Assembly and Comparative Analysis of the Complete Mitochondrial Genome of Hippophae salicifolia

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Sequencing
2.2. Mitochondrial Genome Assembly and Annotation
2.3. Repetitive Sequence Analysis
2.4. Codon Usage Bias Analysis
2.5. RNA Editing Site Prediction
2.6. Analysis of Homologous Fragments Between Organelle Genomes
2.7. Phylogenetic Analysis
2.8. Ka/Ks Ratio Evaluation
2.9. Nucleotide Diversity (Pi) Analysis
2.10. Comparative Analysis of Mitochondrial Genome Structures
3. Results
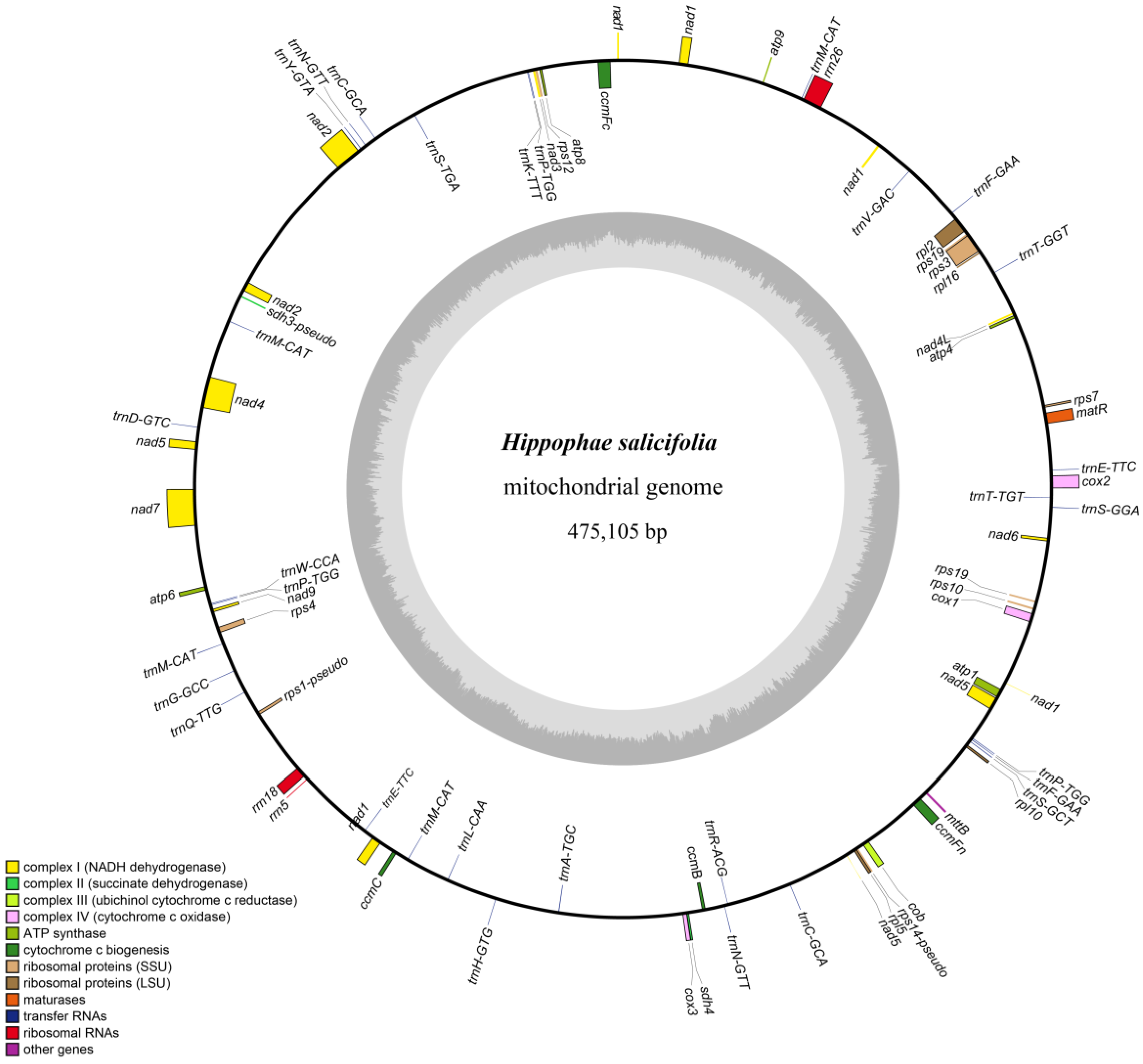
3.1. Characteristics of the Mitochondrial Genome of H. salicifolia
3.2. Analysis of Repetitive Sequences
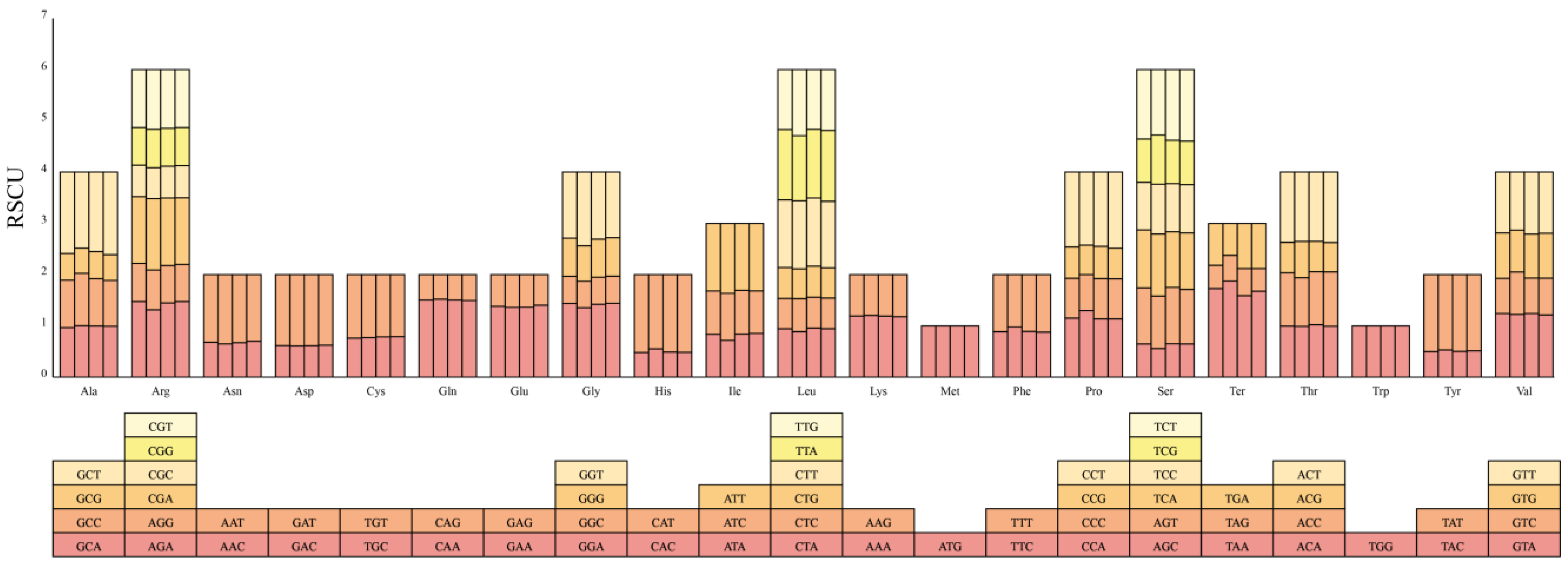
3.3. Codon Usage Analysis of Protein-Coding Genes
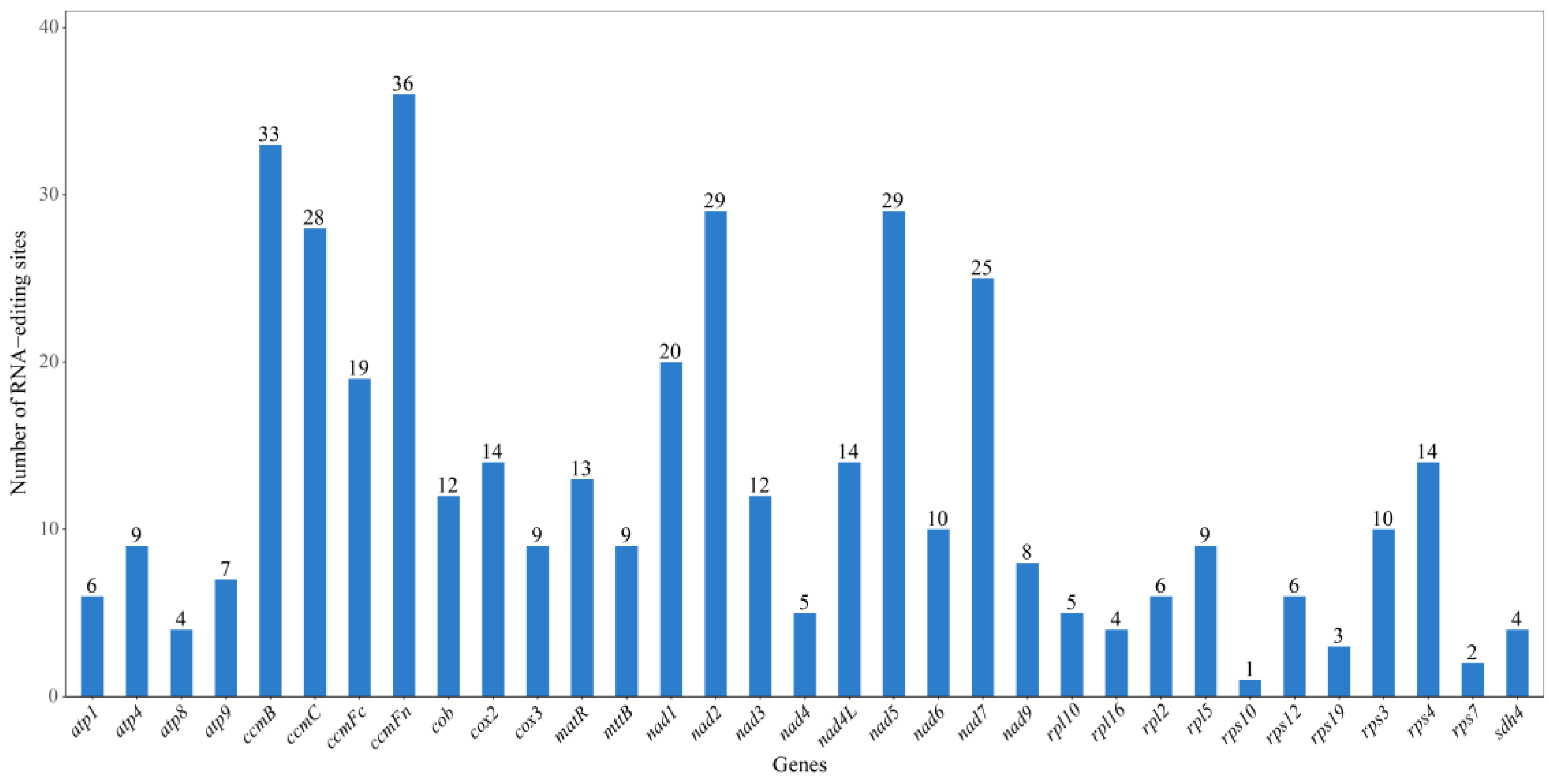
3.4. RNA Editing Prediction
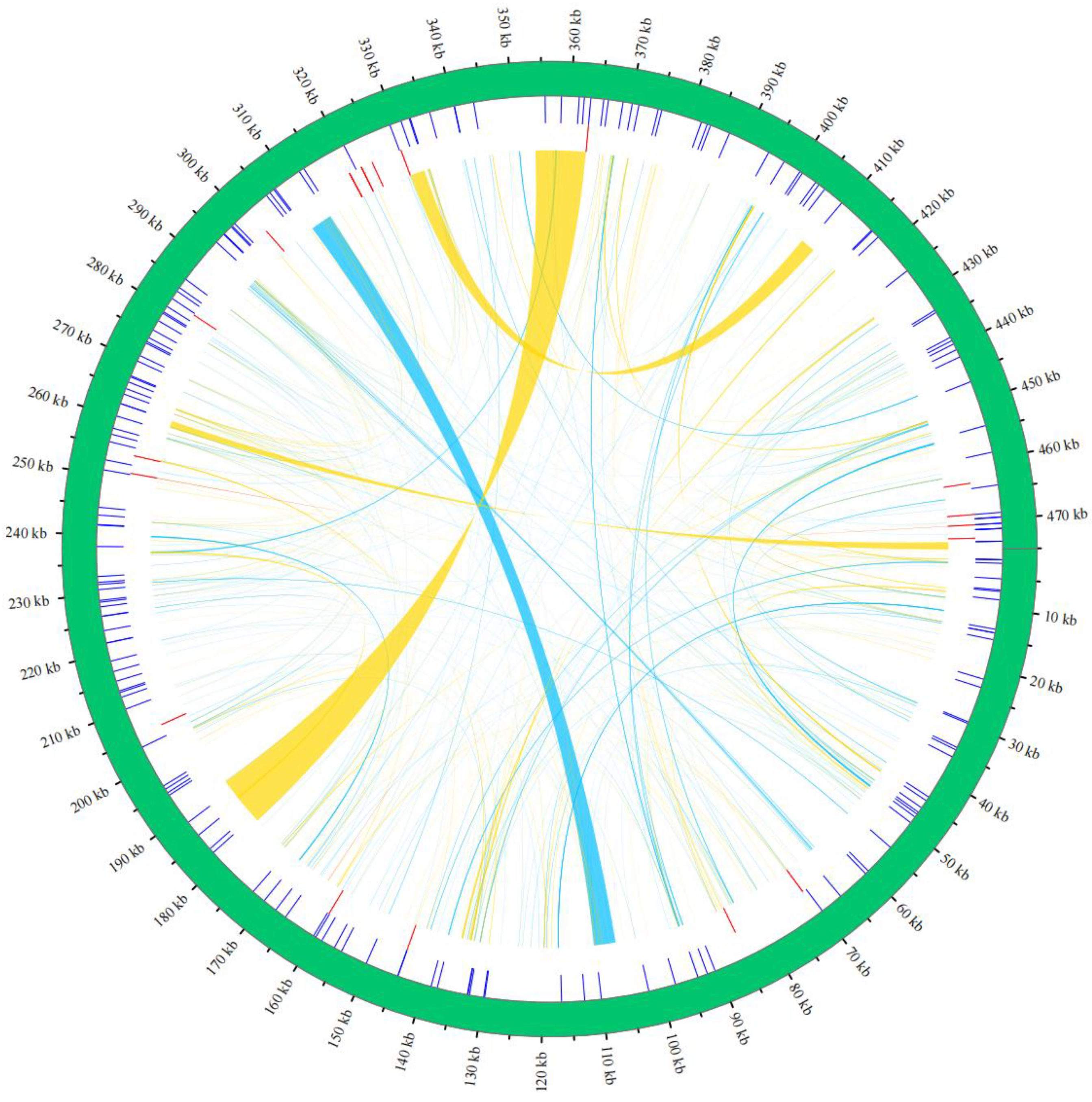
3.5. Homologous Fragment Analysis
3.6. Phylogenetic Relationships
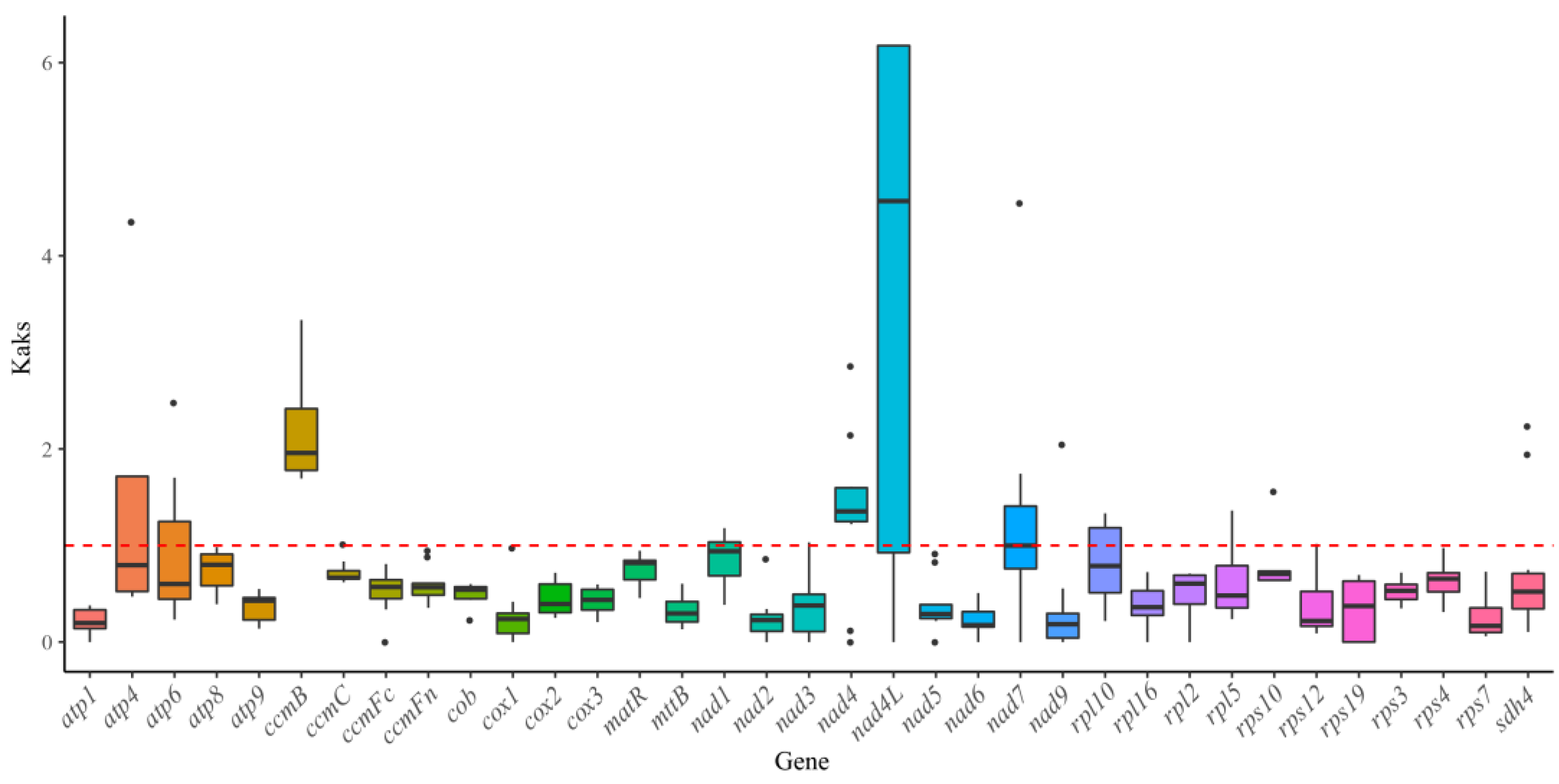
3.7. Ka/Ks Ratio Analysis
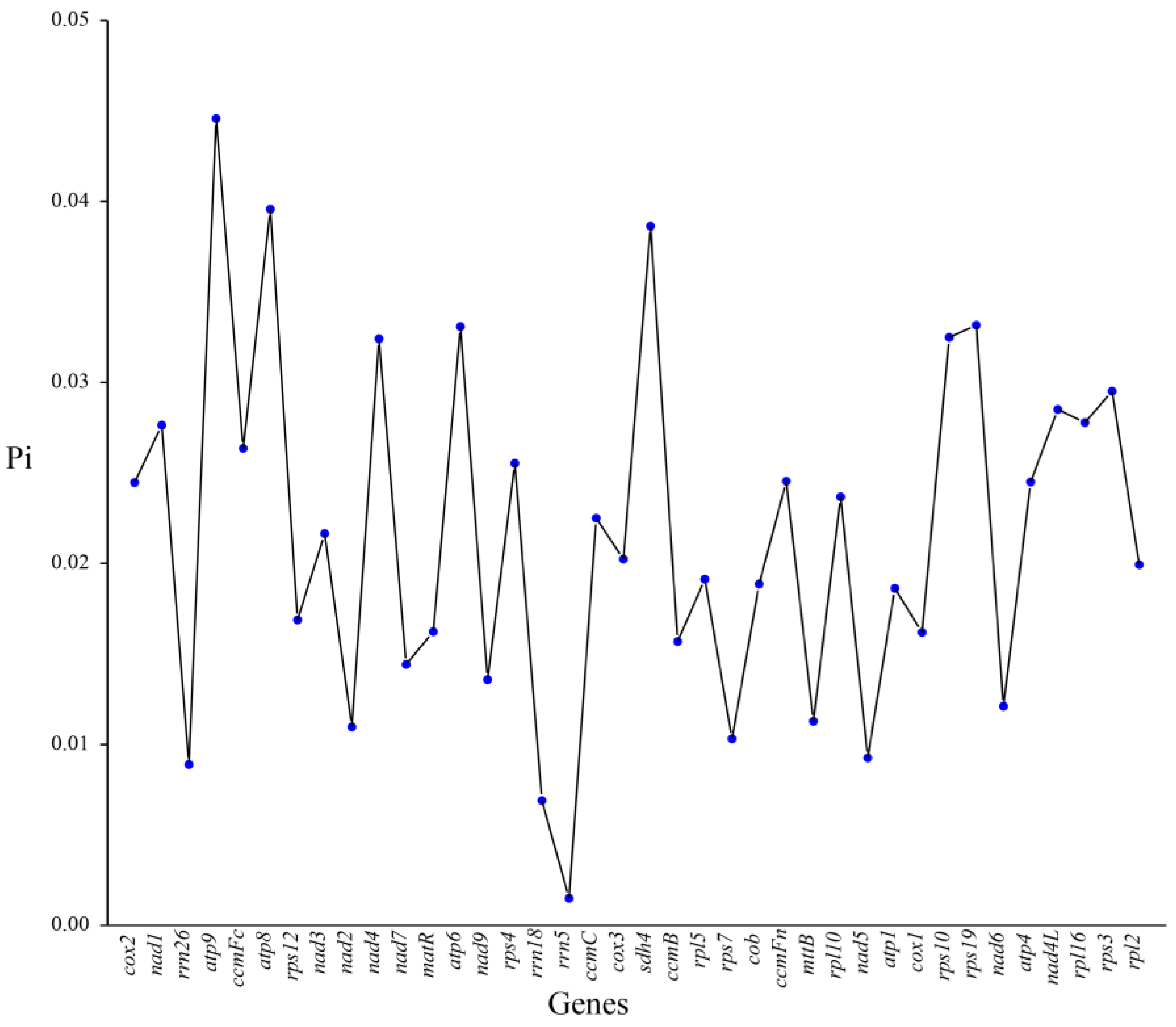
3.8. Nucleotide Diversity Analysis
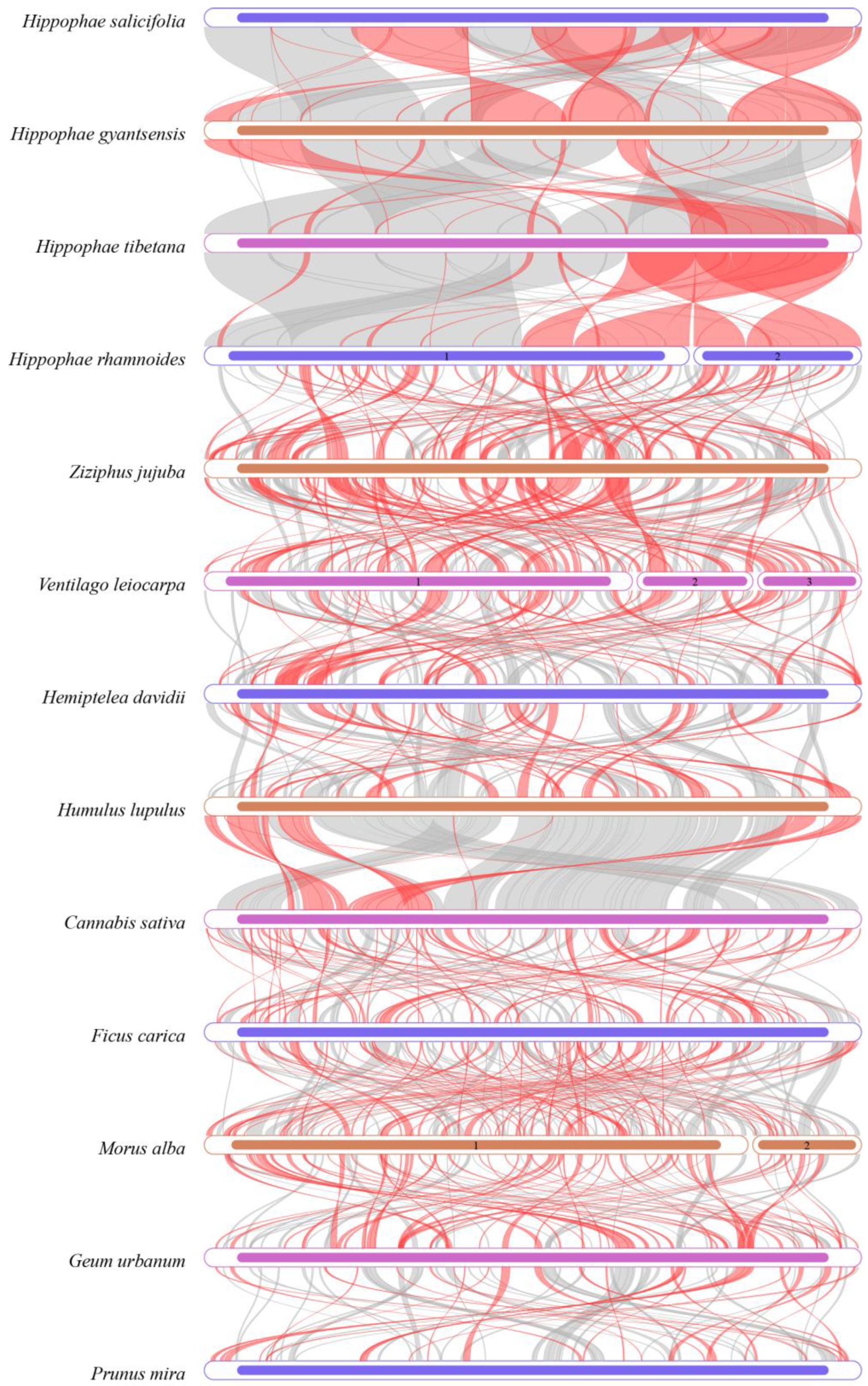
3.9. Collinearity Analysis
4. Discussion
4.1. Characterization of the H. salicifolia Mitogenome
4.2. The Role of Repetitive Sequences in Genome Structure and Evolution
4.3. Codon Usage Bias and Adaptive Evolution
4.4. Characteristics and Functional Significance of RNA Editing
4.5. Phylogenetic Relationships, Collinearity Analysis, and Organelle Gene Transfer
