
Vascular Calcification: A Passive Process That Requires Active Inhibition


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
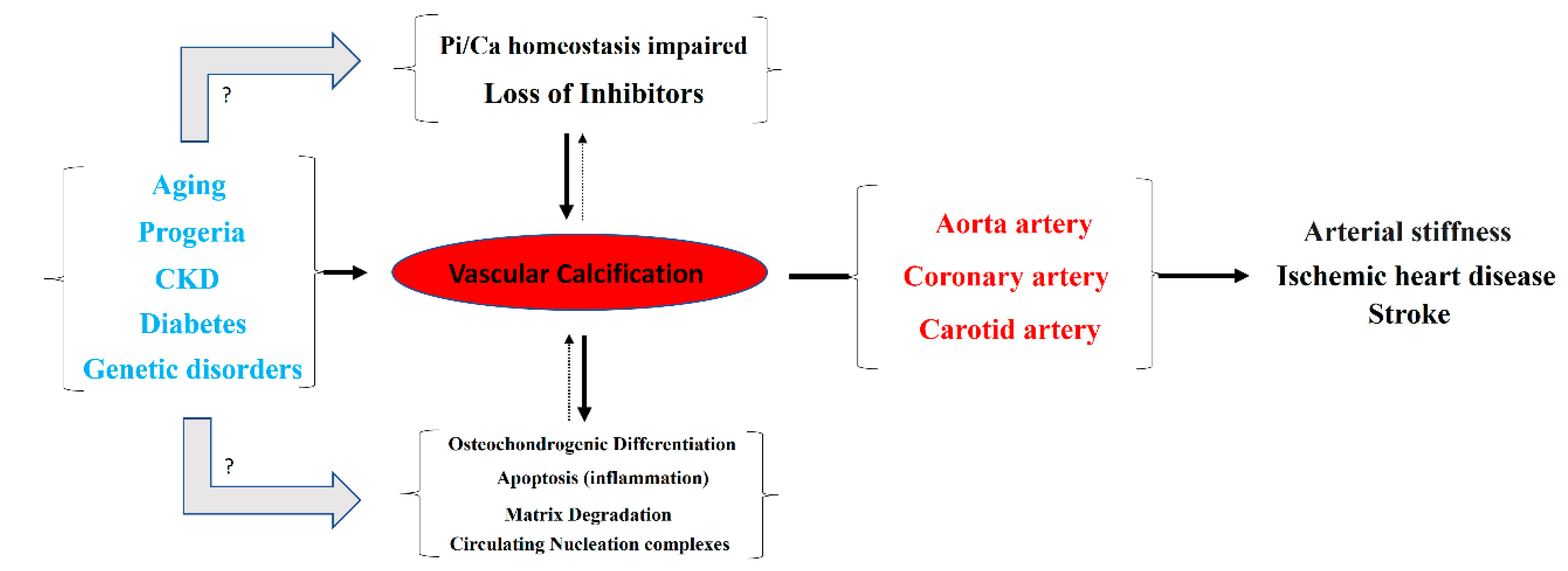
1. Introduction
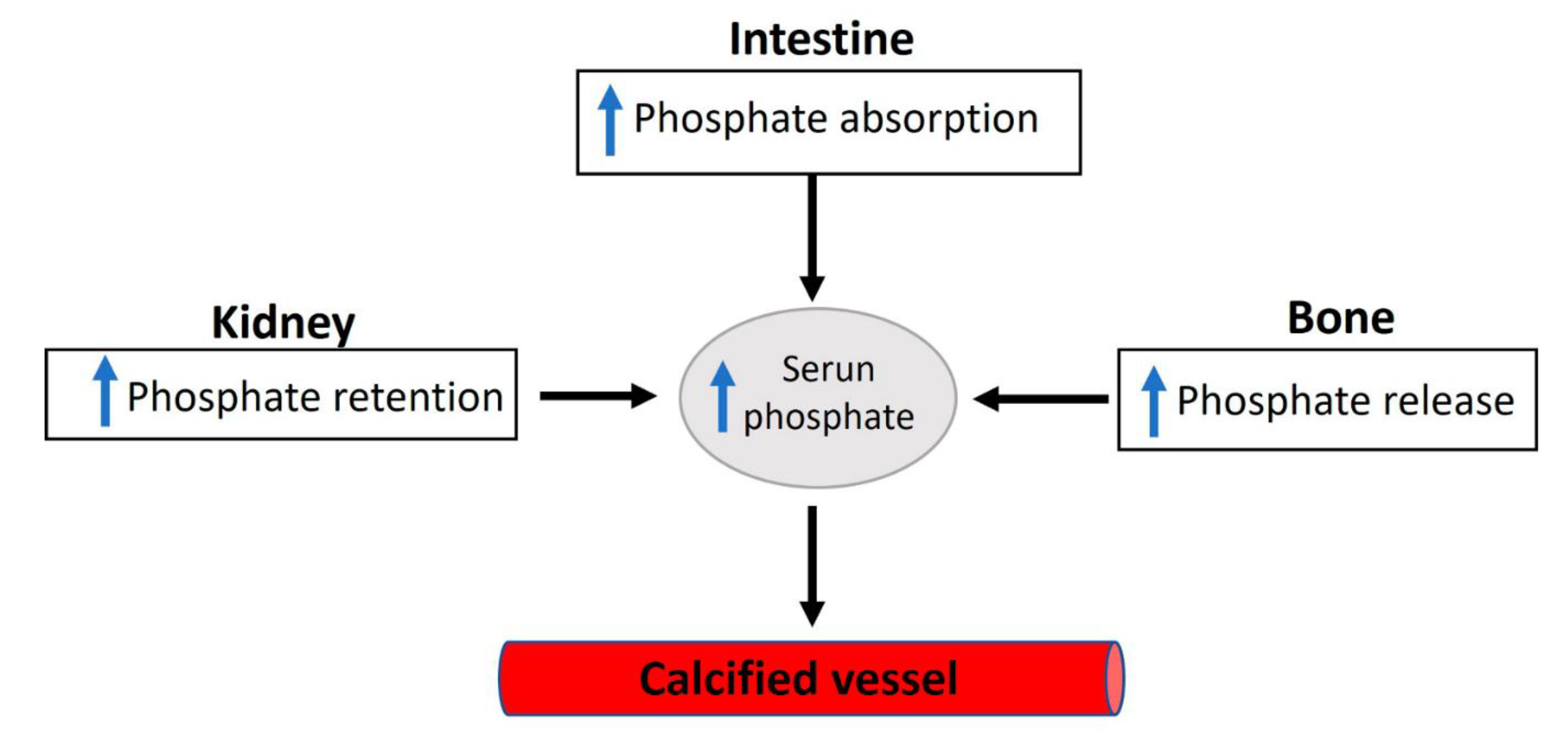
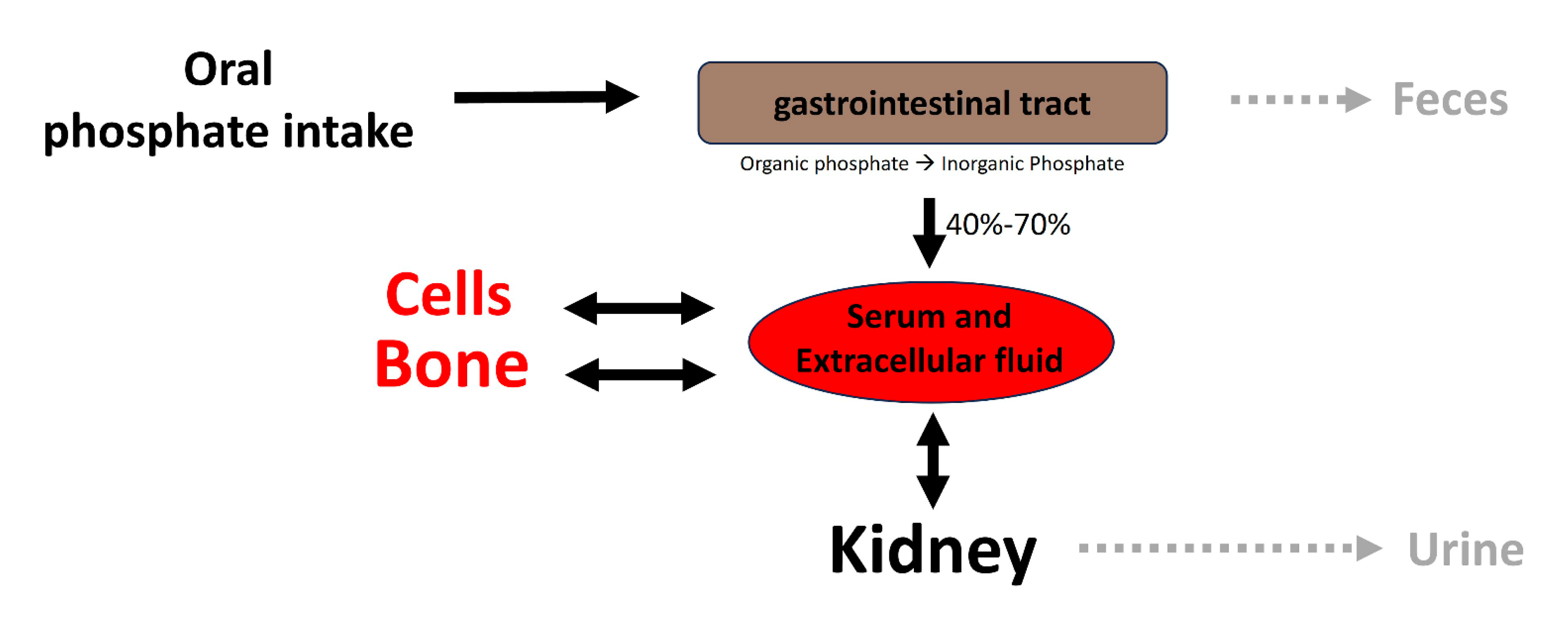
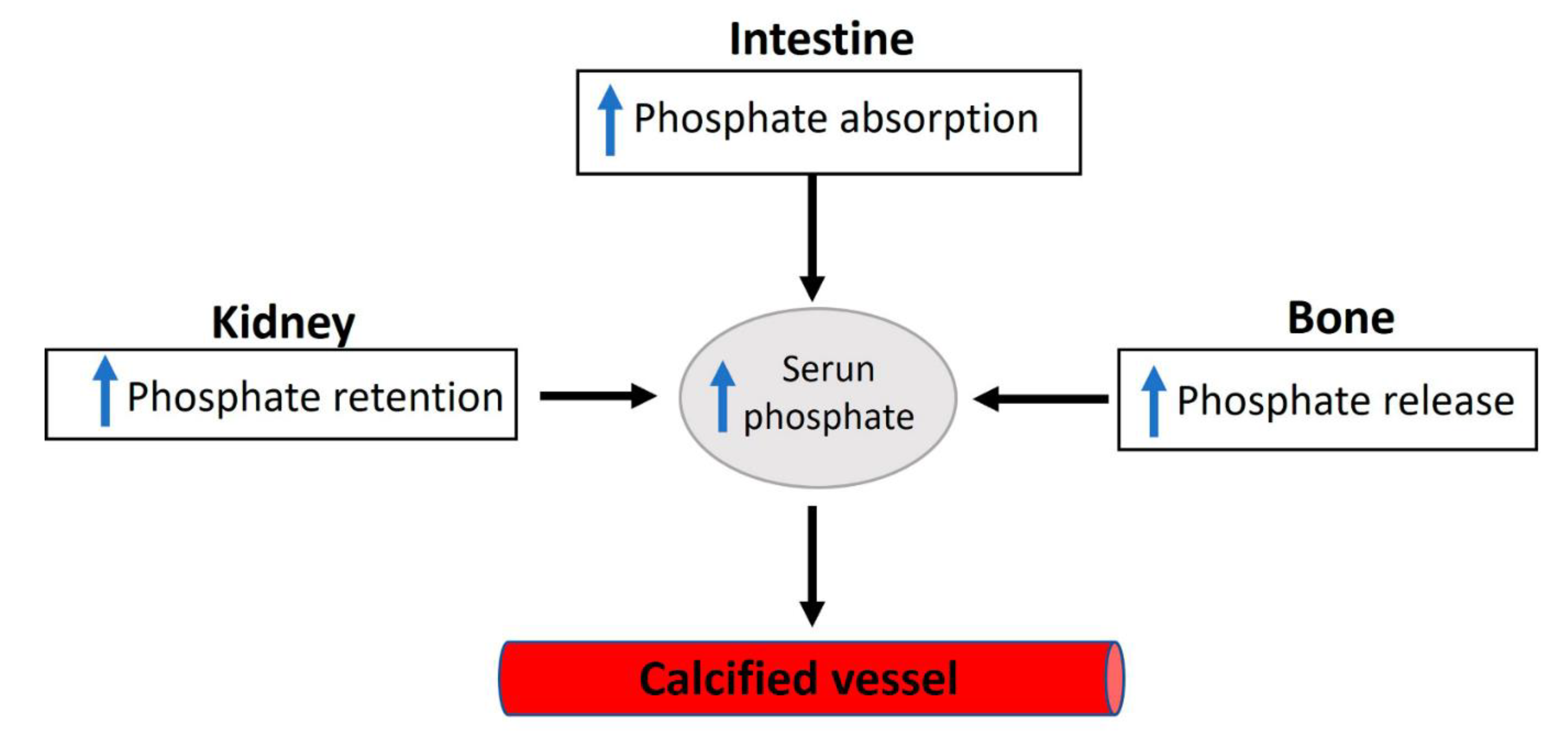
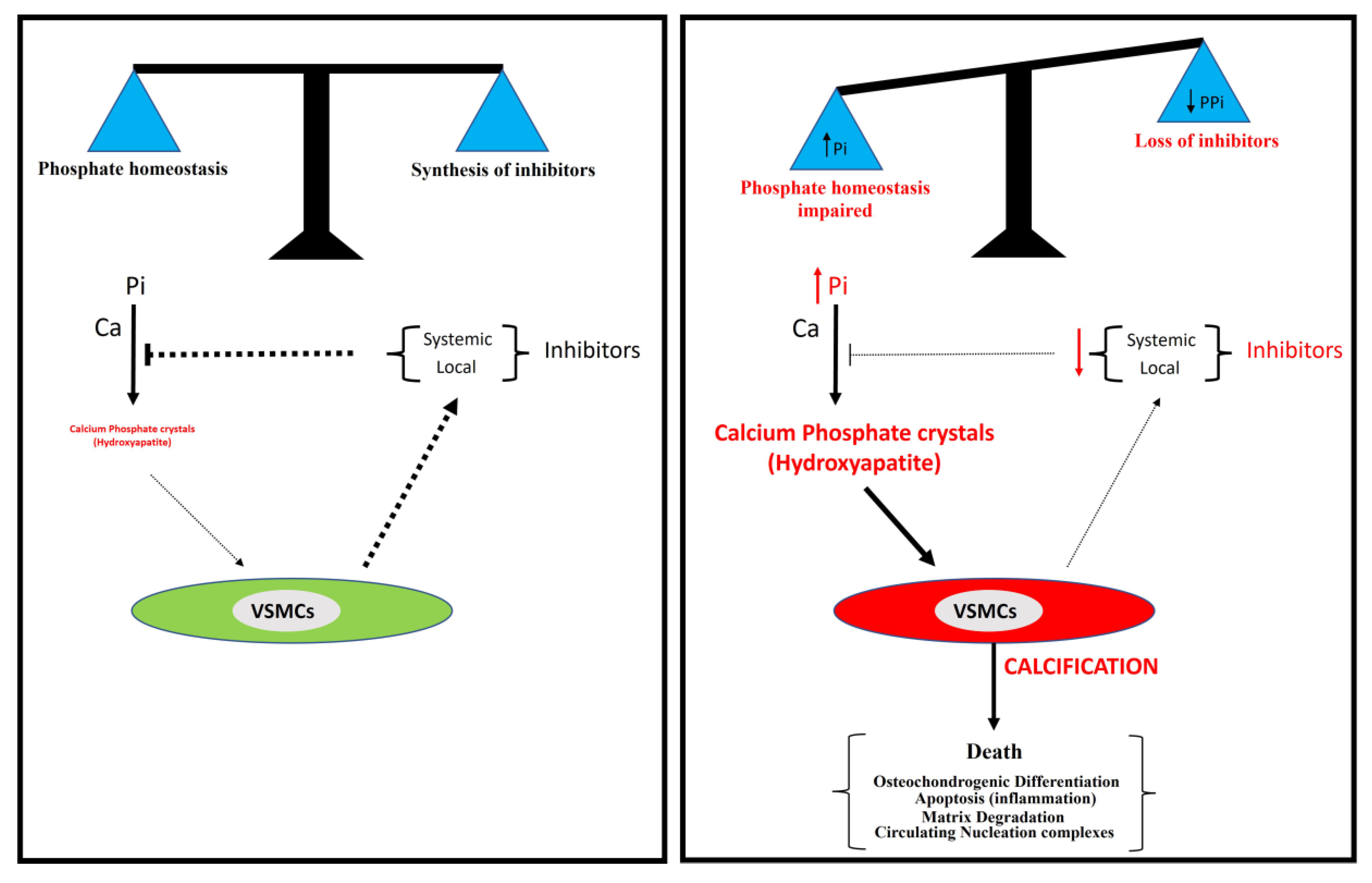
2. Phosphate Homeostasis and Vascular Calcification
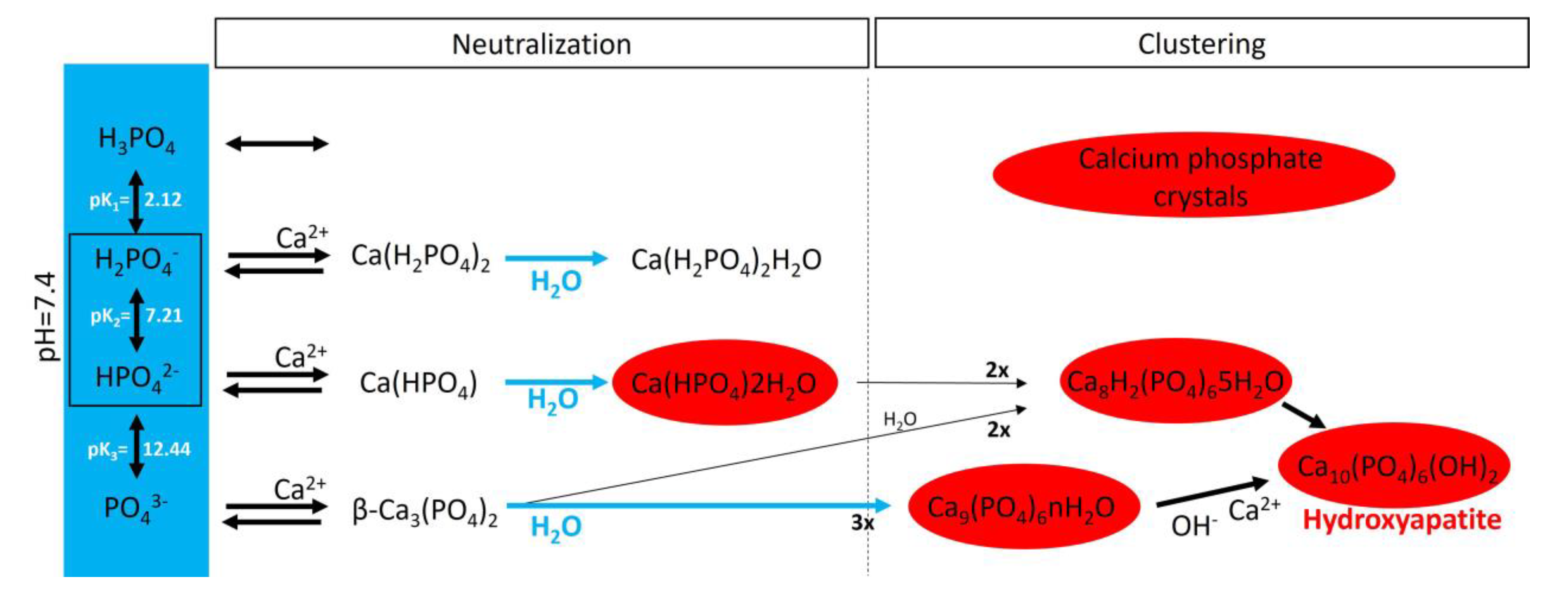
3. Passive Process: Biological Mineralization
4. Active Process: Synthesis of Inhibitors
5. Extracellular Pyrophosphate Metabolism and Vascular Calcification
6. Role of Lipoproteins in Atherosclerosis and Aortic Valve Sclerosis
7. Conclusions
Funding
Acknowledgments
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Villa-Bellosta, R.; Egido, J. Phosphate, pyrophosphate, and vascular calcification: A question of balance. Eur. Heart J. 2017, 38, 1801–1804. [Google Scholar] [CrossRef] [PubMed]
- Rutsch, F.; Nitschke, Y.; Terkeltaub, R. Genetics in arterial calcification: Pieces of a puzzle and cogs in a wheel. Circ. Res. 2011, 109, 578–592. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef] [PubMed]
