google Scholar] [CrossRef] [PubMed]
- Schmeiser, S.; Mast, J.; Thiel, H.J.; Konig, M. Morphogenesis of pestiviruses: New insights from ultrastructural studies of strain giraffe-1. J. Virol. 2014, 88, 2717–2724. [Google Scholar] [CrossRef] [PubMed]
- Ivashkina, N.; Wolk, B.; Lohmann, V.; Bartenschlager, R.; Blum, H.E.; Penin, F.; Moradpour, D. The hepatitis C virus RNA-dependent RNA polymerase mambrane insertion sequence is a transmembrane segment. J. Virol. 2002, 76, 13088–13093. [Google Scholar] [CrossRef] [PubMed]
- Berger, K.L.; Cooper, J.D.; Heaton, N.S.; Yoon, R.; Oakland, T.E.; Jordan, T.X.; Mateu, G.; Grakoui, A.; Randall, G. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 7577–7582. [Google Scholar] [CrossRef] [PubMed]
- Stone, M.; Jia, S.; Heo, W.D.; Meyer, T.; Konan, K.V. Participation of rab5, an early endosome protein, in hepatitis C virus RNA replication machinery. J. Virol. 2007, 81, 4551–4563. [Google Scholar] [CrossRef] [PubMed]
- Tai, A.W.; Benita, Y.; Peng, L.F.; Kim, S.S.; Sakamoto, N.; Xavier, R.J.; Chung, R.T. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell. Host Microbe 2009, 5, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.T.; Lee, K.J.; Aizaki, H.; Hwang, S.B.; Lai, M.M. Hepatitis C virus RNA replication occurs on a detergent-resistant membrane that cofractionates with caveolin-2. J. Virol. 2003, 77, 4160–4168. [Google Scholar] [CrossRef] [PubMed]
- Borawski, J.; Troke, P.; Puyang, X.; Gibaja, V.; Zhao, S.; Mickanin, C.; Leighton-Davies, J.; Wilson, C.J.; Myer, V.; Cornellataracido, I.; et al. Class III phosphatidylinositol 4-kinase alpha and beta are novel host factor regulators of hepatitis C virus replication. J. Virol. 2009, 83, 10058–10074. [Google Scholar]
- Li, Q.; Brass, A.L.; Ng, A.; Hu, Z.; Xavier, R.J.; Liang, T.J.; Elledge, S.J. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc. Natl. Acad. Sci. USA 2009, 106, 16410–16415. [Google Scholar] [CrossRef] [PubMed]
- Trotard, M.; Lepere-Douard, C.; Regeard, M.; Piquet-Pellorce, C.; Lavillette, D.; Cosset, F.L.; Gripon, P.; Le Seyec, J. Kinases required in hepatitis C virus entry and replication highlighted by small interference RNA screening. FASEB J. 2009, 23, 3780–3789. [Google Scholar] [CrossRef] [PubMed]
- Vaillancourt, F.H.; Pilote, L.; Cartier, M.; Lippens, J.; Liuzzi, M.; Bethell, R.C.; Cordingley, M.G.; Kukolj, G. Identification of a lipid kinase as a host factor involved in hepatitis C virus RNA replication. Virology 2009, 387, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Chung, K.S.; Kim, D.U.; Won, M.; Kim, L.; Kim, K.S.; Nam, M.; Choi, S.J.; Kim, H.C.; Yoon, M.; et al. Systematic identification of hepatocellular proteins interacting with NS5A of the hepatitis C virus. J. Biochem. Mol. Biol. 2004, 37, 741–748. [Google Scholar]
- Berger, K.L.; Kelly, S.M.; Jordan, T.X.; Tartell, M.A.; Randall, G. Hepatitis C virus stimulates the phosphatidylinositol 4-kinase III alpha-dependent phosphatidylinositol 4-phosphate production that is essential for its replication. J. Virol. 2011, 85, 8870–8883. [Google Scholar] [CrossRef] [PubMed]
- Reiss, S.; Rebhan, I.; Backes, P.; Romero-Brey, I.; Erfle, H.; Matula, P.; Kaderali, L.; Poenisch, M.; Blankenburg, H.; Hiet, M.S.; et al. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell. Host Microbe 2011, 9, 32–45. [Google Scholar]
- Leivers, A.L.; Tallant, M.; Shotwell, J.B.; Dickerson, S.; Leivers, M.R.; McDonald, O.B.; Gobel, J.; Creech, K.L.; Strum, S.L.; Mathis, A.; et al. Discovery of selective small molecule type III phosphatidylinositol 4-kinase alpha (PI4KIIIalpha) inhibitors as anti hepatitis C (HCV) agents. J. Med. Chem. 2014, 57, 2091–2106. [Google Scholar]
