

H3 Acetylation-Induced Basal Progenitor Generation and Neocortex Expansion Depends on the Transcription Factor Pax6

 ,
,  , ,
, ,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Care and Procedures
2.2. Plasmids
2.3. Antibodies
2.4. Fluorescence-Activated Cell Sorting (FACS)
2.5. Chromatin Immunoprecipitation (ChIP)
2.6. ChIP-Seq
2.7. RNA-Sequencing
2.8. Immunohistochemistry (IHC) Experiment
2.9. qRT-PCR and WB Analyses
2.10. Luciferase Assay
2.11. Assay for Pax6 Interaction Protein (Yeast Two-Hybrid Screening, WB, coIP)
2.12. Cell Counts and Quantitative Analysis of Immunohistochemical Signal Intensity
2.13. Imaging and Statistical Analysis
3. Results
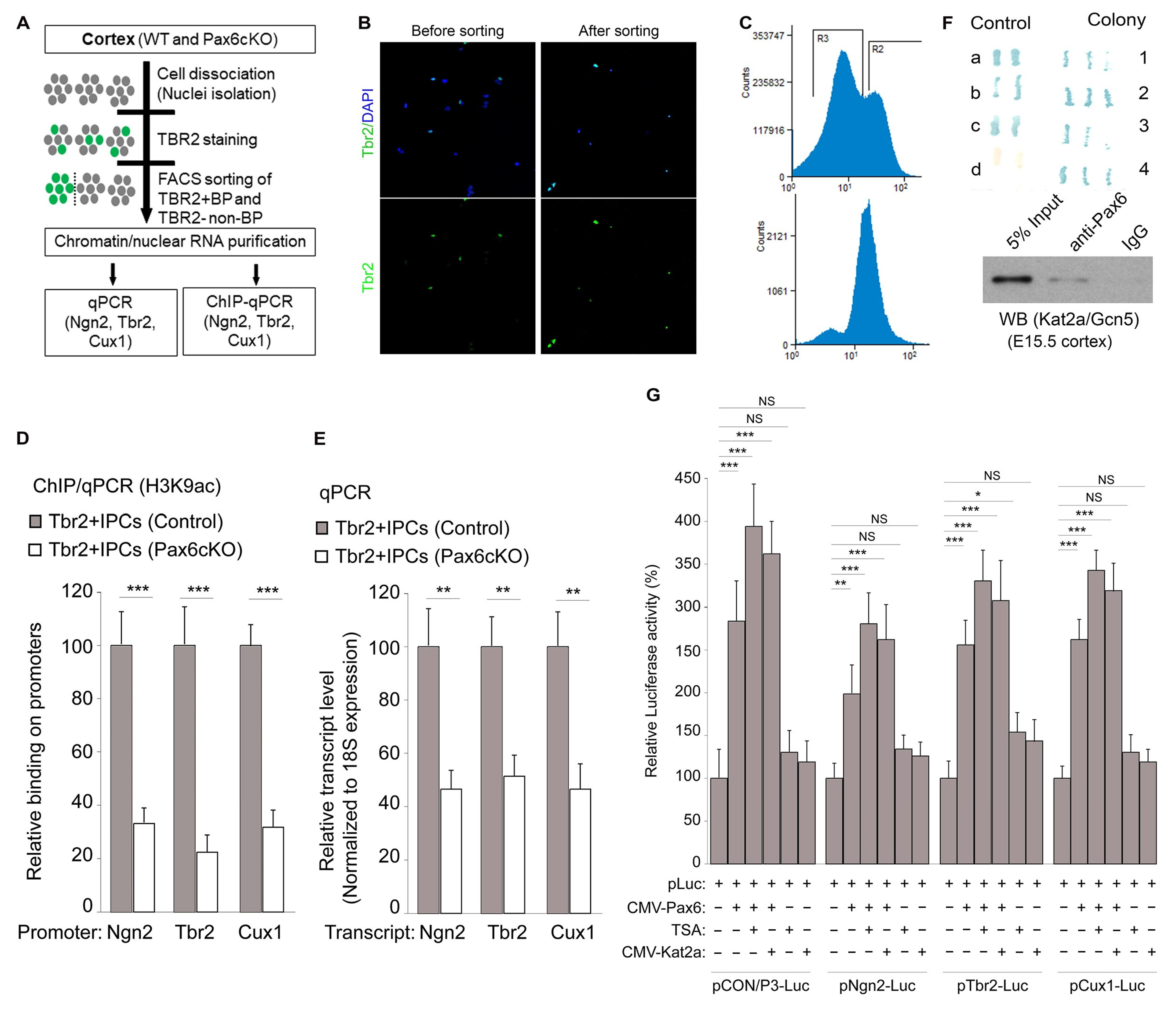
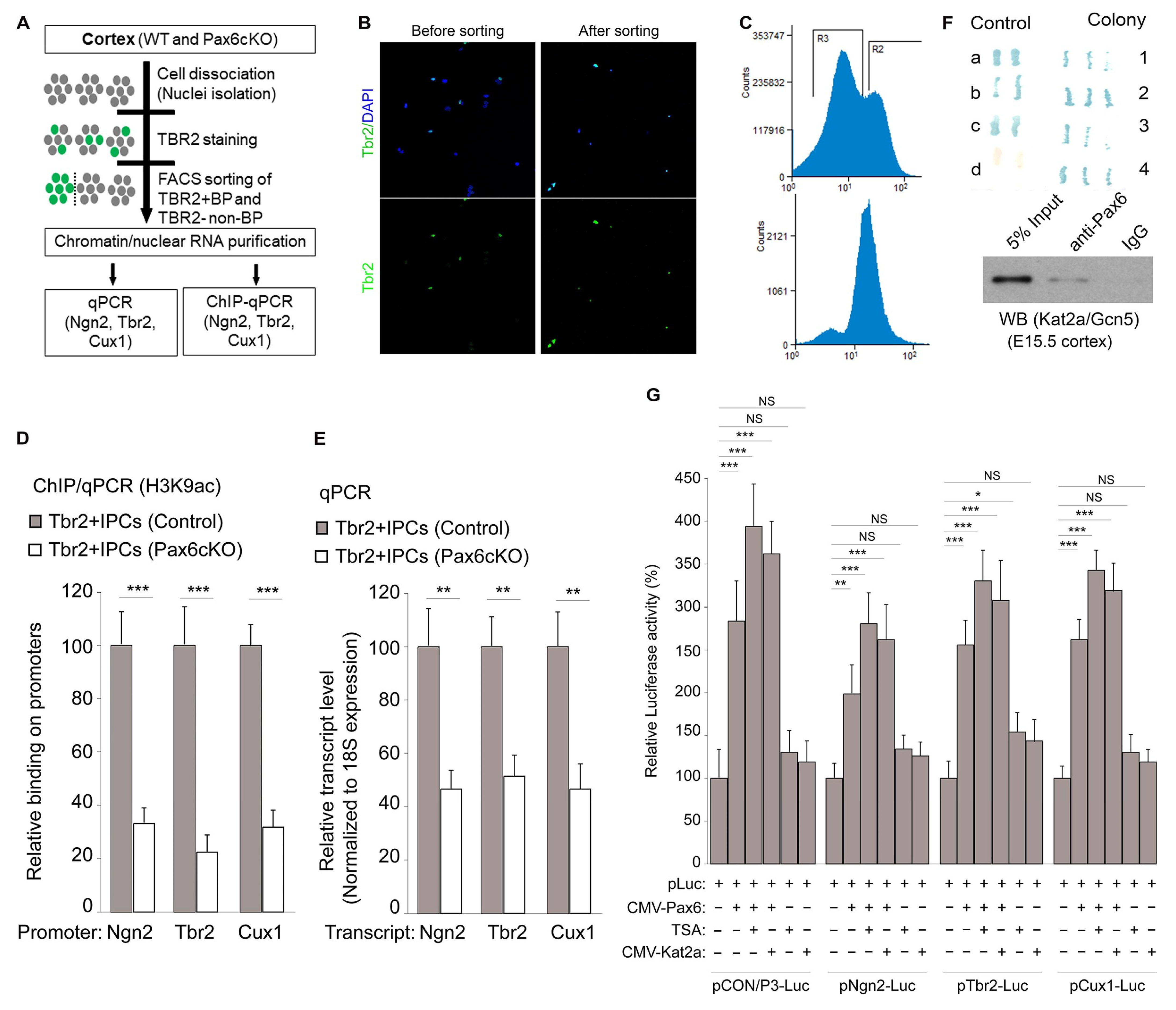
3.1. Pax6 Is Indispensable for H3 Acetylation-Induced bIPC Production
3.2. Pax6 Expression Is Required for H3 Acetylation-Induced Genesis of bRGCs
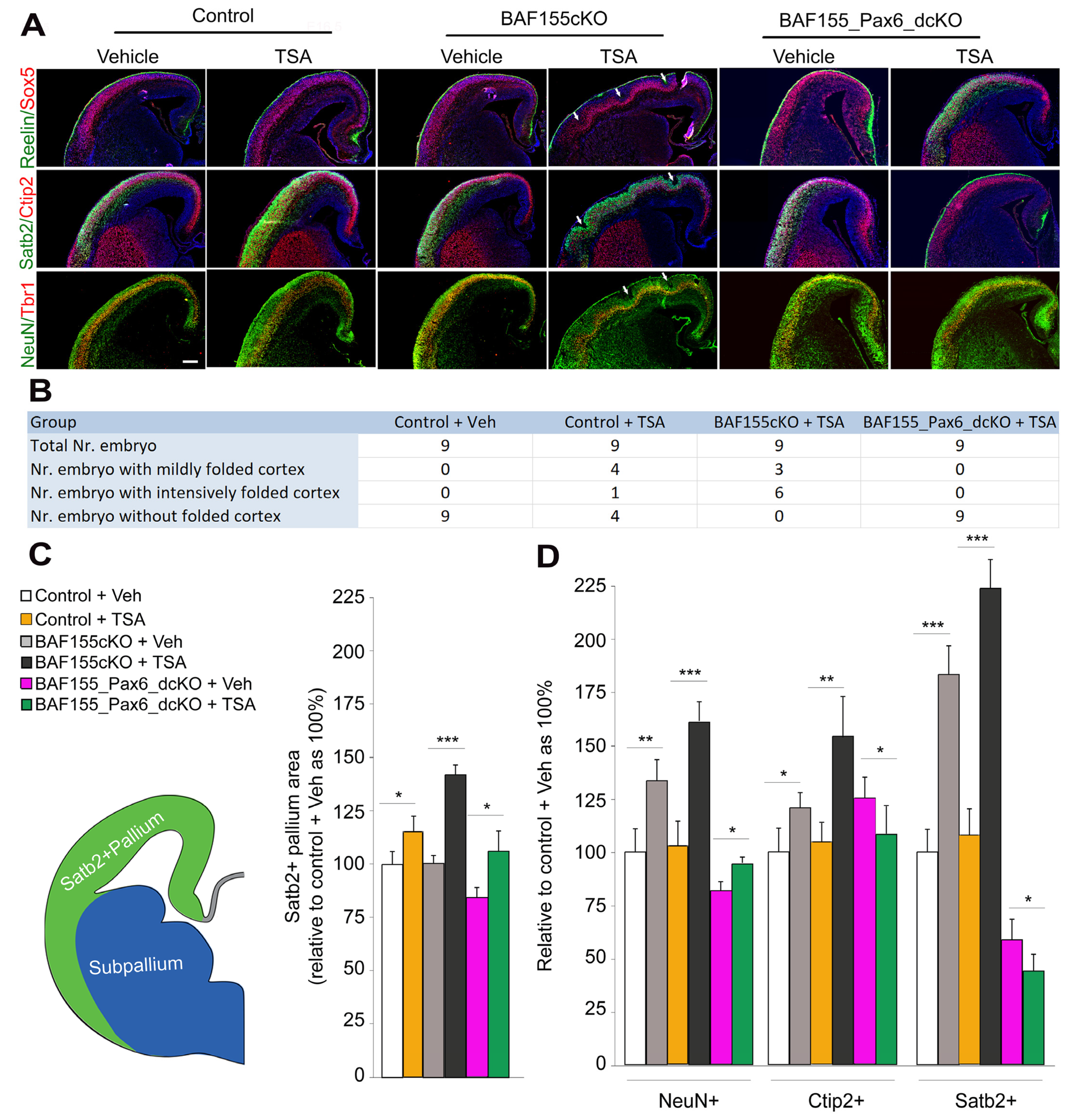
3.3. H3 Acetylation Drives Cortical Expansion and Folding in a Pax6-Dependent Manner
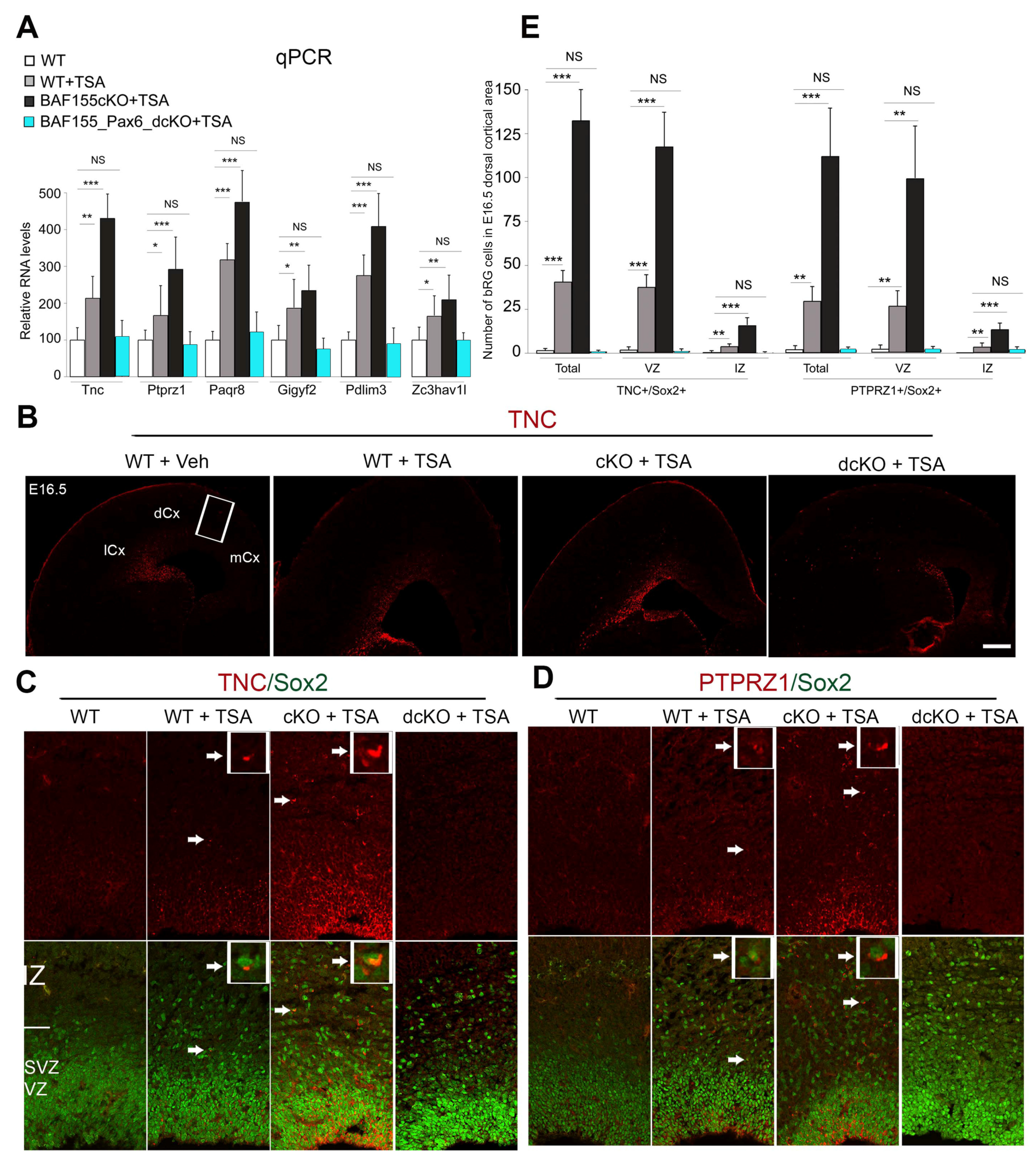
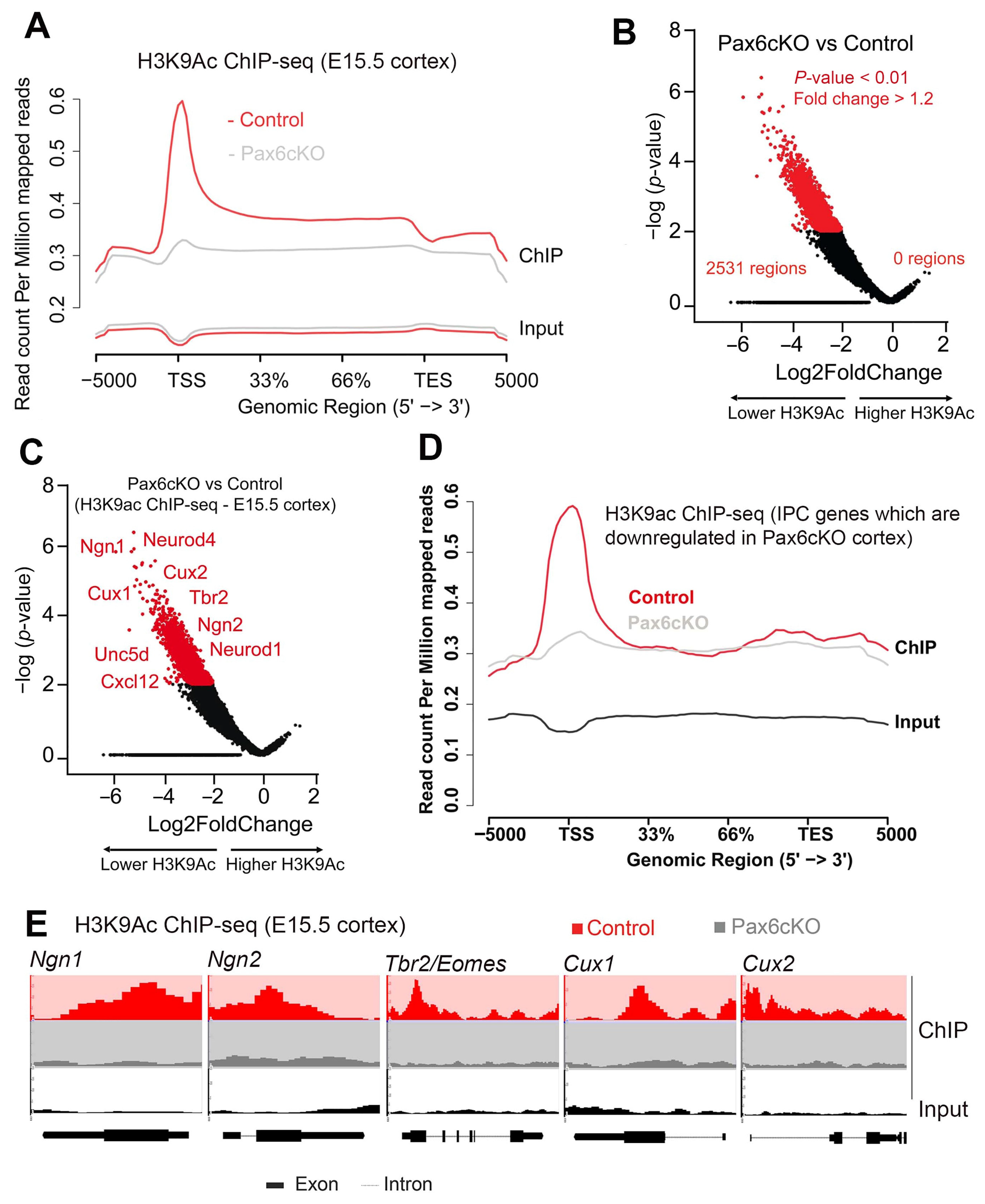
3.4. Global Impairment of the Euchromatin Mark H3K9ac and Gene Expression Programs in Pax6-Deficient Developing Cortex
3.5. Pax6 Interacts with Kat2a to Cause H3K9ac-Linked Activation of BP Genes
4. Discussion
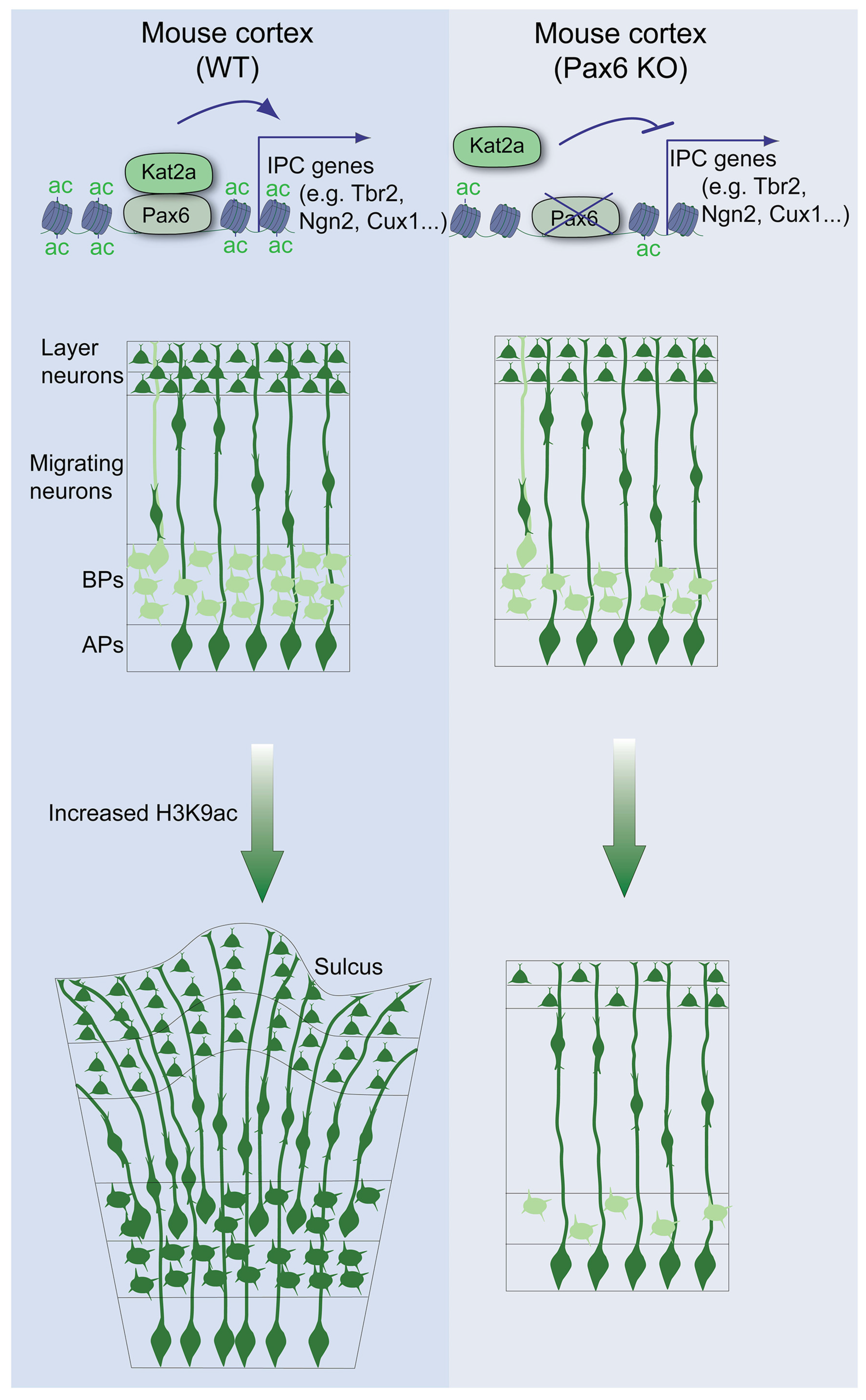
4.1. Cortical BP Pool Expansion Depends on H3 Acetylation in the Presence of Pax6
4.2. Pax6 Interacts with Kat2a to Activate H3K9ac-Dependent Gene Expression Required for BP Genesis and Cortical Expansion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taverna, E.; Gotz, M.; Huttner, W.B. The cell biology of neurogenesis: Toward an understanding of the development and evolution of the neocortex. Annu. Rev. Cell. Dev. Biol. 2014, 30, 465–502. [Google Scholar] [CrossRef] [PubMed]
- Lui, J.H.; Hansen, D.V.; Kriegstein, A.R. Development and evolution of the human neocortex. Cell 2011, 146, 18–36. [Google Scholar] [CrossRef] [PubMed]
- Dehay, C.; Kennedy, H.; Kosik, K.S. The outer subventricular zone and primate-specific cortical complexification. Neuron 2015, 85, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Borrell, V.; Gotz, M. Role of radial glial cells in cerebral cortex folding. Curr. Opin. Neurobiol. 2014, 27, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Molnar, Z.; Clowry, G.J.; Šestan, N.; Alzu’bi, A.; Bakken, T.; Hevner, R.F.; Hüppi, P.S.; Kostović, I.; Rakic, P.; Anton, E.S.; et al. New insights into the development of the human cerebral cortex. J. Anat. 2019, 235, 432–451. [Google Scholar] [CrossRef] [PubMed]
- Llinares-Benadero, C.; Borrell, V. Deconstructing cortical folding: Genetic, cellular and mechanical determinants. Nat. Rev. Neurosci. 2019, 20, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Betizeau, M.; Cortay, V.; Patti, D.; Pfister, S.; Gautier, E.; Bellemin-Menard, A.; Afanassieff, M.; Huissoud, C.; Douglas, R.J.; Kennedy, H.; et al. Precursor diversity and complexity of lineage relationships in the outer subventricular zone of the primate. Neuron 2013, 80, 442–457. [Google Scholar] [CrossRef]