4.6. Ka/Ks Ratio and Nucleotide Diversity Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mackenzie, S.; McIntosh, L. Higher plant mitochondria. Plant Cell 1999, 11, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.; Hoffmeister, M.; Rotte, C.; Henze, K. An overview of endosymbiotic models for the origins of eukaryotes, their ATP-producing organelles (mitochondria and hydrogenosomes), and their heterotrophic lifestyle. J. Biol. Chem. 2001, 382, 1521–1539. [Google Scholar] [CrossRef] [PubMed]
- Barreto, P.; Koltun, A.; Nonato, J.; Yassitepe, J.; Maia, I.D.; Arruda, P. Metabolism and signaling of plant mitochondria in adaptation to environmental stresses. Int. J. Mol. Sci. 2022, 23, 11176. [Google Scholar] [CrossRef]
- Zeng, Z.; Zhang, Z.; Tso, N.; Zhang, S.; Chen, Y.; Shu, Q.; Li, J.; Liang, Z.; Wang, R.; Wang, J.; et al. Complete mitochondrial genome of Hippophae tibetana: Insights into adaptation to high-altitude environments. Front. Plant Sci. 2024, 15, 1449606. [Google Scholar] [CrossRef]
- Wynn, E.L.; Christensen, A.C. Repeats of unusual size in plant mitochondrial genomes: Identification, incidence and evolution. G3 Genes Genomes Genet. 2019, 9, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.F.; Gray, M.W.; Burger, G. Mitochondrial genome evolution and the origin of eukaryotes. Annu. Rev. Genet. 1999, 33, 351–397. [Google Scholar] [CrossRef]
- Maréchal, A.; Brisson, N. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010, 186, 299–317. [Google Scholar] [CrossRef]
