Integrated Analysis of microRNA Targets Reveals New Insights into Transcriptional–Post-Transcriptional Regulatory Cross-Talk



Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Dataset Integration
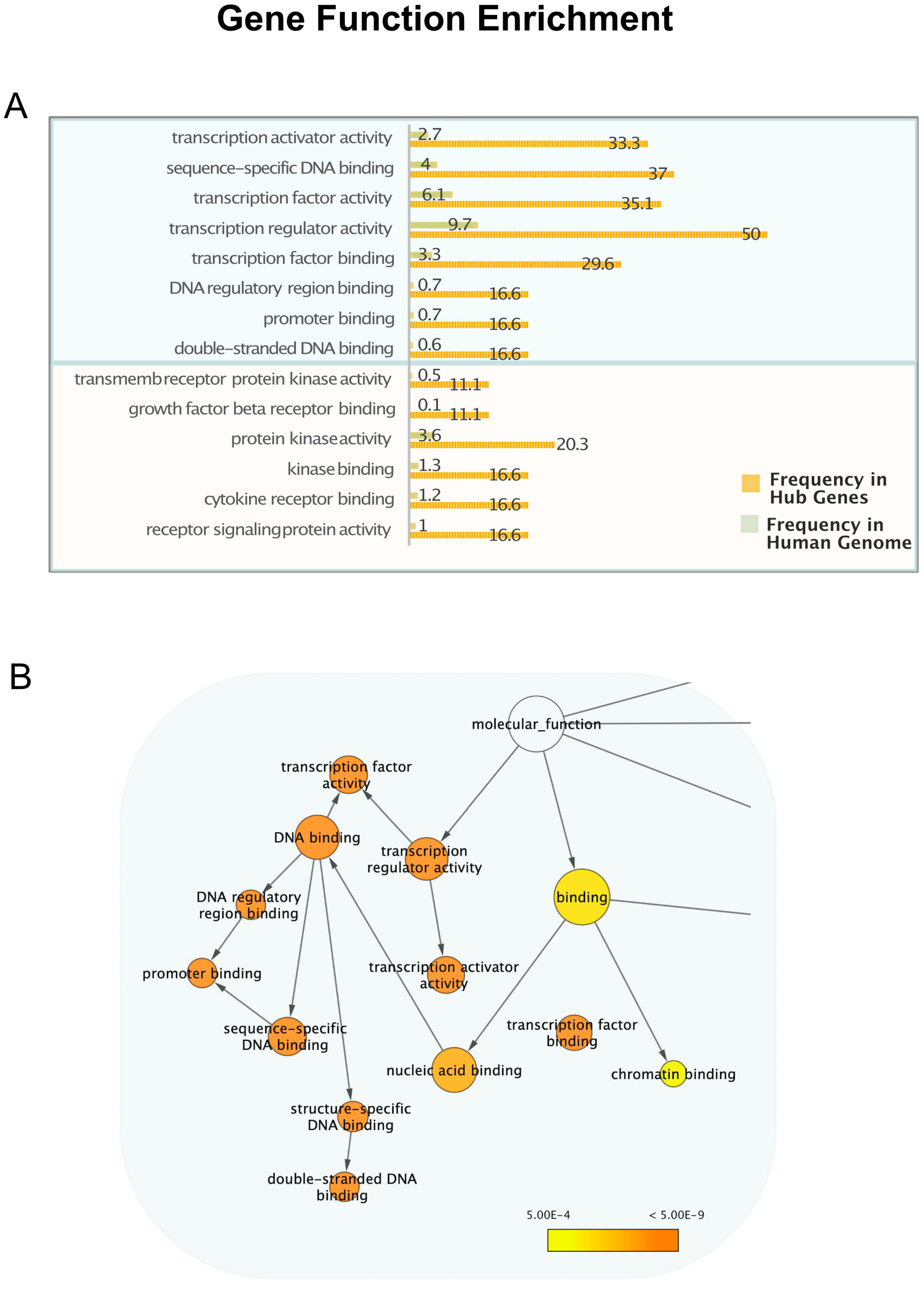
2.2. Gene Ontology and Pathway Enrichment Analysis
2.3. Other Statistical Analysis
3. Results
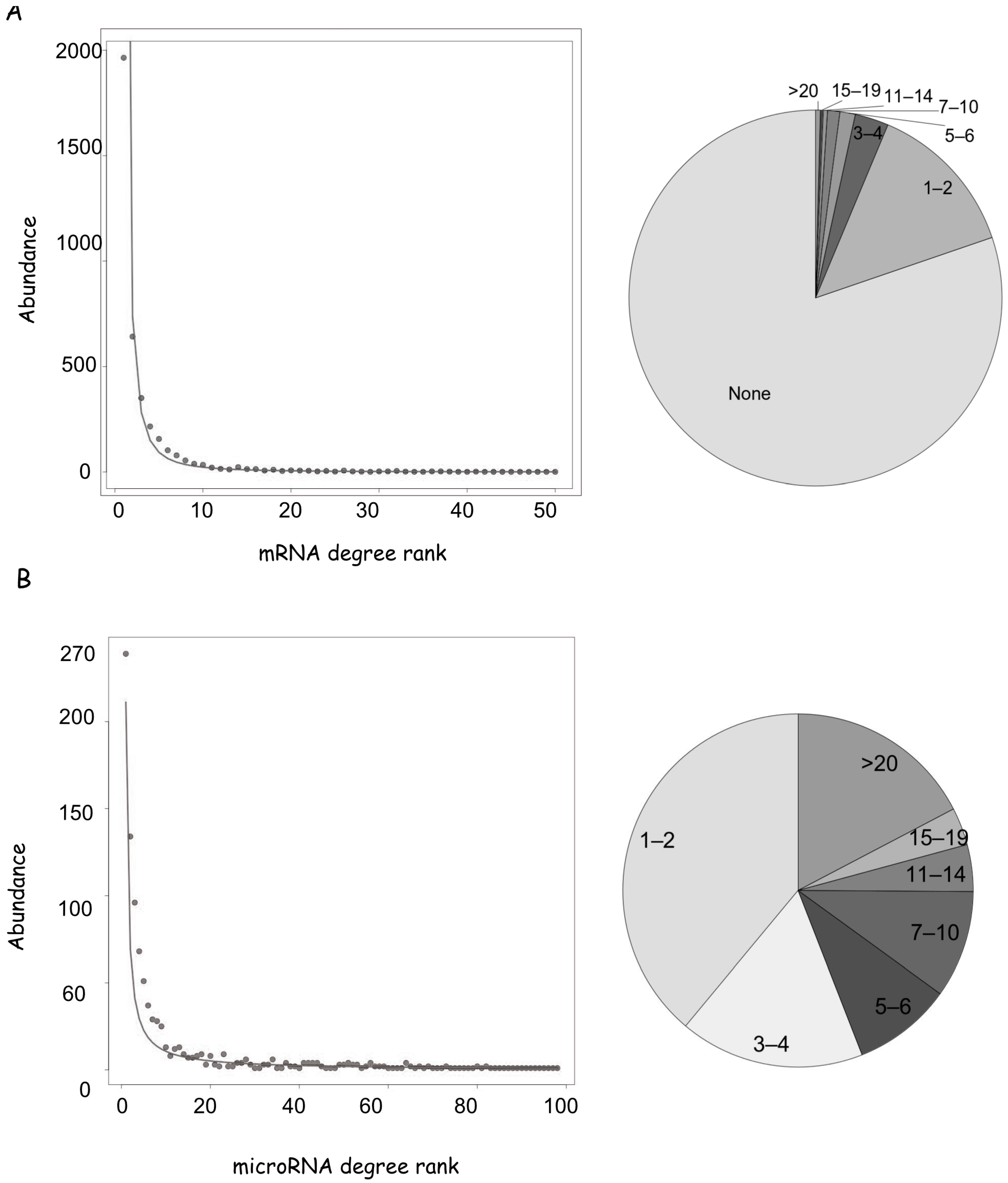
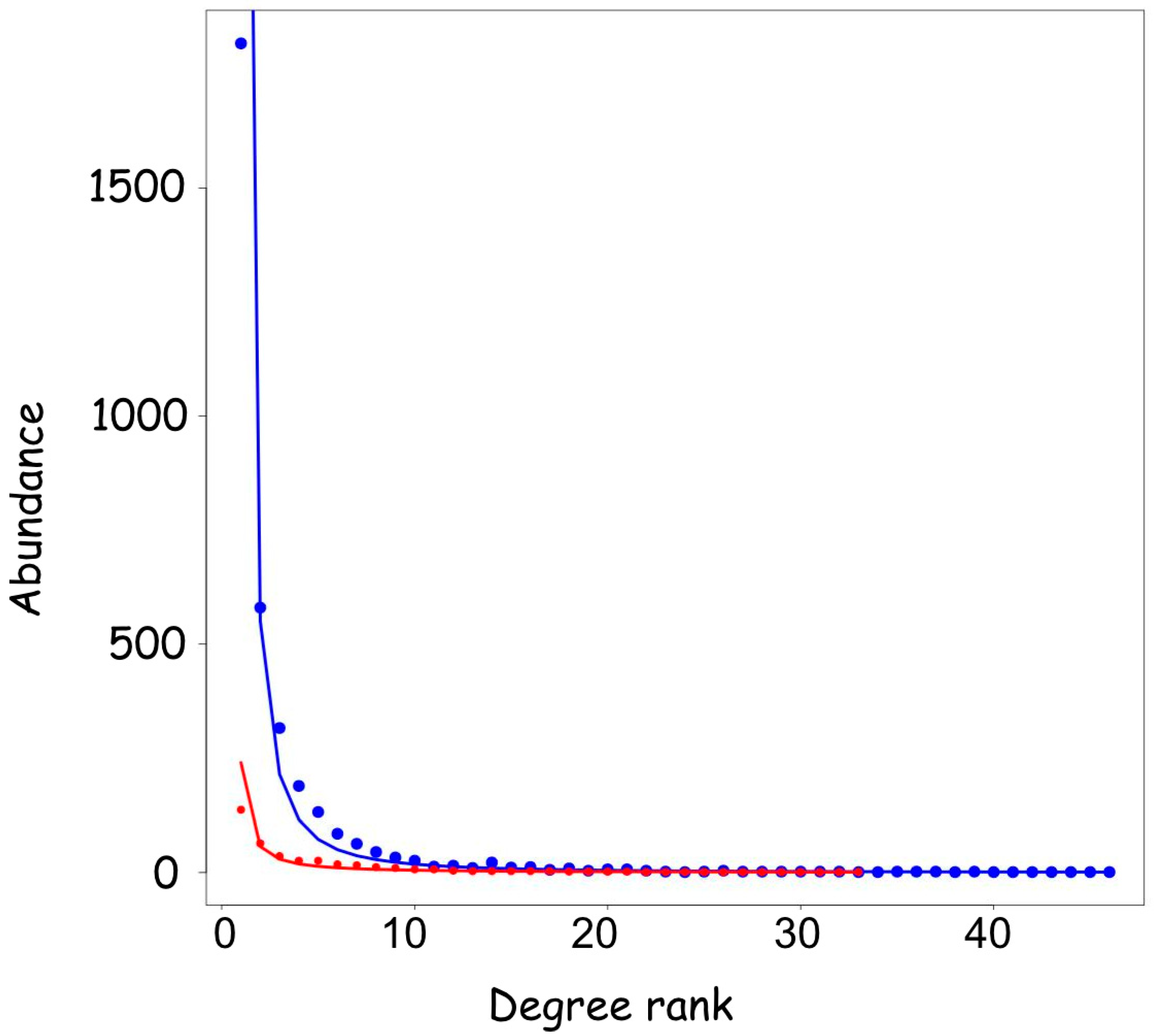
3.1. General Characteristics of the microRNA-mRNA Network
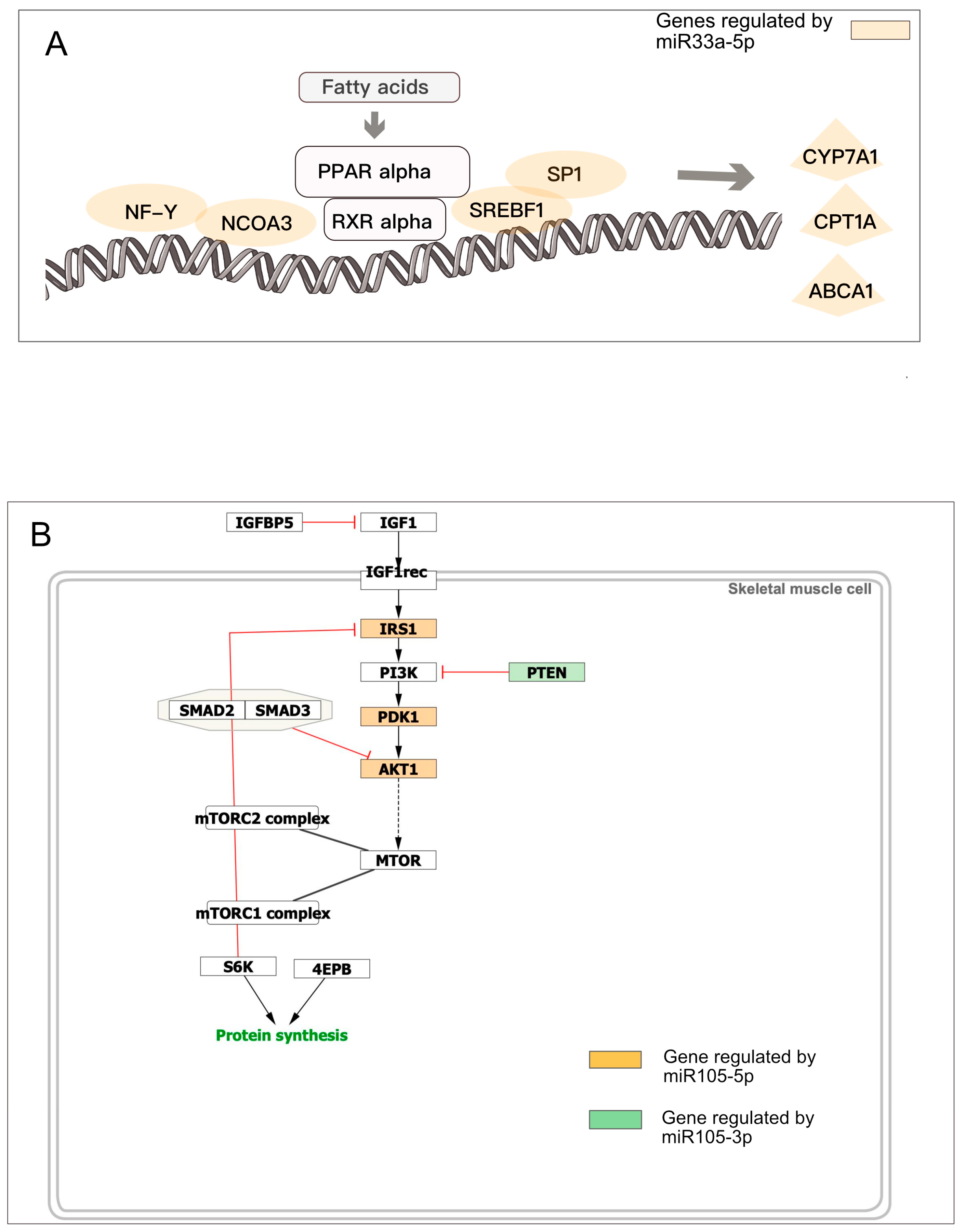
3.2. mRNA Highly Regulated Are Regulators
3.3. microRNAs Regulate Multiple Targets

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| microRNA Name | Number of Interactors | Oncogene | Tumour Suppressor | Reference |
|---|---|---|---|---|
| hsa-miR-155-5p | 262 | YES | [50] | |
| hsa-miR-21-5p | 182 | YES | [51] | |
| hsa-miR-145-5p | 171 | YES | [13] | |
| hsa-miR-34a-5p | 156 | YES | [50] | |
| hsa-miR-125b-5p | 141 | YES | [52] | |
| hsa-miR-124-3p | 138 | YES | [52] | |
| hsa-miR-29b-3p | 135 | YES | [52] | |
| hsa-miR-200c-3p | 134 | YES | [13] | |
| hsa-miR-17-5p | 131 | YES | [53] | |
| hsa-miR-29a-3p | 127 | YES | [52] | |
| hsa-miR-1-3p | 110 | YES | [52] | |
| hsa-miR-20a-5p | 107 | YES | [53] | |
| hsa-miR-9-5p | 103 | YES | [52] |