- Hansen, D.V.; Lui, J.H.; Parker, P.R.; Kriegstein, A.R. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 2010, 464, 554–561. [Google Scholar] [CrossRef]
- Fietz, S.A.; Huttner, W.B. Cortical progenitor expansion, self-renewal and neurogenesis-a polarized perspective. Curr. Opin. Neurobiol. 2011, 21, 23–35. [Google Scholar] [CrossRef]
- Wang, X.; Tsai, J.W.; LaMonica, B.; Kriegstein, A.R. A new subtype of progenitor cell in the mouse embryonic neocortex. Nat. Neurosci. 2011, 14, 555–561. [Google Scholar] [CrossRef]
- Hevner, R.F. Intermediate progenitors and Tbr2 in cortical development. J. Anat. 2019, 235, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Fietz, S.A.; Kelava, I.; Vogt, J.; Wilsch-Brauninger, M.; Stenzel, D.; Fish, J.L.; Corbeil, D.; Riehn, A.; Distler, W.; Nitsch, R.; et al. OSVZ progenitors of human and ferret neocortex are epithelial-like and expand by integrin signaling. Nat. Neurosci. 2010, 13, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Espinós, A.; Fernández-Ortuño, E.; Negri, E.; Borrell, V. Evolution of genetic mechanisms regulating cortical neurogenesis. Dev. Neurobiol. 2022, 82, 428–453. [Google Scholar] [CrossRef] [PubMed]
- Manuel, M.N.; Mi, D.; Mason, J.O.; Price, D.J. Regulation of cerebral cortical neurogenesis by the Pax6 transcription factor. Front. Cell. Neurosci. 2015, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.C.; Molinek, M.; Martynoga, B.S.; Zaki, P.A.; Faedo, A.; Bulfone, A.; Hevner, R.F.; West, J.D.; Price, D.J. Pax6 controls cerebral cortical cell number by regulating exit from the cell cycle and specifies cortical cell identity by a cell autonomous mechanism. Dev. Biol. 2006, 22, 22. [Google Scholar] [CrossRef] [PubMed]
- Sansom, S.N.; Griffiths, D.S.; Faedo, A.; Kleinjan, D.J.; Ruan, Y.; Smith, J.; van Heyningen, V.; Rubenstein, J.L.; Livesey, F.J. The level of the transcription factor Pax6 is essential for controlling the balance between neural stem cell self-renewal and neurogenesis. PLoS Genet. 2009, 5, e1000511. [Google Scholar] [CrossRef] [PubMed]
- Georgala, P.A.; Manuel, M.; Price, D.J. The generation of superficial cortical layers is regulated by levels of the transcription factor Pax6. Cereb. Cortex 2011, 21, 81–94. [Google Scholar] [CrossRef]
- Gotz, M.; Stoykova, A.; Gruss, P. Pax6 controls radial glia differentiation in the cerebral cortex. Neuron 1998, 21, 1031–1044. [Google Scholar] [CrossRef]
- Asami, M.; Pilz, G.A.; Ninkovic, J.; Godinho, L.; Schroeder, T.; Huttner, W.B.; Götz, M. The role of Pax6 in regulating the orientation and mode of cell division of progenitors in the mouse cerebral cortex. Development 2011, 138, 5067–5078. [Google Scholar] [CrossRef]
- Heins, N.; Malatesta, P.; Cecconi, F.; Nakafuku, M.; Tucker, K.L.; Hack, M.A.; Chapouton, P.; Barde, Y.A.; Gotz, M. Glial cells generate neurons: The role of the transcription factor Pax6. Nat. Neurosci. 2002, 5, 308–315. [Google Scholar] [CrossRef]