- Oda, K.; Yamato, K.; Ohta, E.; Nakamura, Y.; Takemura, M.; Nozato, N.; Akashi, K.; Kanegae, T.; Ogura, Y.; Kohchi, T.; et al. Gene organization deduced from the complete sequence of liverwort Marchantia polymorpha mitochondrial DNA: A primitive form of plant mitochondrial genome. J. Mol. Biol. 1992, 223, 1–7. [Google Scholar] [CrossRef]
- Sloan, D.B.; Alverson, A.J.; Chuckalovcak, J.P.; Wu, M.; McCauley, D.E.; Palmer, J.D.; Taylor, D.R. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 2012, 10, e1001241. [Google Scholar] [CrossRef]
- Li, X.; Han, Q.; Li, M.; Luo, Q.; Zhu, S.; Zheng, Y.; Tan, G. Complete Mitochondrial Genome Sequence, Characteristics, and Phylogenetic Analysis of Oenanthe javanica. Agronomy 2023, 13, 2103. [Google Scholar] [CrossRef]
- Møller, I.M.; Rasmusson, A.G.; Van Aken, O. Plant mitochondria–past, present and future. Plant J. 2021, 108, 912–959. [Google Scholar] [CrossRef]
- Smith, D.R. Mutation rates in plastid genomes: They are lower than you might think. Genome Biol. Evol. 2015, 7, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kan, S.; Liao, X.; Zhou, J.; Tembrock, L.R.; Daniell, H.; Jin, S.; Wu, Z. Plant organellar genomes: Much done, much more to do. Trends Plant Sci. 2024, 29, 754–769. [Google Scholar] [CrossRef]
- Yuan, Y.; Bayer, P.E.; Batley, J.; Edwards, D. Improvements in genomic technologies: Application to crop genomics. Trends Biotechnol. 2017, 35, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.; Zhang, S.; Ren, P.; Liang, F.; Sun, Z.; Meng, G.; Tan, Y.; Li, X.; Lai, Q.; Han, L.; et al. Comparison of the two up-to-date sequencing technologies for genome assembly: HiFi reads of Pacific Biosciences Sequel II system and ultralong reads of Oxford Nanopore. Gigascience 2020, 9, giaa123. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Luo, Y.; Gao, L.M.; Yi, T.S.; Li, H.T.; Yang, J.B.; Li, D.Z. Phylogenomics and the flowering plant tree of life. J. Integr. Plant Biol. 2023, 65, 299–323. [Google Scholar] [CrossRef]
- Ranjana, S.; Goyal, A.; Jena, G.; Tikoo, K.; Bansal, A.K.; Singh, I.P.; Jachak, S.M. Hippophae salicifolia D. Don, a Fascinating Medicinal Plant: An Update on its Traditional Medicinal Uses, Ethnopharmacology and Phytochemistry. Curr. Tradit. Med. 2023, 9, 19–32. [Google Scholar]
- Basistha, B.C.; Sharma, N.P.; Lepcha, L.; Arrawatia, M.L.; Sen, A. Ecology of Hippophae salicifolia D. Don of temperate and sub-alpine forests of North Sikkim Himalayas—A case study. Symbiosis 2010, 50, 87–95. [Google Scholar] [CrossRef]
- Nybom, H.; Ruan, C.; Rumpunen, K. The Systematics, Reproductive Biology, Biochemistry, and Breeding of Sea Buckthorn—A Review. Genes 2023, 14, 2120. [Google Scholar] [CrossRef]
- Dhyani, D.; Maikhuri, R.K.; Dhyani, S. Seabuckthorn: An underutilized resource for the nutritional security and livelihood improvement of rural communities in Uttarakhand Himalaya. Ecol. Food Nutr. 2011, 50, 168–180. [Google Scholar] [CrossRef]
- Suryakumar, G.; Gupta, A. Medicinal and therapeutic potential of Sea buckthorn (Hippophae rhamnoides L.). J. Ethnopharmacol. 2011, 138, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wang, R.; La, Q.; Zhang, W. Characterization of the complete chloroplast genome of Hippophae salicifolia D. Don (Elaeagnaceae). Mitochondrial DNA Part B 2021, 6, 1818–1820. [Google Scholar] [CrossRef]