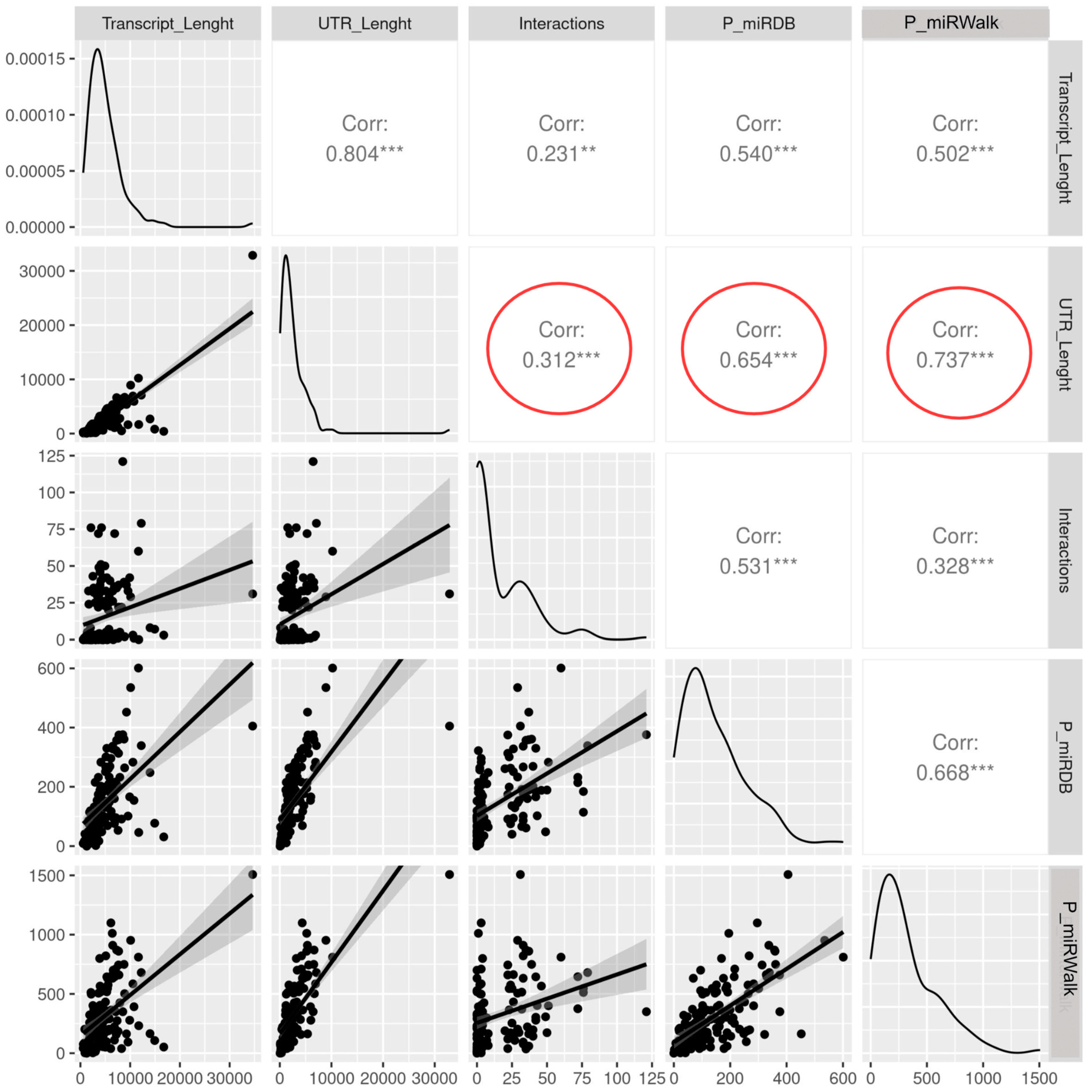
3.4. The Length of the Untranslated Regions Do Not Correlate with the Abundance of Interactors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. Elegans Heterochronic Gene Lin-4 Encodes Small RNAs with Antisense Complementarity to Lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Ambros, V.; Ruvkun, G. Recent Molecular Genetic Explorations of Caenorhabditis Elegans MicroRNAs. Genetics 2018, 209, 651–673. [Google Scholar] [CrossRef]
- Duchaine, T.F.; Fabian, M.R. Mechanistic Insights into MicroRNA-Mediated Gene Silencing. Cold Spring Harb. Perspect. Biol. 2019, 11, a032771. [Google Scholar] [CrossRef] [PubMed]
- Diener, C.; Keller, A.; Meese, E. The miRNA-Target Interactions: An Underestimated Intricacy. Nucleic Acids Res. 2024, 52, 1544–1557. [Google Scholar] [CrossRef] [PubMed]
- Quillet, A.; Anouar, Y.; Lecroq, T.; Dubessy, C. Prediction Methods for microRNA Targets in Bilaterian Animals: Toward a Better Understanding by Biologists. Comput. Struct. Biotechnol. J. 2021, 19, 5811–5825. [Google Scholar] [CrossRef]
- Schäfer, M.; Ciaudo, C. Prediction of the miRNA Interactome—Established Methods and Upcoming Perspectives. Comput. Struct. Biotechnol. J. 2020, 18, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Luna