- Bianco, A.; Reghellin, V.; Donnici, L.; Fenu, S.; Alvarez, R.; Baruffa, C.; Peri, F.; Pagani, M.; Abrignani, S.; Neddermann, P.; et al. Metabolism of phosphatidylinositol 4-kinase IIIalpha-dependent PI4P is subverted by HCV and is targeted by a 4-anilino quinazoline with antiviral activity. PLOS Pathog. 2012, 8, e1002576. [Google Scholar]
- Lee, C.; Ma, H.; Hang, J.Q.; Leveque, V.; Sklan, E.H.; Elazar, M.; Klumpp, K.; Glenn, J.S. The hepatitis C virus NS5A inhibitor (BMS-790052) alters the subcellular localization of the NS5A non-structural viral protein. Virology 2011, 414, 10–18. [Google Scholar] [CrossRef] [PubMed]
- McGivern, D.R.; Masaki, T.; Williford, S.; Ingravallo, P.; Feng, Z.; Lahser, F.; Asante-Appiah, E.; Neddermann, P.; de Francesco, R.; Howe, A.Y.; et al. Kinetic analyses reveal potent and early blockade of hepatitis C virus assembly by NS5A inhibitors. Gastroenterology 2014, 147, 453.e7–462.e7. [Google Scholar]
- Reghellin, V.; Donnici, L.; Fenu, S.; Berno, V.; Calabrese, V.; Pagani, M.; Abrignani, S.; Peri, F.; de Francesco, R.; Neddermann, P. NS5A inhibitors impair NS5A- PI4KIIIalpha complex formation and cause a decrease of PI4P and cholesterol levels in HCV-associated membranes. Antimicrob. Agents Chemother. 2014. [Google Scholar] [CrossRef]
- Targett-Adams, P.; Graham, E.J.; Middleton, J.; Palmer, A.; Shaw, S.M.; Lavender, H.; Brain, P.; Tran, T.D.; Jones, L.H.; Wakenhut, F.; et al. Small molecules targeting hepatitis C virus-encoded NS5A cause subcellular redistribution of their target: Insights into compound modes of action. J. Virol. 2011, 85, 6353–6368. [Google Scholar]
- Hsu, N.Y.; Ilnytska, O.; Belov, G.; Santiana, M.; Chen, Y.H.; Takvorian, P.M.; Pau, C.; van der Schaar, H.; Kaushik-Basu, N.; Balla, T.; et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010, 141, 799–811. [Google Scholar]
- Zhang, L.; Hong, Z.; Lin, W.; Shao, R.X.; Goto, K.; Hsu, V.W.; Chung, R.T. ARF1 and GBF1 generate a PI4P-enriched environment supportive of hepatitis C virus replication. PLOS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Balla, T. Phosphoinositides: Tiny lipids with giant impact on cell regulation. Physiol. Rev. 2013, 93, 1019–1137. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Katikaneni, D.S.; Han, Q.; Sanchez-Felipe, L.; Hanada, K.; Ambrose, R.L.; Mackenzie, J.M.; Konan, K.V. Modulation of hepatitis C virus genome replication by glycosphingolipids and four-phosphate adaptor protein 2. J. Virol. 2014, 88, 12276–12295. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Perry, J.W.; Lauring, A.S.; Neddermann, P.; de Francesco, R.; Tai, A.W. Oxysterol-binding protein is a phosphatidylinositol 4-kinase effector required for HCV replication membrane integrity and cholesterol trafficking. Gastroenterology 2014, 146. [Google Scholar] [CrossRef]
- Mesmin, B.; Bigay, J.; Moser von Filseck, J.; Lacas-Gervais, S.; Drin, G.; Antonny, B. A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell 2013, 155, 830–843. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Aizaki, H.; He, J.W.; Lai, M.M. Interactions between viral nonstructural proteins and host protein hVAP-33 mediate the formation of hepatitis C virus RNA replication complex on lipid raft. J. Virol. 2004, 78, 3480–3488. [Google Scholar] [CrossRef] [PubMed]