- Narayanan, R.; Pham, L.; Kerimoglu, C.; Watanabe, T.; Castro Hernandez, R.; Sokpor, G.; Ulmke, P.A.; Kiszka, K.A.; Tonchev, A.B.; Rosenbusch, J.; et al. Chromatin Remodeling BAF155 Subunit Regulates the Genesis of Basal Progenitors in Developing Cortex. iScience 2018, 4, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Tuoc, T.C.; Radyushkin, K.; Tonchev, A.B.; Pinon, M.C.; Ashery-Padan, R.; Molnar, Z.; Davidoff, M.S.; Stoykova, A. Selective cortical layering abnormalities and behavioral deficits in cortex-specific Pax6 knock-out mice. J. Neurosci. 2009, 29, 8335–8349. [Google Scholar] [CrossRef] [PubMed]
- Wong, F.K.; Fei, J.F.; Mora-Bermudez, F.; Taverna, E.; Haffner, C.; Fu, J.; Anastassiadis, K.; Stewart, A.F.; Huttner, W.B. Sustained Pax6 Expression Generates Primate-like Basal Radial Glia in Developing Mouse Neocortex. PLoS Biol. 2015, 13, e1002217. [Google Scholar] [CrossRef] [PubMed]
- Holm, P.C.; Mader, M.T.; Haubst, N.; Wizenmann, A.; Sigvardsson, M.; Gotz, M. Loss- and gain-of-function analyses reveal targets of Pax6 in the developing mouse telencephalon. Mol. Cell Neurosci. 2007, 34, 99–119. [Google Scholar] [CrossRef] [PubMed]
- Scardigli, R.; Bäumer, N.; Gruss, P.; Guillemot, F.; Le Roux, I. Direct and concentration-dependent regulation of the proneural gene Neurogenin2 by Pax6. Development 2003, 130, 3269–3281. [Google Scholar] [CrossRef] [PubMed]
- Ninkovic, J.; Steiner-Mezzadri, A.; Jawerka, M.; Akinci, U.; Masserdotti, G.; Petricca, S.; Fischer, J.; von Holst, A.; Beckers, J.; Lie, C.D.; et al. The BAF Complex Interacts with Pax6 in Adult Neural Progenitors to Establish a Neurogenic Cross-Regulatory Transcriptional Network. Cell Stem Cell 2013, 13, 403–418. [Google Scholar] [CrossRef]
- Xie, Y.; Castro-Hernández, R.; Sokpor, G.; Pham, L.; Narayanan, R.; Rosenbusch, J.; Staiger, J.F.; Tuoc, T. RBM15 Modulates the Function of Chromatin Remodeling Factor BAF155 Through RNA Methylation in Developing Cortex. Mol. Neurobiol. 2019, 56, 7305–7320. [Google Scholar] [CrossRef]
- Kerimoglu, C.; Pham, L.; Tonchev, A.B.; Sakib, M.S.; Xie, Y.; Sokpor, G.; Ulmke, P.A.; Kaurani, L.; Abbas, E.; Nguyen, H.; et al. H3 acetylation selectively promotes basal progenitor proliferation and neocortex expansion. Sci. Adv. 2021, 7, eabc6792. [Google Scholar] [CrossRef]
- Ashery-Padan, R.; Marquardt, T.; Zhou, X.; Gruss, P. Pax6 activity in the lens primordium is required for lens formation and for correct placement of a single retina in the eye. Genes Dev. 2000, 14, 2701–2711. [Google Scholar] [CrossRef]
- Gorski, J.A.; Talley, T.; Qiu, M.; Puelles, L.; Rubenstein, J.L.; Jones, K.R. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J. Neurosci. 2002, 22, 6309–6314. [Google Scholar] [CrossRef]
- Choi, J.; Ko, M.; Jeon, S.; Jeon, Y.; Park, K.; Lee, C.; Lee, H.; Seong, R.H. The SWI/SNF-like BAF complex is essential for early B cell development. J. Immunol. 2012, 188, 3791–3803. [Google Scholar] [CrossRef] [PubMed]
- Tuoc, T.C.; Stoykova, A. Trim11 modulates the function of neurogenic transcription factor Pax6 through ubiquitin-proteosome system. Genes Dev. 2008, 22, 1972–1986. [Google Scholar] [CrossRef] [PubMed]