Buitrago, D.; Lovering, R.C.; Caporali, A. Insights into Online microRNA Bioinformatics Tools. Noncoding RNA 2023, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs Predominantly Act to Decrease Target mRNA Levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Shih, I.H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of Mammalian microRNA Targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Tomasello, L.; Cluts, L.; Croce, C.M. Experimental Validation of MicroRNA Targets: Luciferase Reporter Assay. Methods Mol. Biol. 2019, 1970, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Kakumani, P.K. AGO-RBP Crosstalk on Target mRNAs: Implications in miRNA-Guided Gene Silencing and Cancer. Transl. Oncol. 2022, 21, 101434. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Slack, F.J. MicroRNA Therapeutics: Towards a New Era for the Management of Cancer and Other Diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Huntley, R.P.; Kramarz, B.; Sawford, T.; Umrao, Z.; Kalea, A.; Acquaah, V.; Martin, M.J.; Mayr, M.; Lovering, R.C. Expanding the Horizons of microRNA Bioinformatics. RNA 2018, 24, 1005–1017. [Google Scholar] [CrossRef]
- Simona, P.; Panneerselvam, K.; Porras, P.; Duesbury, M.; Perfetto, L.; Licata, L.; Hermjakob, H.; Orchard, S. The Landscape of microRNA Interaction Annotation: Analysis of Three Rare Disorders as a Case Study. Database 2023, 2023, baad066. [Google Scholar] [CrossRef]
- RNAcentral Consortium. RNAcentral 2021: Secondary Structure Integration, Improved Sequence Search and New Member Databases. Nucleic Acids Res. 2021, 49, D212–D220. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.W.; Zang, J.B.; Mele, A.; Darnell, R.B. Argonaute HITS-CLIP Decodes microRNA-mRNA Interaction Maps. Nature 2009, 460, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the Human miRNA Interactome by CLASH Reveals Frequent Noncanonical Binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef]
- Hejret, V.; Varadarajan, N.M.; Klimentova, E.; Gresova, K.; Giassa, I.-C.; Vanacova, S.; Alexiou, P. Analysis of Chimeric Reads Characterises the Diverse Targetome of AGO2-Mediated Regulation. Sci. Rep. 2023, 13, 22895. [Google Scholar] [CrossRef]
- Huntley, R.P.; Sitnikov, D.; Orlic-Milacic, M.; Balakrishnan, R.; D’Eustachio, P.; Gillespie, M.E.; Howe, D.; Kalea, A.Z.; Maegdefessel, L.; Osumi-Sutherland, D.; et al. Guidelines for the Functional Annotation of microRNAs Using the Gene Ontology. RNA 2016, 22, 667–676. [Google Scholar] [CrossRef]