- Matto, M.; Sklan, E.H.; David, N.; Melamed-Book, N.; Casanova, J.E.; Glenn, J.S.; Aroeti, B. Role for ADP ribosylation factor 1 in the regulation of hepatitis C virus replication. J. Virol. 2011, 85, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Arita, M.; Kojima, H.; Nagano, T.; Okabe, T.; Wakita, T.; Shimizu, H. Phosphatidylinositol 4-kinase III beta is a target of enviroxime-like compounds for antipoliovirus activity. J. Virol. 2011, 85, 2364–2372. [Google Scholar] [CrossRef] [PubMed]
- Arita, M. Phosphatidylinositol-4 kinase III beta and oxysterol-binding protein accumulate unesterified cholesterol on poliovirus-induced membrane structure. Microbiol. Immunol. 2014, 58, 239–256. [Google Scholar] [CrossRef] [PubMed]
- Amako, Y.; Sarkeshik, A.; Hotta, H.; Yates, J., 3rd. Siddiqui, A.Role of oxysterol binding protein in hepatitis C virus infection. J. Virol. 2009, 83, 9237–9246. [Google Scholar]
- D’Angelo, G.; Polishchuk, E.; di Tullio, G.; Santoro, M.; di Campli, A.; Godi, A.; West, G.; Bielawski, J.; Chuang, C.C.; van der Spoel, A.C.; et al. Glycosphingolipid synthesis requires FAPP2 transfer of glucosylceramide. Nature 2007, 449, 62–67. [Google Scholar]
- Diamond, D.L.; Syder, A.J.; Jacobs, J.M.; Sorensen, C.M.; Walters, K.A.; Proll, S.C.; McDermott, J.E.; Gritsenko, M.A.; Zhang, Q.; Zhao, R.; et al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLOS Pathog. 2010, 6. [Google Scholar] [CrossRef]
- Kapadia, S.B.; Chisari, F.V. Hepatitis C virus rna replication is regulated by host geranylgeranylation and fatty acids. Proc. Natl. Acad. Sci. USA 2005, 102, 2561–2566. [Google Scholar] [CrossRef] [PubMed]
- Su, A.I.; Pezacki, J.P.; Wodicka, L.; Brideau, A.D.; Supekova, L.; Thimme, R.; Wieland, S.; Bukh, J.; Purcell, R.H.; Schultz, P.G.; et al. Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl. Acad. Sci. USA 2002, 99, 15669–15674. [Google Scholar]
- Waris, G.; Felmlee, D.J.; Negro, F.; Siddiqui, A. Hepatitis C virus induces proteolytic cleavage of sterol regulatory element binding proteins and stimulates their phosphorylation via oxidative stress. J. Virol. 2007, 81, 8122–8130. [Google Scholar] [CrossRef] [PubMed]
- Olmstead, A.D.; Knecht, W.; Lazarov, I.; Dixit, S.B.; Jean, F. Human subtilase SKI-1/S1P is a master regulator of the HCV lifecycle and a potential host cell target for developing indirect-acting antiviral agents. PLOS Pathog. 2012, 8, e1002468. [Google Scholar] [CrossRef] [PubMed]
- Nasheri, N.; Joyce, M.; Rouleau, Y.; Yang, P.; Yao, S.; Tyrrell, D.L.; Pezacki, J.P. Modulation of fatty acid synthase enzyme activity and expression during hepatitis C virus replication. Chem. Biol. 2013, 20, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Hood, B.L.; Chadwick, S.L.; Liu, S.; Watkins, S.C.; Luo, G.; Conrads, T.P.; Wang, T. Fatty acid synthase is up-regulated during hepatitis C virus infection and regulates hepatitis C virus entry and production. Hepatology 2008, 48, 1396–1403. [Google Scholar] [CrossRef] [PubMed]
- Lyn, R.K.; Singaravelu, R.; Kargman, S.; O’Hara, S.; Chan, H.; Oballa, R.; Huang, Z.; Jones, D.M.; Ridsdale, A.; Russell, R.S.; et al. Stearoyl-CoA desaturase inhibition blocks formation of hepatitis C virus-induced specialized membranes. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef]
- Nguyen, L.N.; Lim, Y.S.; Pham, L.V.; Shin, H.Y.; Kim, Y.S.; Hwang, S.B. Stearoyl coenzyme A desaturase 1 is associated with hepatitis C virus replication complex and regulates viral replication. J. Virol. 2014, 88, 12311–12325. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, A.L.; Diamond, D.L.; McDermott, J.E.; Gao, X.; Metz, T.O.; Matzke, M.M.; Carter, V.S.; Belisle, S.E.; Korth, M.J.; Waters, K.M.; et al. Systems virology identifies a mitochondrial fatty acid oxidation enzyme, dodecenoyl coenzyme A delta isomerase, required for hepatitis C virus replication and likely pathogenesis. J. Virol. 2011, 85, 11646–11654. [Google Scholar]