- Sokpor, G.; Kerimoglu, C.; Nguyen, H.; Pham, L.; Rosenbusch, J.; Wagener, R.; Nguyen, H.P.; Fischer, A.; Staiger, J.F.; Tuoc, T. Loss of BAF Complex in Developing Cortex Perturbs Radial Neuronal Migration in a WNT Signaling-Dependent Manner. Front. Mol. Neurosci. 2021, 14, 687581. [Google Scholar] [CrossRef] [PubMed]
- Tuoc, T.C.; Boretius, S.; Sansom, S.N.; Pitulescu, M.E.; Frahm, J.; Livesey, F.J.; Stoykova, A. Chromatin regulation by BAF170 controls cerebral cortical size and thickness. Dev. Cell. 2013, 25, 256–269. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, W.; Nakatani, S.; Takahara, T.; Kainuma, M.; Masaoka, M.; Minobe, S.; Namihira, M.; Nakashima, K.; Sakakibara, A.; Ogawa, M.; et al. Periventricular notch activation and asymmetric Ngn2 and Tbr2 expression in pair-generated neocortical daughter cells. Mol. Cell. Neurosci. 2009, 40, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Sakib, M.S.; Sokpor, G.; Nguyen, H.P.; Fischer, A.; Tuoc, T. Intranuclear immunostaining-based FACS protocol from embryonic cortical tissue. STAR Protoc. 2021, 2, 100318. [Google Scholar] [CrossRef]
- Ulmke, P.A.; Sakib, M.S.; Ditte, P.; Sokpor, G.; Kerimoglu, C.; Pham, L.; Xie, Y.; Mao, X.; Rosenbusch, J.; Teichmann, U.; et al. Molecular Profiling Reveals Involvement of ESCO2 in Intermediate Progenitor Cell Maintenance in the Developing Mouse Cortex. Stem Cell Rep. 2021, 16, 968–984. [Google Scholar] [CrossRef]
- Narayanan, R.; Pirouz, M.; Kerimoglu, C.; Pham, L.; Wagener, R.J.; Kiszka, K.A.; Rosenbusch, J.; Seong, R.H.; Kessel, M.; Fischer, A.; et al. Loss of BAF (mSWI/SNF) Complexes Causes Global Transcriptional and Chromatin State Changes in Forebrain Development. Cell Rep. 2015, 13, 1842–1854. [Google Scholar] [CrossRef]
- Stilling, R.M.; Ronicke, R.; Benito, E.; Urbanke, H.; Capece, V.; Burkhardt, S.; Bahari-Javan, S.; Barth, J.; Sananbenesi, F.; Schutz, A.L.; et al. K-Lysine acetyltransferase 2a regulates a hippocampal gene expression network linked to memory formation. EMBO J. 2014, 33, 1912–1927. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup; et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Shao, N.; Liu, X.; Nestler, E. ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genom. 2014, 15, 284. [Google Scholar] [CrossRef]
- Nicol, J.W.; Helt, G.A.; Blanchard, S.G., Jr.; Raja, A.; Loraine, A.E. The Integrated Genome Browser: Free software for distribution and exploration of genome-scale datasets. Bioinformatics 2009, 25, 2730–2731. [Google Scholar] [CrossRef]
- Lienhard, M.; Grimm, C.; Morkel, M.; Herwig, R.; Chavez, L. MEDIPS: Genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics 2014, 30, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Liu, T.; Zhang, Y. Using MACS to identify peaks from ChIP-Seq data. Curr. Protoc. Bioinform. 2011, 34, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Ross-Innes, C.S.; Stark, R.; Teschendorff, A.E.; Holmes, K.A.; Ali, H.R.; Dunning, M.J.; Brown, G.D.; Gojis, O.; Ellis, I.O.; Green, A.R.; et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012, 481, 389–393. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K.; et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 2014, 15, 1. [Google Scholar] [CrossRef]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef]