- Ahmed, S.H.; Deng, A.T.; Huntley, R.P.; Campbell, N.H.; Lovering, R.C. Capturing Heart Valve Development with Gene Ontology. Front. Genet. 2023, 14, 1251902. [Google Scholar] [CrossRef] [PubMed]
- Del Toro, N.; Shrivastava, A.; Ragueneau, E.; Meldal, B.; Combe, C.; Barrera, E.; Perfetto, L.; How, K.; Ratan, P.; Shirodkar, G.; et al. The IntAct Database: Efficient Access to Fine-Grained Molecular Interaction Data. Nucleic Acids Res. 2022, 50, D648–D653. [Google Scholar] [CrossRef]
- Cui, S.; Yu, S.; Huang, H.-Y.; Lin, Y.-C.-D.; Huang, Y.; Zhang, B.; Xiao, J.; Zuo, H.; Wang, J.; Li, Z.; et al. miRTarBase 2025: Updates to the Collection of Experimentally Validated microRNA–Target Interactions. Nucleic Acids Res. 2024, gkae1072. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Tang, Q.; He, J.; Li, L.; Yang, N.; Yu, S.; Wang, M.; Zhang, Y.; Lin, J.; Cui, T.; et al. RNAInter v4.0: RNA Interactome Repository with Redefined Confidence Scoring System and Improved Accessibility. Nucleic Acids Res. 2022, 50, D326–D332. [Google Scholar] [CrossRef]
- Huang, H.-Y.; Lin, Y.-C.-D.; Cui, S.; Huang, Y.; Tang, Y.; Xu, J.; Bao, J.; Li, Y.; Wen, J.; Zuo, H.; et al. miRTarBase Update 2022: An Informative Resource for Experimentally Validated miRNA-Target Interactions. Nucleic Acids Res. 2022, 50, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Binns, D.; Dimmer, E.; Huntley, R.; Barrell, D.; O’Donovan, C.; Apweiler, R. QuickGO: A Web-Based Tool for Gene Ontology Searching. Bioinformatics 2009, 25, 3045–3046. [Google Scholar] [CrossRef] [PubMed]
- Porras, P.; Barrera, E.; Bridge, A.; Del-Toro, N.; Cesareni, G.; Duesbury, M.; Hermjakob, H.; Iannuccelli, M.; Jurisica, I.; Kotlyar, M.; et al. Towards a Unified Open Access Dataset of Molecular Interactions. Nat. Commun. 2020, 11, 6144. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Lovering, R.C.; Gaudet, P.; Acencio, M.L.; Ignatchenko, A.; Jolma, A.; Fornes, O.; Kuiper, M.; Kulakovskiy, I.V.; Lægreid, A.; Martin, M.J.; et al. A GO Catalogue of Human DNA-Binding Transcription Factors. Biochim. Biophys. Acta Gene Regul. Mech. 2021, 1864, 194765. [Google Scholar] [CrossRef] [PubMed]
- Liska, O.; Bohár, B.; Hidas, A.; Korcsmáros, T.; Papp, B.; Fazekas, D.; Ari, E. TFLink: An Integrated Gateway to Access Transcription Factor-Target Gene Interactions for Multiple Species. Database 2022, 2022, baac083. [Google Scholar] [CrossRef] [PubMed]
- Prado, P.I.; Miranda, M.D.; Chalom, A. Sads: Maximum Likelihood Models for Species Abundance Distributions. R Package Version 0.6.3 2024. Available online: https://CRAN.R-Project.Org/package=Sads (accessed on 1 July 2024).