- Majeau, N.; Fromentin, R.; Savard, C.; Duval, M.; Tremblay, M.J.; Leclerc, D. Palmitoylation of hepatitis C virus core protein is important for virion production. J. Biol. Chem. 2009, 284, 33915–33925. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.Y.; Lee, K.J.; Gao, L.; Lai, M.M. Palmitoylation and polymerization of hepatitis C virus NS4B protein. J. Virol. 2006, 80, 6013–6023. [Google Scholar] [CrossRef] [PubMed]
- Chao, T.C.; Su, W.C.; Huang, J.Y.; Chen, Y.C.; Jeng, K.S.; Wang, H.D.; Lai, M.M. Proline-serine-threonine phosphatase-interacting protein 2 (PSTPIP2), a host membrane-deforming protein, is critical for membranous web formation in hepatitis C virus replication. J. Virol. 2012, 86, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Pei, R.; Guo, M.; Han, Q.; Lai, J.; Wang, Y.; Wu, C.; Zhou, Y.; Lu, M.; Chen, X. Cytosolic phospholipase A2 gamma is involved in hepatitis C virus replication and assembly. J. Virol. 2012, 86, 13025–13037. [Google Scholar] [CrossRef]
- Hirata, Y.; Ikeda, K.; Sudoh, M.; Tokunaga, Y.; Suzuki, A.; Weng, L.; Ohta, M.; Tobita, Y.; Okano, K.; Ozeki, K.; et al. Self-enhancement of hepatitis C virus replication by promotion of specific sphingolipid biosynthesis. PLOS Pathog. 2012, 8, e1002860. [Google Scholar]
- Katsume, A.; Tokunaga, Y.; Hirata, Y.; Munakata, T.; Saito, M.; Hayashi, H.; Okamoto, K.; Ohmori, Y.; Kusanagi, I.; Fujiwara, S.; et al. A serine palmitoyltransferase inhibitor blocks hepatitis C virus replication in human hepatocytes. Gastroenterology 2013, 145, 865–873. [Google Scholar]
- Sakamoto, H.; Okamoto, K.; Aoki, M.; Kato, H.; Katsume, A.; Ohta, A.; Tsukuda, T.; Shimma, N.; Aoki, Y.; Arisawa, M.; et al. Host sphingolipid biosynthesis as a target for hepatitis C virus therapy. Nat. Chem. Biol. 2005, 1, 333–337. [Google Scholar]
- Huang, H.; Chen, Y.; Ye, J. Inhibition of hepatitis C virus replication by peroxidation of arachidonate and restoration by vitamin E. Proc. Natl. Acad. Sci. USA 2007, 104, 18666–18670. [Google Scholar] [CrossRef] [PubMed]
- Pollock, S.; Nichita, N.B.; Bohmer, A.; Radulescu, C.; Dwek, R.A.; Zitzmann, N. Polyunsaturated liposomes are antiviral against hepatitis B and C viruses and hiv by decreasing cholesterol levels in infected cells. Proc. Natl. Acad. Sci. USA 2010, 107, 17176–17181. [Google Scholar] [CrossRef] [PubMed]
- Yamane, D.; McGivern, D.R.; Wauthier, E.; Yi, M.; Madden, V.J.; Welsch, C.; Antes, I.; Wen, Y.; Chugh, P.E.; McGee, C.E.; et al. Regulation of the hepatitis C virus RNA replicase by endogenous lipid peroxidation. Nat. Med. 2014, 20, 927–935. [Google Scholar]
- Goueslain, L.; Alsaleh, K.; Horellou, P.; Roingeard, P.; Descamps, V.; Duverlie, G.; Ciczora, Y.; Wychowski, C.; Dubuisson, J.; Rouille, Y. Identification of GBF1 as a cellular factor required for hepatitis C virus RNA replication. J. Virol. 2010, 84, 773–787. [Google Scholar] [CrossRef] [PubMed]
- Belov, G.A.; Feng, Q.; Nikovics, K.; Jackson, C.L.; Ehrenfeld, E. A critical role of a cellular membrane traffic protein in poliovirus RNA replication. PLOS Pathog. 2008, 4, e1000216. [Google Scholar] [CrossRef] [PubMed]
- Lanke, K.H.; van der Schaar, H.M.; Belov, G.A.; Feng, Q.; Duijsings, D.; Jackson, C.L.; Ehrenfeld, E.; van Kuppeveld, F.J. GBF1, a guanine nucleotide exchange factor for Arf, is crucial for coxsackievirus B3 RNA replication. J. Virol. 2009, 83, 11940–11949. [Google Scholar] [CrossRef] [PubMed]