- Aboul-Maaty, N.A.; Oraby, H.A. Extraction of high-quality genomic DNA from different plant orders applying a modified CTAB-based method. Bull. Natl. Res. Cent. 2019, 43, 25. [Google Scholar] [CrossRef]
- Bi, C.; Shen, F.; Han, F.; Qu, Y.; Hou, J.; Xu, K.; Xu, L.A.; He, W.; Wu, Z.; Yin, T. PMAT: An efficient plant mitogenome assembly toolkit using low-coverage HiFi sequencing data. Hortic. Res. 2024, 11, uhae023. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Li, J.; Ni, Y.; Lu, Q.; Chen, H.; Liu, C. PMGA: A Plant Mitochondrial Genome Annotator. Plant Commun. 2025, 6, 101191. [Google Scholar] [CrossRef]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High speed BLASTN: An accelerated MegaBLAST search tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Lee, E.; Harris, N.; Gibson, M.; Chetty, R.; Lewis, S. Apollo: A community resource for genome annotation editing. Bioinformatics 2009, 25, 1836–1837. [Google Scholar] [CrossRef]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and high-performance computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. GPB 2010, 8, 77–80. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar]
- Ahmad, A.; Wang, J.D.; Pan, Y.B.; Sharif, R.; Gao, S.J. Development and use of simple sequence repeats (SSRs) markers for sugarcane breeding and genetic studies. Agronomy 2018, 8, 260. [Google Scholar] [CrossRef]