- Jin, Q.; Yu, L.R.; Wang, L.; Zhang, Z.; Kasper, L.H.; Lee, J.E.; Wang, C.; Brindle, P.K.; Dent, S.Y.; Ge, K. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011, 30, 249–262. [Google Scholar] [CrossRef]
- Tjeertes, J.V.; Miller, K.M.; Jackson, S.P. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J. 2009, 28, 1878–1889. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Zhuang, L.; Lai, B.; Wang, C.; Li, W.; Dolan, B.; Lu, Y.; Wang, Z.; Zhao, K.; Peng, W.; et al. Gcn5 and PCAF negatively regulate interferon-beta production through HAT-independent inhibition of TBK1. EMBO Rep. 2014, 15, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, C.; Nguyen, H.; Rosenbusch, J.; Pham, L.; Rabe, T.; Patwa, M.; Sokpor, G.; Seong, R.H.; Ashery-Padan, R.; Mansouri, A.; et al. mSWI/SNF (BAF) Complexes Are Indispensable for the Neurogenesis and Development of Embryonic Olfactory Epithelium. PLoS Genet. 2016, 12, e1006274. [Google Scholar] [CrossRef] [PubMed]
- Englund, C.; Fink, A.; Lau, C.; Pham, D.; Daza, R.A.; Bulfone, A.; Kowalczyk, T.; Hevner, R.F. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J. Neurosci. 2005, 25, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.; Zecevic, N. Is Pax6 critical for neurogenesis in the human fetal brain? Cereb. Cortex 2008, 18, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xi, J.; Wang, G.; Guo, Z.; Sun, Q.; Lu, C.; Ma, L.; Wu, Y.; Jia, W.; Zhu, S.; et al. PAUPAR and PAX6 sequentially regulate human embryonic stem cell cortical differentiation. Nucleic Acids Res. 2021, 49, 1935–1950. [Google Scholar] [CrossRef]
- Vaid, S.; Camp, J.G.; Hersemann, L.; Eugster Oegema, C.; Heninger, A.K.; Winkler, S.; Brandl, H.; Sarov, M.; Treutlein, B.; Huttner, W.B.; et al. A novel population of Hopx-dependent basal radial glial cells in the developing mouse neocortex. Development 2018, 145, dev169276. [Google Scholar] [CrossRef]
- Pollen, A.A.; Nowakowski, T.J.; Chen, J.; Retallack, H.; Sandoval-Espinosa, C.; Nicholas, C.R.; Shuga, J.; Liu, S.J.; Oldham, M.C.; Diaz, A.; et al. Molecular identity of human outer radial glia during cortical development. Cell 2015, 163, 55–67. [Google Scholar] [CrossRef]
- Thomsen, E.R.; Mich, J.K.; Yao, Z.; Hodge, R.D.; Doyle, A.M.; Jang, S.; Shehata, S.I.; Nelson, A.M.; Shapovalova, N.V.; Levi, B.P.; et al. Fixed single-cell transcriptomic characterization of human radial glial diversity. Nat. Methods 2016, 13, 87–93. [Google Scholar] [CrossRef]
- Sun, T.; Hevner, R.F. Growth and folding of the mammalian cerebral cortex: From molecules to malformations. Nat. Rev. Neurosci. 2014, 15, 217–232. [Google Scholar] [CrossRef]