- Verheije, M.H.; Raaben, M.; Mari, M.; te Lintelo, E.G.; Reggiori, F.; van Kuppeveld, F.J.; Rottier, P.J.; de Haan, C.A. Mouse hepatitis coronavirus RNA replication depends on GBF1-mediated ARF1 activation. PLOS Pathog. 2008, 4, e1000088. [Google Scholar] [CrossRef] [PubMed]
- Carpp, L.N.; Rogers, R.S.; Moritz, R.L.; Aitchison, J.D. Quantitative proteomic analysis of host-virus interactions reveals a role for GBF1 in dengue infection. Mol. Cell. Proteom. 2014, 13, 2836–2854. [Google Scholar]
- Donaldson, J.G.; Jackson, C.L. ARF family G proteins and their regulators: Roles in membrane transport, development and disease. Nat. Rev. Mol. Cell. Biol. 2011, 12, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.; Kahn, R.A.; Sztul, E. Regulating the large Sec7 ARF guanine nucleotide exchange factors: The when, where and how of activation. Cell. Mol. Life Sci. 2014, 71, 3419–3438. [Google Scholar] [CrossRef] [PubMed]
- Cherry, S.; Kunte, A.; Wang, H.; Coyne, C.; Rawson, R.B.; Perrimon, N. COPI activity coupled with fatty acid biosynthesis is required for viral replication. PLOS Pathog. 2006, 2, e102. [Google Scholar] [CrossRef] [PubMed]
- Gazina, E.V.; Mackenzie, J.M.; Gorrell, R.J.; Anderson, D.A. Differential requireents for COPI coats in formation of replication complexes among three genera of picornaviridae. J. Virol. 2002, 76, 11113–11122. [Google Scholar] [CrossRef] [PubMed]
- Farhat, R.; Goueslain, L.; Wychowski, C.; Belouzard, S.; Feneant, L.; Jackson, C.L.; Dubuisson, J.; Rouille, Y. Hepatitis C virus replication and Golgi function in brefeldin A-resistant hepatoma-derived cells. PLOS ONE 2013, 8, e74491. [Google Scholar] [CrossRef] [PubMed]
- Ellong, E.N.; Soni, K.G.; Bui, Q.T.; Sougrat, R.; Golinelli-Cohen, M.P.; Jackson, C.L. Interaction between the triglyceride lipase ATGL and the Arf1 activator GBF1. PLOS ONE 2011, 6, e21889. [Google Scholar] [CrossRef] [PubMed]
- Beller, M.; Sztalryd, C.; Southall, N.; Bell, M.; Jackle, H.; Auld, D.S.; Oliver, B. Copi complex is a regulator of lipid homeostasis. PLOS Biol. 2008, 6, e292. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Walther, T.C.; Rao, M.; Stuurman, N.; Goshima, G.; Terayama, K.; Wong, J.S.; Vale, R.D.; Walter, P.; Farese, R.V. Functional genomic screen reveals genes involved in lipid-droplet formation and utilization. Nature 2008, 453, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, X.; Yang, G.; Hong, Z.; Zhou, L.; Yin, P.; Xiao, Y.; Chen, L.; Chung, R.T.; Zhang, L. Hepatitis C virus NS5A hijacks ARFGAP1 to maintain a phosphatidylinositol 4-phosphate-enriched microenvironment. J. Virol. 2014, 88, 5956–5966. [Google Scholar] [CrossRef] [PubMed]
- Belov, G.A.; Kovtunovych, G.; Jackson, C.L.; Ehrenfeld, E. Poliovirus replication requires the N-terminus but not the catalytic sec7 domain of ARFGEF GBF1. Cell. MicroBiol. 2010, 12, 1463–1479. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.L.; Jones, C.T.; Rice, C.M. Architects of assembly: Roles of Flaviviridae non-structural proteins in virion morphogenesis. Nat. Rev. MicroBiol. 2008, 6, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell. Biol. 2007, 9, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Liefhebber, J.M.; Hague, C.V.; Zhang, Q.; Wakelam, M.J.; McLauchlan, J. Modulation of triglyceride and cholesterol ester synthesis impairs assembly of infectious hepatitis C virus. J. Biol. Chem. 2014, 289, 21276–21288. [Google Scholar] [CrossRef] [PubMed]