- Mehrotra, S.; Goyal, V. Repetitive sequences in plant nuclear DNA: Types, distribution, evolution and function. GPB 2014, 12, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Gualberto, J.M.; Mileshina, D.; Wallet, C.; Niazi, A.K.; Weber-Lotfi, F.; Dietrich, A. The plant mitochondrial genome: Dynamics and maintenance. Biochimie 2014, 100, 107–120. [Google Scholar] [CrossRef]
- Li, J.; Xu, Y.; Shan, Y.; Pei, X.; Yong, S.; Liu, C.; Yu, J. Assembly of the complete mitochondrial genome of an endemic plant, Scutellaria tsinyunensis, revealed the existence of two conformations generated by a repeat-mediated recombination. Planta 2021, 254, 36. [Google Scholar] [CrossRef]
- Small, I.D.; Schallenberg-Rüdinger, M.; Takenaka, M.; Mireau, H.; Ostersetzer-Biran, O. Plant organellar RNA editing: What 30 years of research has revealed. Plant J. 2020, 101, 1040–1056. [Google Scholar] [CrossRef] [PubMed]
- Knoop, V. C-to-U and U-to-C: RNA editing in plant organelles and beyond. J. Exp. Bot. 2023, 74, 2273–2294. [Google Scholar] [CrossRef]
- Wang, L.; Liu, X.; Xu, Y.; Zhang, Z.; Wei, Y.; Hu, Y.; Zheng, C.; Qu, X. Assembly and comparative analysis of the first complete mitochondrial genome of a traditional Chinese medicine Angelica biserrata (Shan et Yuan) Yuan et Shan. Int. J. Biol. Macromol. 2024, 257, 128571. [Google Scholar] [CrossRef]
- Jia, D.R.; Bartish, I.V. Climatic changes and orogeneses in the Late Miocene of Eurasia: The main triggers of an expansion at a continental scale? Front. Plant Sci. 2018, 9, 1400. [Google Scholar] [CrossRef]