- Pinson, A.; Huttner, W.B. Neocortex expansion in development and evolution-from genes to progenitor cell biology. Curr. Opin. Cell Biol. 2021, 73, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Fernández, V.; Borrell, V. Developmental mechanisms of gyrification. Curr. Opin. Neurobiol. 2023, 80, 102711. [Google Scholar] [CrossRef] [PubMed]
- Garcia, K.E.; Kroenke, C.D.; Bayly, P.V. Mechanics of cortical folding: Stress, growth and stability. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170321. [Google Scholar] [CrossRef]
- Pinto, L.; Drechsel, D.; Schmid, M.T.; Ninkovic, J.; Irmler, M.; Brill, M.S.; Restani, L.; Gianfranceschi, L.; Cerri, C.; Weber, S.N.; et al. AP2gamma regulates basal progenitor fate in a region- and layer-specific manner in the developing cortex. Nat. Neurosci. 2009, 12, 1229–1237. [Google Scholar] [CrossRef] [PubMed]
- Sessa, A.; Mao, C.A.; Hadjantonakis, A.K.; Klein, W.H.; Broccoli, V. Tbr2 directs conversion of radial glia into basal precursors and guides neuronal amplification by indirect neurogenesis in the developing neocortex. Neuron 2008, 60, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.J.; Huang, G.J.; Cheung, A.F.; Era, T.; Nishikawa, S.; Bikoff, E.K.; Molnar, Z.; Robertson, E.J.; Groszer, M. The T-box transcription factor Eomes/Tbr2 regulates neurogenesis in the cortical subventricular zone. Genes Dev. 2008, 22, 2479–2484. [Google Scholar] [CrossRef] [PubMed]
- Hevner, R.F.; Hodge, R.D.; Daza, R.A.; Englund, C. Transcription factors in glutamatergic neurogenesis: Conserved programs in neocortex, cerebellum, and adult hippocampus. Neurosci. Res. 2006, 55, 223–233. [Google Scholar] [CrossRef]
- Schuurmans, C.; Armant, O.; Nieto, M.; Stenman, J.M.; Britz, O.; Klenin, N.; Brown, C.; Langevin, L.M.; Seibt, J.; Tang, H.; et al. Sequential phases of cortical specification involve Neurogenin-dependent and -independent pathways. EMBO J. 2004, 23, 2892–2902. [Google Scholar] [CrossRef]
- Nieto, M.; Monuki, E.S.; Tang, H.; Imitola, J.; Haubst, N.; Khoury, S.J.; Cunningham, J.; Gotz, M.; Walsh, C.A. Expression of Cux-1 and Cux-2 in the subventricular zone and upper layers II-IV of the cerebral cortex. J. Comp. Neurol. 2004, 479, 168–180. [Google Scholar] [CrossRef]
- Zimmer, C.; Tiveron, M.C.; Bodmer, R.; Cremer, H. Dynamics of Cux2 expression suggests that an early pool of SVZ precursors is fated to become upper cortical layer neurons. Cereb. Cortex. 2004, 14, 1408–1420. [Google Scholar] [CrossRef]
- Cubelos, B.; Sebastian-Serrano, A.; Kim, S.; Moreno-Ortiz, C.; Redondo, J.M.; Walsh, C.A.; Nieto, M. Cux-2 controls the proliferation of neuronal intermediate precursors of the cortical subventricular zone. Cereb. Cortex 2008, 18, 1758–1770. [Google Scholar] [CrossRef] [PubMed]
- Domingues, A.F.; Kulkarni, R.; Giotopoulos, G.; Gupta, S.; Vinnenberg, L.; Arede, L.; Foerner, E.; Khalili, M.; Adao, R.R.; Johns, A.; et al. Loss of Kat2a enhances transcriptional noise and depletes acute myeloid leukemia stem-like cells. Elife 2020, 9, e51754. [Google Scholar] [CrossRef]
- Borrell, V. How Cells Fold the Cerebral Cortex. J. Neurosci. 2018, 38, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Van Essen, D.C. A 2020 view of tension-based cortical morphogenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 32868–32879. [Google Scholar] [CrossRef] [PubMed]