- Pol, A.; Gross, S.P.; Parton, R.G. Review: Biogenesis of the multifunctional lipid droplet: Lipids, proteins, and sites. J. Cell. Biol. 2014, 204, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Welte, M.A. Fat on the move: Intracellular motion of lipid droplets. BioChem. Soc. Trans. 2009, 37, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Bartz, R.; Zehmer, J.K.; Zhu, M.; Chen, Y.; Serrero, G.; Zhao, Y.; Liu, P. Dynamic activity of lipid droplets: Protein phosphorylation and GTP-mediated protein translocation. J. Proteome Res. 2007, 6, 3256–3265. [Google Scholar] [CrossRef] [PubMed]
- Marcinkiewicz, A.; Gauthier, D.; Garcia, A.; Brasaemle, D.L. The phosphorylation of serine 492 of perilipin a directs lipid droplet fragmentation and dispersion. J. Biol. Chem. 2006, 281, 11901–11909. [Google Scholar] [CrossRef] [PubMed]
- Boulant, S.; Vanbelle, C.; Ebel, C.; Penin, F.; Lavergne, J.P. Hepatitis C virus core protein is a dimeric alpha-helical protein exhibiting membrane protein features. J. Virol. 2005, 79, 11353–11365. [Google Scholar] [CrossRef] [PubMed]
- Boulant, S.; Montserret, R.; Hope, G.; Ratinier, M.; Targett-Adams, P.; Lavergne, J.P.; Penin, F.; McLauchlan, J. Structural determinants that target the hepatitis C virus core protein to lipid droplets. J. Biol. Chem. 2006, 281, 22236–22247. [Google Scholar] [CrossRef] [PubMed]
- Jirasko, V.; Montserret, R.; Lee, J.Y.; Gouttenoire, J.; Moradpour, D.; Penin, F.; Bartenschlager, R. Structural and functional studies of nonstructural protein 2 of the hepatitis C virus reveal its key role as organizer of virion assembly. PLOS Pathog. 2010, 6, e1001233. [Google Scholar] [CrossRef] [PubMed]
- Popescu, C.I.; Callens, N.; Trinel, D.; Roingeard, P.; Moradpour, D.; Descamps, V.; Duverlie, G.; Penin, F.; Heliot, L.; Rouille, Y.; et al. NS2 protein of hepatitis C virus interacts with structural and non-structural proteins towards virus assembly. PLOS Pathog. 2011, 7, e1001278. [Google Scholar]
- Tellinghuisen, T.L.; Marcotrigiano, J.; Rice, C.M. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 2005, 435, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Hwang, J.; Sharma, S.D.; Hargittai, M.R.; Chen, Y.; Arnold, J.J.; Raney, K.D.; Cameron, C.E. Hepatitis C virus nonstructural protein 5A (NS5A) is an RNA-binding protein. J. Biol. Chem. 2005, 280, 36417–36428. [Google Scholar] [CrossRef] [PubMed]
- Appel, N.; Zayas, M.; Miller, S.; Krijnse-Locker, J.; Schaller, T.; Friebe, P.; Kallis, S.; Engel, U.; Bartenschlager, R. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLOS Pathog. 2008, 4, e1000035. [Google Scholar] [CrossRef] [PubMed]
- Tanji, Y.; Kaneko, T.; Satoh, S.; Shimotohno, K. Phosphorylation of hepatitis C virus-encoded nonstructural protein NS5A. J. Virol. 1995, 69, 3980–3986. [Google Scholar] [PubMed]
- Tellinghuisen, T.L.; Foss, K.L.; Treadaway, J. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLOS Pathog. 2008, 4, e1000032. [Google Scholar] [CrossRef] [PubMed]
- Masaki, T.; Suzuki, R.; Murakami, K.; Aizaki, H.; Ishii, K.; Murayama, A.; Date, T.; Matsuura, Y.; Miyamura, T.; Wakita, T.; et al. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J. Virol. 2008, 82, 7964–7976. [Google Scholar]
- Boulant, S.; Douglas, M.W.; Moody, L.; Budkowska, A.; Targett-Adams, P.; McLauchlan, J. Hepatitis C virus core protein induces lipid droplet redistribution in a microtubule- and dynein-dependent manner. Traffic 2008, 9, 1268–1282. [Google Scholar] [CrossRef] [PubMed]
- Counihan, N.A.; Rawlinson, S.M.; Lindenbach, B.D. Trafficking of hepatitis C virus core protein during virus particle assembly. PLOS Pathog. 2011, 7, e1002302. [Google Scholar] [CrossRef] [PubMed]