- Hausser, J.; Strimmer, K. Entropy Inference and the James-Stein Estimator, with Application to Nonlinear Gene Association Networks. J. Mach. Learn. Res. 2009, 10, 1469–1484. [Google Scholar]
- Zheng, L. Using Mutual Information as a Cocitation Similarity Measure. Scientometrics 2019, 119, 1695–1713. [Google Scholar] [CrossRef]
- Jeuken, G.S.; Käll, L. Pathway Analysis through Mutual Information. Bioinformatics 2024, 40, btad776. [Google Scholar] [CrossRef]
- Neeson, T.M.; Mandelik, Y. Pairwise Measures of Species Co-Occurrence for Choosing Indicator Species and Quantifying Overlap. Ecol. Indic. 2014, 45, 721–727. [Google Scholar] [CrossRef]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Adler, P.; Vilo, J.; Peterson, H. G: Profiler-Interoperable Web Service for Functional Enrichment Analysis and Gene Identifier Mapping (2023 Update). Nucleic Acids Res. 2023, 51, W207–W212. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.E.; Bult, C.J. Visual Annotation Display (VLAD): A Tool for Finding Functional Themes in Lists of Genes. Mamm. Genome 2015, 26, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape Plugin to Assess Overrepresentation of Gene Ontology Categories in Biological Networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef] [PubMed]
- Griss, J.; Viteri, G.; Sidiropoulos, K.; Nguyen, V.; Fabregat, A.; Hermjakob, H. ReactomeGSA—Efficient Multi-Omics Comparative Pathway Analysis. Mol. Cell. Proteom. 2020, 19, 2115–2125. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X. miRDB: An Online Database for Prediction of Functional microRNA Targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. miRWalk: An Online Resource for Prediction of microRNA Binding Sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef] [PubMed]
- Schloerke, B.; Cook, D.; Larmarange, J.; Briatte, F.; Marbach, M.; Thoen, E.; Elberg, A.; Crowley, J. GGally: Extension to ‘ggplot2’_. R Package Version 2.1.2. 2024. Available online: https://CRAN.R-Project.Org/package=GGally (accessed on 1 July 2024).
- R Core Team. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2023. [Google Scholar]
- Shalgi, R.; Lieber, D.; Oren, M.; Pilpel, Y. Global and Local Architecture of the Mammalian microRNA-Transcription Factor Regulatory Network. PLoS Comput. Biol. 2007, 3, e131. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting Effective microRNA Target Sites in Mammalian mRNAs. Elife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri-Fard, S.; Abak, A.; Shoorei, H.; Mohaqiq, M.; Majidpoor, J.; Sayad, A.; Taheri, M. Regulatory Role of microRNAs on PTEN Signaling. Biomed. Pharmacother. 2021, 133, 110986. [Google Scholar] [CrossRef] [PubMed]
- Lall, S.; Grün, D.; Krek, A.; Chen, K.; Wang, Y.-L.; Dewey, C.N.; Sood, P.; Colombo, T.; Bray, N.; MacMenamin, P.; et al. A Genome-Wide Map of Conserved MicroRNA Targets in C. Elegans. Curr. Biol. 2006, 16, 460–471. [Google Scholar] [CrossRef]
- Kehl, T.; Backes, C.; Kern, F.; Fehlmann, T.; Ludwig, N.; Meese, E.; Lenhof, H.-P.; Keller, A. About miRNAs, miRNA Seeds, Target Genes and Target Pathways. Oncotarget 2017, 8, 107167–107175. [Google Scholar] [CrossRef]
- Kim, T.; Croce, C.M. MicroRNA: Trends in Clinical Trials of Cancer Diagnosis and Therapy Strategies. Exp. Mol. Med. 2023, 55, 1314–1321. [Google Scholar] [CrossRef]
- Inoue, J.; Inazawa, J. Cancer-Associated miRNAs and Their Therapeutic Potential. J. Hum. Genet. 2021, 66, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Gambari, R.; Brognara, E.; Spandidos, D.A.; Fabbri, E. Targeting oncomiRNAs and Mimicking Tumor Suppressor miRNAs: Νew Trends in the Development of miRNA Therapeutic Strategies in Oncology (Review). Int. J. Oncol. 2016, 49, 5–32. [Google Scholar] [CrossRef]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A microRNA Polycistron as a Potential Human Oncogene. Nature 2005, 435, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Kehl, T.; Kern, F.; Backes, C.; Fehlmann, T.; Stöckel, D.; Meese, E.; Lenhof, H.-P.; Keller, A. miRPathDB 2.0: A Novel Release of the miRNA Pathway Dictionary Database. Nucleic Acids Res. 2020, 48, D142–D147. [Google Scholar] [CrossRef]
- Kern, F.; Krammes, L.; Danz, K.; Diener, C.; Kehl, T.; Küchler, O.; Fehlmann, T.; Kahraman, M.; Rheinheimer, S.; Aparicio-Puerta, E.; et al. Validation of Human microRNA Target Pathways Enables Evaluation of Target Prediction Tools. Nucleic Acids Res. 2021, 49, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Doran, G. The Short and the Long of UTRs. J. RNAi Gene Silenc. 2008, 4, 264–265. [Google Scholar]