- Jono, S.; McKee, M.D.; Murry, C.E.; Shioi, A.; Nishizawa, Y.; Mori, K.; Morii, H.; Giachelli, C.M. Phosphate regulation of vascular smooth muscle cell calcification. Circ. Res. 2000, 87, E10–E17. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R.; Millan, A.; Sorribas, V. Role of calcium-phosphate deposition in vascular smooth muscle cell calcification. Am. J. Physiol. Cell Physiol. 2011, 300, C210–C220. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R.; Sorribas, V. Calcium phosphate deposition with normal phosphate concentration. Role of pyrophosphate. Circ. J. Off. J. Jpn. Circ. Soc. 2011, 75, 2705–2710. [Google Scholar] [CrossRef] [PubMed]
- Carfagna, F.; Del Vecchio, L.; Pontoriero, G.; Locatelli, F. Current and potential treatment options for hyperphosphatemia. Expert Opin. Drug Saf. 2018, 17, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, H.-Y.; Giachelli, C.M. Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ. Res. 2006, 98, 905–912. [Google Scholar] [CrossRef]
- Sage, A.P.; Lu, J.; Tintut, Y.; Demer, L.L. Hyperphosphatemia-induced nanocrystals upregulate the expression of bone morphogenetic protein-2 and osteopontin genes in mouse smooth muscle cells in vitro. Kidney Int. 2011, 79, 414–422. [Google Scholar] [CrossRef]
- Villa-Bellosta, R. Synthesis of Extracellular Pyrophosphate Increases in Vascular Smooth Muscle Cells During Phosphate-Induced Calcification. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2137–2147. [Google Scholar] [CrossRef]