- Boson, B.; Granio, O.; Bartenschlager, R.; Cosset, F.L. A concerted action of hepatitis C virus p7 and nonstructural protein 2 regulates core localization at the endoplasmic reticulum and virus assembly. PLOS Pathog. 2011, 7, e1002144. [Google Scholar] [CrossRef] [PubMed]
- Shavinskaya, A.; Boulant, S.; Penin, F.; McLauchlan, J.; Bartenschlager, R. The lipid droplet binding domain of hepatitis C virus core protein is a major determinant for efficient virus assembly. J. Biol. Chem. 2007, 282, 37158–37169. [Google Scholar] [CrossRef] [PubMed]
- Coller, K.E.; Heaton, N.S.; Berger, K.L.; Cooper, J.D.; Saunders, J.L.; Randall, G. Molecular determinants and dynamics of hepatitis C virus secretion. PLOS Pathog. 2012, 8, e1002466. [Google Scholar] [CrossRef] [PubMed]
- Herker, E.; Harris, C.; Hernandez, C.; Carpentier, A.; Kaehlcke, K.; Rosenberg, A.R.; Farese, R.V., Jr.; Ott, M. Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat. Med. 2010, 16, 1295–1298. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Pene, V.; Krishnamurthy, S.; Cha, H.; Liang, T.J. Hepatitis C virus infection activates an innate pathway involving IKK-alpha in lipogenesis and viral assembly. Nat. Med. 2013, 19, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Menzel, N.; Fischl, W.; Hueging, K.; Bankwitz, D.; Frentzen, A.; Haid, S.; Gentzsch, J.; Kaderali, L.; Bartenschlager, R.; Pietschmann, T. Map-kinase regulated cytosolic phospholipase A2 activity is essential for production of infectious hepatitis C virus particles. PLOS Pathog. 2012, 8, e1002829. [Google Scholar] [CrossRef] [PubMed]
- Nevo-Yassaf, I.; Yaffe, Y.; Asher, M.; Ravid, O.; Eizenberg, S.; Henis, Y.I.; Nahmias, Y.; Hirschberg, K.; Sklan, E.H. Role for TBC1D20 and rab1 in hepatitis C virus replication via interaction with lipid droplet-bound nonstructural protein 5A. J. Virol. 2012, 86, 6491–6502. [Google Scholar] [CrossRef] [PubMed]
- Masaki, T.; Matsunaga, S.; Takahashi, H.; Nakashima, K.; Kimura, Y.; Ito, M.; Matsuda, M.; Murayama, A.; Kato, T.; Hirano, H.; et al. Involvement of hepatitis C virus NS5A hyperphosphorylation mediated by casein kinase I-alpha in infectious virus production. J. Virol. 2014, 88, 7541–7555. [Google Scholar]
- Evans, M.J.; Rice, C.M.; Goff, S.P. Phosphorylation of hepatitis C virus nonstructural protein 5A modulates its protein interactions and viral RNA replication. Proc. Natl. Acad. Sci. USA 2004, 101, 13038–13043. [Google Scholar] [CrossRef] [PubMed]
- Salloum, S.; Wang, H.; Ferguson, C.; Parton, R.G.; Tai, A.W. Rab18 binds to hepatitis C virus NS5A and promotes interaction between sites of viral replication and lipid droplets. PLOS Pathog. 2013, 9, e1003513. [Google Scholar] [CrossRef] [PubMed]
- Camus, G.; Herker, E.; Modi, A.A.; Haas, J.T.; Ramage, H.R.; Farese, R.V., Jr.; Ott, M. Diacylglycerol acyltransferase-1 localizes hepatitis C virus NS5A protein to lipid droplets and enhances NS5A interaction with the viral capsid core. J. Biol. Chem. 2013, 288, 9915–9923. [Google Scholar] [CrossRef] [PubMed]
- Benga, W.J.; Krieger, S.E.; Dimitrova, M.; Zeisel, M.B.; Parnot, M.; Lupberger, J.; Hildt, E.; Luo, G.; McLauchlan, J.; Baumert, T.F.; et al. Apolipoprotein E interacts with hepatitis C virus nonstructural protein 5A and determines assembly of infectious particles. Hepatology 2010, 51, 43–53. [Google Scholar]
- Cun, W.; Jiang, J.; Luo, G. The C-terminal alpha-helix domain of apolipoprotein E is required for interaction with nonstructural protein 5A and assembly of hepatitis C virus. J. Virol. 2010, 84, 11532–11541. [Google Scholar] [CrossRef] [PubMed]