- Angiosperm Phylogeny Group; Chase, M.W.; Christenhusz, M.J.; Fay, M.F.; Byng, J.W.; Judd, W.S.; Soltis, D.E.; Mabberley, D.J.; Sennikov, A.N.; Soltis, P.S.; et al. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 2002, 18, 486–487. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Zhang, Q.; Li, F.; Huang, J.; Zhang, M. Assembly and comparative analysis of the complete mitochondrial genome of Ilex metabaptista (Aquifoliaceae), a Chinese endemic species with a narrow distribution. BMC Plant Biol. 2023, 23, 393. [Google Scholar] [CrossRef] [PubMed]
- Dwiningsih, Y.; Rahmaningsih, M.; Alkahtani, J. Development of single nucleotide polymorphism (SNP) markers in tropical crops. Adv. Sustain. Sci. Eng. Technol. 2020, 2, 343558. [Google Scholar] [CrossRef]
- Jacquier, A.; Dujon, B. An intron-encoded protein is active in a gene conversion process that spreads an intron into a mitochondrial gene. Cell 1985, 41, 383–394. [Google Scholar] [CrossRef]
- Unseld, M.; Marienfeld, J.R.; Brandt, P.; Brennicke, A. The mitochondrial genome of Arabidopsis thaliana contains 57 genes in 366,924 nucleotides. Nat. Genet. 1997, 15, 57–61. [Google Scholar] [CrossRef]
- Hu, H.L.; Zhang, F.; Wang, P.; Lu, F.H. Evolutionary genetics of wheat mitochondrial genomes. Crop J. 2023, 11, 1774–1781. [Google Scholar] [CrossRef]
- Islam, M.S.; Studer, B.; Byrne, S.L.; Farrell, J.D.; Panitz, F.; Bendixen, C.; Møller, I.M.; Asp, T. The genome and transcriptome of perennial ryegrass mitochondria. BMC Genom. 2013, 14, 202. [Google Scholar] [CrossRef]
- Panchy, N.; Lehti-Shiu, M.; Shiu, S.H. Evolution of gene duplication in plants. Plant Physiol. 2016, 171, 2294–2316. [Google Scholar] [CrossRef]