- Steitz, S.A.; Speer, M.Y.; Curinga, G.; Yang, H.Y.; Haynes, P.; Aebersold, R.; Schinke, T.; Karsenty, G.; Giachelli, C.M. Smooth muscle cell phenotypic transition associated with calcification: Upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ. Res. 2001, 89, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, H.-Y.; Giachelli, C.M. BMP-2 promotes phosphate uptake, phenotypic modulation, and calcification of human vascular smooth muscle cells. Atherosclerosis 2008, 199, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Speer, M.Y.; Li, X.; Hiremath, P.G.; Giachelli, C.M. Runx2/Cbfa1, but not loss of myocardin, is required for smooth muscle cell lineage reprogramming toward osteochondrogenesis. J. Cell. Biochem. 2010, 110, 935–947. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.C.; McNair, R.; Figg, N.; Skepper, J.N.; Schurgers, L.; Gupta, A.; Hiorns, M.; Donald, A.E.; Deanfield, J.; Rees, L.; et al. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation 2008, 118, 1748–1757. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis regulates human vascular calcification in vitro: Evidence for initiation of vascular calcification by apoptotic bodies. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Ewence, A.E.; Bootman, M.; Roderick, H.L.; Skepper, J.N.; McCarthy, G.; Epple, M.; Neumann, M.; Shanahan, C.M.; Proudfoot, D. Calcium phosphate crystals induce cell death in human vascular smooth muscle cells: A potential mechanism in atherosclerotic plaque destabilization. Circ. Res. 2008, 103, e28–e34. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Sinha, A.; Nosoudi, N.; Grover, A.; Vyavahare, N. Hydroxyapatite and calcified elastin induce osteoblast-like differentiation in rat aortic smooth muscle cells. Exp. Cell Res. 2014, 323, 198–208. [Google Scholar] [CrossRef]
- Zimmermann, H. Extracellular metabolism of ATP and other nucleotides. Naunyn. Schmiedebergs Arch. Pharmacol. 2000, 362, 299–309. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; O’Neill, W.C. Pyrophosphate deficiency in vascular calcification. Kidney Int. 2018, 93, 1293–1297. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Wang, X.; Millán, J.L.; Dubyak, G.R.; O’Neill, W.C. Extracellular pyrophosphate metabolism and calcification in vascular smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H61–H68. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Sorribas, V. Prevention of vascular calcification by polyphosphates and nucleotides—Role of ATP. Circ. J. Off. J. Jpn. Circ. Soc. 2013, 77, 2145–2151. [Google Scholar] [CrossRef]
- Schibler, D.; Russell, R.G.; Fleisch, H. Inhibition by pyrophosphate and polyphosphate of aortic calcification induced by vitamin D3 in rats. Clin. Sci. 1968, 35, 363–372. [Google Scholar] [PubMed]