- Backes, P.; Quinkert, D.; Reiss, S.; Binder, M.; Zayas, M.; Rescher, U.; Gerke, V.; Bartenschlager, R.; Lohmann, V. Role of annexin A2 in the production of infectious hepatitis C virus particles. J. Virol. 2010, 84, 5775–5789. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Rice, C.M. The ins and outs of hepatitis C virus entry and assembly. Nat. Rev. MicroBiol. 2013, 11, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Anantpadma, M.; Timpe, J.M.; Shanmugam, S.; Singh, S.M.; Lemon, S.M.; Yi, M. Hepatitis C virus NS2 protein serves as a scaffold for virus assembly by interacting with both structural and nonstructural proteins. J. Virol. 2011, 85, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Stapleford, K.A.; Lindenbach, B.D. Hepatitis C virus NS2 coordinates virus particle assembly through physical interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J. Virol. 2011, 85, 1706–1717. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Matsuda, M.; Watashi, K.; Aizaki, H.; Matsuura, Y.; Wakita, T.; Suzuki, T. Signal peptidase complex subunit 1 participates in the assembly of hepatitis C virus through an interaction with E2 and NS2. PLOS Pathog. 2013, 9, e1003589. [Google Scholar] [CrossRef] [PubMed]
- Bishe, B.; Syed, G.H.; Field, S.J.; Siddiqui, A. Role of phosphatidylinositol 4-phosphate (PI4P) and its binding protein GOLPH3 in hepatitis C virus secretion. J. Biol. Chem. 2012, 287, 27637–27647. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jiang, H.; Qu, L.; Yao, W.; Cai, H.; Chen, L.; Peng, T. Hepatocyte nuclear factor 4alpha and downstream secreted phospholipase A2 GXIIB regulate production of infectious hepatitis C virus. J. Virol. 2014, 88, 612–627. [Google Scholar] [CrossRef] [PubMed]
- Parent, R.; Qu, X.; Petit, M.A.; Beretta, L. The heat shock cognate protein 70 is associated with hepatitis C virus particles and modulates virus infectivity. Hepatology 2009, 49, 1798–1809. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Sun, F.; Owen, D.M.; Li, W.; Chen, Y.; Gale, M., Jr.; Ye, J. Hepatitis c virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. USA 2007, 104, 5848–5853. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, D.; Turek, M.; Felmlee, D.J.; Girardi, E.; Pfeffer, S.; Long, G.; Bartenschlager, R.; Zeisel, M.B.; Baumert, T.F. Reconstitution of the entire hepatitis C virus life cycle in nonhepatic cells. J. Virol. 2012, 86, 11919–11925. [Google Scholar] [CrossRef] [PubMed]
- Hueging, K.; Doepke, M.; Vieyres, G.; Bankwitz, D.; Frentzen, A.; Doerrbecker, J.; Gumz, F.; Haid, S.; Wolk, B.; Kaderali, L.; et al. Apolipoprotein E codetermines tissue tropism of hepatitis C virus and is crucial for viral cell-to-cell transmission by contributing to a postenvelopment step of assembly. J. Virol. 2014, 88, 1433–1446. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popescu, C.-I.; Riva, L.; Vlaicu, O.; Farhat, R.; Rouillé, Y.; Dubuisson, J. Hepatitis C Virus Life Cycle and Lipid Metabolism. Biology 2014, 3, 892-921. https://doi.org/10.3390/biology3040892
Popescu C-I, Riva L, Vlaicu O, Farhat R, Rouillé Y, Dubuisson J. Hepatitis C Virus Life Cycle and Lipid Metabolism. Biology. 2014; 3(4):892-921. https://doi.org/10.3390/biology3040892
Chicago/Turabian StylePopescu, Costin-Ioan, Laura Riva, Ovidiu Vlaicu, Rayan Farhat, Yves Rouillé, and Jean Dubuisson. 2014. "Hepatitis C Virus Life Cycle and Lipid Metabolism" Biology 3, no. 4: 892-921. https://doi.org/10.3390/biology3040892
APA StylePopescu, C.-I., Riva, L., Vlaicu, O., Farhat, R., Rouillé, Y., & Dubuisson, J. (2014). Hepatitis C Virus Life Cycle and Lipid Metabolism. Biology, 3(4), 892-921. https://doi.org/10.3390/biology3040892