- Qiao, Y.; Zhang, X.; Li, Z.; Song, Y.; Sun, Z. Assembly and comparative analysis of the complete mitochondrial genome of Bupleurum chinense DC. BMC Genom. 2022, 23, 664. [Google Scholar] [CrossRef]
- Balcão, V.M.; Vila, M.M. Structural and functional stabilization of protein entities: State-of-the-art. Adv. Drug Deliv. Rev. 2015, 93, 25–41. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, Z.; Mao, C.; Shang, Z.; Norbu, N.; Bonjor, N.; Jia, X.; Li, W.; Zhang, W.; Wang, J.; Qiong, L. Assembly and Comparative Analysis of the Complete Mitochondrial Genome of Hippophae salicifolia. Biology 2025, 14, 448. https://doi.org/10.3390/biology14040448
Zeng Z, Mao C, Shang Z, Norbu N, Bonjor N, Jia X, Li W, Zhang W, Wang J, Qiong L. Assembly and Comparative Analysis of the Complete Mitochondrial Genome of Hippophae salicifolia. Biology. 2025; 14(4):448. https://doi.org/10.3390/biology14040448
Chicago/Turabian StyleZeng, Zhefei, Chunmin Mao, Zhuo Shang, Ngawang Norbu, Ngawang Bonjor, Xiaoyan Jia, Wei Li, Wenju Zhang, Junwei Wang, and La Qiong. 2025. "Assembly and Comparative Analysis of the Complete Mitochondrial Genome of Hippophae salicifolia" Biology 14, no. 4: 448. https://doi.org/10.3390/biology14040448
APA StyleZeng, Z., Mao, C., Shang, Z., Norbu, N., Bonjor, N., Jia, X., Li, W., Zhang, W., Wang, J., & Qiong, L. (2025). Assembly and Comparative Analysis of the Complete Mitochondrial Genome of Hippophae salicifolia. Biology, 14(4), 448. https://doi.org/10.3390/biology14040448