- Villa-Bellosta, R.; Sorribas, V. Phosphonoformic acid prevents vascular smooth muscle cell calcification by inhibiting calcium-phosphate deposition. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Lomashvili, K.A.; Monier-Faugere, M.-C.; Wang, X.; Malluche, H.H.; O’Neill, W.C. Effect of bisphosphonates on vascular calcification and bone metabolism in experimental renal failure. Kidney Int. 2009, 75, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Warraich, S.; Bone, D.B.J.; Quinonez, D.; Ii, H.; Choi, D.-S.; Holdsworth, D.W.; Drangova, M.; Dixon, S.J.; Séguin, C.A.; Hammond, J.R. Loss of equilibrative nucleoside transporter 1 in mice leads to progressive ectopic mineralization of spinal tissues resembling diffuse idiopathic skeletal hyperostosis in humans. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2013, 28, 1135–1149. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, Y.; Shi, L.; Ren, J.; Patti, M.; Wang, T.; de Oliveira, J.R.M.; Sobrido, M.-J.; Quintáns, B.; Baquero, M.; et al. Mutations in SLC20A2 link familial idiopathic basal ganglia calcification with phosphate homeostasis. Nat. Genet. 2012, 44, 254–256. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R.; Bogaert, Y.E.; Levi, M.; Sorribas, V. Characterization of phosphate transport in rat vascular smooth muscle cells: Implications for vascular calcification. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1030–1036. [Google Scholar] [CrossRef]
- Lohman, A.W.; Billaud, M.; Isakson, B.E. Mechanisms of ATP release and signalling in the blood vessel wall. Cardiovasc. Res. 2012, 95, 269–280. [Google Scholar] [CrossRef]
- Jansen, R.S.; Duijst, S.; Mahakena, S.; Sommer, D.; Szeri, F.; Váradi, A.; Plomp, A.; Bergen, A.A.; Oude Elferink, R.P.J.; Borst, P.; et al. ABCC6-mediated ATP secretion by the liver is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation-brief report. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1985–1989. [Google Scholar] [CrossRef]
- Michigami, T.; Yamazaki, M.; Razzaque, M.S. Extracellular Phosphate, Inflammation and Cytotoxicity. Adv. Exp. Med. Biol. 2022, 1362, 15–25. [Google Scholar] [CrossRef]
- Portales-Castillo, I.; Rieg, T.; Khalid, S.B.; Nigwekar, S.U.; Neyra, J.A. Physiopathology of Phosphate Disorders. Adv. Kidney Dis. Health 2023, 30, 177–188. [Google Scholar] [CrossRef]
- Kritmetapak, K.; Kumar, R. Phosphate as a Signaling Molecule. Calcif. Tissue Int. 2021, 108, 16–31. [Google Scholar] [CrossRef]