- Mazumder, B.; Seshadri, V.; Fox, P.L. Translational Control by the 3’-UTR: The Ends Specify the Means. Trends Biochem. Sci. 2003, 28, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.-W.; Rissland, O.S.; Koppstein, D.; Abreu-Goodger, C.; Jan, C.H.; Agarwal, V.; Yildirim, M.A.; Rodriguez, A.; Bartel, D.P. Global Analyses of the Effect of Different Cellular Contexts on microRNA Targeting. Mol. Cell 2014, 53, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Varendi, K.; Kumar, A.; Härma, M.-A.; Andressoo, J.-O. miR-1, miR-10b, miR-155, and miR-191 Are Novel Regulators of BDNF. Cell. Mol. Life Sci. 2014, 71, 4443–4456. [Google Scholar] [CrossRef] [PubMed]
- Selbach, M.; Schwanhäusser, B.; Thierfelder, N.; Fang, Z.; Khanin, R.; Rajewsky, N. Widespread Changes in Protein Synthesis Induced by microRNAs. Nature 2008, 455, 58–63. [Google Scholar] [CrossRef]
- Panni, S.; Corbelli, A.; Sztuba-Solinska, J. Regulation of Non-Coding RNAs. In Navigating Non-Coding RNA; Elsevier: Amsterdam, The Netherlands, 2023; pp. 209–271. ISBN 978-0-323-90406-3. [Google Scholar]
- O’Donnell, K.A.; Wentzel, E.A.; Zeller, K.I.; Dang, C.V.; Mendell, J.T. C-Myc-Regulated microRNAs Modulate E2F1 Expression. Nature 2005, 435, 839–843. [Google Scholar] [CrossRef]
- Martinez, N.J.; Walhout, A.J.M. The Interplay between Transcription Factors and microRNAs in Genome-Scale Regulatory Networks. BioEssays 2009, 31, 435–445. [Google Scholar] [CrossRef]
- Alam, T.; Agrawal, S.; Severin, J.; Young, R.S.; Andersson, R.; Arner, E.; Hasegawa, A.; Lizio, M.; Ramilowski, J.A.; Abugessaisa, I.; et al. Comparative Transcriptomics of Primary Cells in Vertebrates. Genome Res. 2020, 30, 951–961. [Google Scholar] [CrossRef]
- Migault, M.; Sapkota, S.; Bracken, C.P. Transcriptional and Post-Transcriptional Control of Epithelial-Mesenchymal Plasticity: Why so Many Regulators? Cell. Mol. Life Sci. 2022, 79, 182. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Chiba, T.; Ozawa, R.; Yoshida, M.; Hattori, M.; Sakaki, Y. A Comprehensive Two-Hybrid Analysis to Explore the Yeast Protein Interactome. Proc. Natl. Acad. Sci. USA 2001, 98, 4569–4574. [Google Scholar] [CrossRef]
- Villaveces, J.M.; Jiménez, R.C.; Porras, P.; Del-Toro, N.; Duesbury, M.; Dumousseau, M.; Orchard, S.; Choi, H.; Ping, P.; Zong, N.C.; et al. Merging and Scoring Molecular Interactions Utilising Existing Community Standards: Tools, Use-Cases and a Case Study. Database 2015, 2015, bau131. [Google Scholar] [CrossRef] [PubMed]
- Antonazzo, G.; Gaudet, P.; Lovering, R.C.; Attrill, H. Representation of Non-Coding RNA-Mediated Regulation of Gene Expression Using the Gene Ontology. RNA Biol. 2024, 21, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, M.; Bonello, J.; Fernández-Breis, J.T.; Bucher, P.; Futschik, M.E.; Gaudet, P.; Kulakovskiy, I.V.; Licata, L.; Logie, C.; Lovering, R.C.; et al. The Gene Regulation Knowledge Commons: The Action Area of GREEKC. Biochim. Biophys. Acta Gene Regul. Mech. 2022, 1865, 194768. [Google Scholar] [CrossRef]
- Seitz, H. A New Perspective on microRNA-Guided Gene Regulation Specificity, and Its Potential Generalization to Transcription Factors and RNA-Binding Proteins. Nucleic Acids Res. 2024, 52, 9360–9368. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panni, S.; Pizzolotto, R. Integrated Analysis of microRNA Targets Reveals New Insights into Transcriptional–Post-Transcriptional Regulatory Cross-Talk. Biology 2025, 14, 43. https://doi.org/10.3390/biology14010043
Panni S, Pizzolotto R. Integrated Analysis of microRNA Targets Reveals New Insights into Transcriptional–Post-Transcriptional Regulatory Cross-Talk. Biology. 2025; 14(1):43. https://doi.org/10.3390/biology14010043
Chicago/Turabian StylePanni, Simona, and Roberto Pizzolotto. 2025. "Integrated Analysis of microRNA Targets Reveals New Insights into Transcriptional–Post-Transcriptional Regulatory Cross-Talk" Biology 14, no. 1: 43. https://doi.org/10.3390/biology14010043
APA StylePanni, S., & Pizzolotto, R. (2025). Integrated Analysis of microRNA Targets Reveals New Insights into Transcriptional–Post-Transcriptional Regulatory Cross-Talk. Biology, 14(1), 43. https://doi.org/10.3390/biology14010043