- Penisson, M.; Ladewig, J.; Belvindrah, R.; Francis, F. Genes and Mechanisms Involved in the Generation and Amplification of Basal Radial Glial Cells. Front. Cell. Neurosci. 2019, 13, 381. [Google Scholar] [CrossRef]
- Fenlon, L.R. Timing as a Mechanism of Development and Evolution in the Cerebral Cortex. Brain Behav. Evol. 2021, 97, 8–32. [Google Scholar] [CrossRef] [PubMed]
- Del-Valle-Anton, L.; Borrell, V. Folding brains: From development to disease modeling. Physiol. Rev. 2022, 102, 511–550. [Google Scholar] [CrossRef]
- de Juan Romero, C.; Borrell, V. Genetic maps and patterns of cerebral cortex folding. Curr. Opin. Cell Biol. 2017, 49, 31–37. [Google Scholar] [CrossRef]
- Reillo, I.; de Juan Romero, C.; García-Cabezas, M.; Borrell, V. A role for intermediate radial glia in the tangential expansion of the mammalian cerebral cortex. Cereb. Cortex 2011, 21, 1674–1694. [Google Scholar] [CrossRef]
- Nonaka-Kinoshita, M.; Reillo, I.; Artegiani, B.; Martinez-Martinez, M.A.; Nelson, M.; Borrell, V.; Calegari, F. Regulation of cerebral cortex size and folding by expansion of basal progenitors. EMBO J. 2013, 32, 1817–1828. [Google Scholar] [CrossRef]
- Matsumoto, N.; Shinmyo, Y.; Ichikawa, Y.; Kawasaki, H. Gyrification of the cerebral cortex requires FGF signaling in the mammalian brain. Elife 2017, 6, e29285. [Google Scholar] [CrossRef] [PubMed]
- Stahl, R.; Walcher, T.; De Juan Romero, C.; Pilz, G.A.; Cappello, S.; Irmler, M.; Sanz-Aquela, J.M.; Beckers, J.; Blum, R.; Borrell, V.; et al. Trnp1 regulates expansion and folding of the mammalian cerebral cortex by control of radial glial fate. Cell 2013, 153, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Ju, X.C.; Hou, Q.Q.; Sheng, A.L.; Wu, K.Y.; Zhou, Y.; Jin, Y.; Wen, T.; Yang, Z.; Wang, X.; Luo, Z.G. The hominoid-specific gene TBC1D3 promotes generation of basal neural progenitors and induces cortical folding in mice. Elife 2016, 5, e18197. [Google Scholar] [CrossRef] [PubMed]
- Del Toro, D.; Ruff, T.; Cederfjall, E.; Villalba, A.; Seyit-Bremer, G.; Borrell, V.; Klein, R. Regulation of Cerebral Cortex Folding by Controlling Neuronal Migration via FLRT Adhesion Molecules. Cell 2017, 169, 621–635.e616. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hou, S.; Han, Y.G. Hedgehog signaling promotes basal progenitor expansion and the growth and folding of the neocortex. Nat. Neurosci. 2016, 19, 888–896. [Google Scholar] [CrossRef]
- Lewitus, E.; Kelava, I.; Huttner, W.B. Conical expansion of the outer subventricular zone and the role of neocortical folding in evolution and development. Front. Hum. Neurosci. 2013, 7, 424. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Cerdeno, V.; Cunningham, C.L.; Camacho, J.; Antczak, J.L.; Prakash, A.N.; Cziep, M.E.; Walker, A.I.; Noctor, S.C. Comparative analysis of the subventricular zone in rat, ferret and macaque: Evidence for an outer subventricular zone in rodents. PLoS ONE 2012, 7, e30178. [Google Scholar] [CrossRef]