- Bhadada, S.K.; Rao, S.D. Role of Phosphate in Biomineralization. Calcif. Tissue Int. 2021, 108, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Peacock, M. Phosphate Metabolism in Health and Disease. Calcif. Tissue Int. 2021, 108, 3–15. [Google Scholar] [CrossRef] [PubMed]
- McClure, S.T.; Chang, A.R.; Selvin, E.; Rebholz, C.M.; Appel, L.J. Dietary Sources of Phosphorus among Adults in the United States: Results from NHANES 2001–2014. Nutrients 2017, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Calvo, M.S.; Moshfegh, A.J.; Tucker, K.L. Assessing the health impact of phosphorus in the food supply: Issues and considerations. Adv. Nutr. 2014, 5, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine (US) Standing Committee on the Scientific Evaluationof Dietary Reference Intakes. Dietary Reference Intakes for Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride [Internet]; National Academies Press: Washington, DC, USA, 1997; The National Academies Collection: Reports funded by National Institutes of Health. Available online: http://www.ncbi.nlm.nih.gov/books/NBK109825/ (accessed on 18 December 2023).
- Manghat, P.; Sodi, R.; Swaminathan, R. Phosphate homeostasis and disorders. Ann. Clin. Biochem. 2014, 51, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Imel, E.A. Congenital Conditions of Hypophosphatemia in Children. Calcif. Tissue Int. 2021, 108, 74–90. [Google Scholar] [CrossRef]