- Stoykova, A.; Götz, M.; Gruss, P.; Price, J. Pax6-dependent regulation of adhesive patterning, R-cadherin expression and boundary formation in developing forebrain. Development 1997, 124, 3765–3777. [Google Scholar] [CrossRef]
- Aydin, B.; Kakumanu, A.; Rossillo, M.; Moreno-Estelles, M.; Garipler, G.; Ringstad, N.; Flames, N.; Mahony, S.; Mazzoni, E.O. Proneural factors Ascl1 and Neurog2 contribute to neuronal subtype identities by establishing distinct chromatin landscapes. Nat. Neurosci. 2019, 22, 897–908. [Google Scholar] [CrossRef]
- Noack, F.; Vangelisti, S.; Raffl, G.; Carido, M.; Diwakar, J.; Chong, F.; Bonev, B. Multimodal profiling of the transcriptional regulatory landscape of the developing mouse cortex identifies Neurog2 as a key epigenome remodeler. Nat. Neurosci. 2022, 25, 154–167. [Google Scholar] [CrossRef]
- Anderson, S.A.; Eisenstat, D.D.; Shi, L.; Rubenstein, J.L. Interneuron migration from basal forebrain to neocortex: Dependence on Dlx genes. Science 1997, 278, 474–476. [Google Scholar] [CrossRef] [PubMed]
- Sussel, L.; Marin, O.; Kimura, S.; Rubenstein, J.L. Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: Evidence for a transformation of the pallidum into the striatum. Development 1999, 126, 3359–3370. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Tam, M.; Anderson, S.A. Fate mapping Nkx2.1-lineage cells in the mouse telencephalon. J. Comp. Neurol. 2008, 506, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.A.; Marín, O.; Horn, C.; Jennings, K.; Rubenstein, J.L. Distinct cortical migrations from the medial and lateral ganglionic eminences. Development 2001, 128, 353–363. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sokpor, G.; Kerimoglu, C.; Ulmke, P.A.; Pham, L.; Nguyen, H.D.; Brand-Saberi, B.; Staiger, J.F.; Fischer, A.; Nguyen, H.P.; Tuoc, T. H3 Acetylation-Induced Basal Progenitor Generation and Neocortex Expansion Depends on the Transcription Factor Pax6. Biology 2024, 13, 68. https://doi.org/10.3390/biology13020068
Sokpor G, Kerimoglu C, Ulmke PA, Pham L, Nguyen HD, Brand-Saberi B, Staiger JF, Fischer A, Nguyen HP, Tuoc T. H3 Acetylation-Induced Basal Progenitor Generation and Neocortex Expansion Depends on the Transcription Factor Pax6. Biology. 2024; 13(2):68. https://doi.org/10.3390/biology13020068
Chicago/Turabian StyleSokpor, Godwin, Cemil Kerimoglu, Pauline Antonie Ulmke, Linh Pham, Hoang Duy Nguyen, Beate Brand-Saberi, Jochen F. Staiger, Andre Fischer, Huu Phuc Nguyen, and Tran Tuoc. 2024. "H3 Acetylation-Induced Basal Progenitor Generation and Neocortex Expansion Depends on the Transcription Factor Pax6" Biology 13, no. 2: 68. https://doi.org/10.3390/biology13020068
APA StyleSokpor, G., Kerimoglu, C., Ulmke, P. A., Pham, L., Nguyen, H. D., Brand-Saberi, B., Staiger, J. F., Fischer, A., Nguyen, H. P., & Tuoc, T. (2024). H3 Acetylation-Induced Basal Progenitor Generation and Neocortex Expansion Depends on the Transcription Factor Pax6. Biology, 13(2), 68. https://doi.org/10.3390/biology13020068