- Megapanou, E.; Florentin, M.; Milionis, H.; Elisaf, M.; Liamis, G. Drug-Induced Hypophosphatemia: Current Insights. Drug Saf. 2020, 43, 197–210. [Google Scholar] [CrossRef]
- Christov, M.; Jüppner, H. Phosphate homeostasis disorders. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 685–706. [Google Scholar] [CrossRef]
- Johnsson, M.S.; Nancollas, G.H. The role of brushite and octacalcium phosphate in apatite formation. Crit. Rev. Oral Biol. Med. Off. Publ. Am. Assoc. Oral Biol. 1992, 3, 61–82. [Google Scholar] [CrossRef] [PubMed]
- LeGeros, R.Z. Formation and transformation of calcium phosphates: Relevance to vascular calcification. Z. Kardiol. 2001, 90 (Suppl. S3), 116–124. [Google Scholar] [CrossRef] [PubMed]
- Reid, J.D.; Andersen, M.E. Medial calcification (whitlockite) in the aorta. Atherosclerosis 1993, 101, 213–224. [Google Scholar] [CrossRef]
- P’ng, C.H.; Boadle, R.; Horton, M.; Bilous, M.; Bonar, F. Magnesium whitlockite of the aorta. Pathology 2008, 40, 539–540. [Google Scholar] [CrossRef]
- Kay, M.I.; Young, R.A.; Posner, A.S. Crystal structure of hydroxyapatite. Nature 1964, 204, 1050–1052. [Google Scholar] [CrossRef] [PubMed]
- Posner, A.S. The structure of bone apatite surfaces. J. Biomed. Mater. Res. 1985, 19, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Kanzaki, N.; Treboux, G.; Onuma, K.; Tsutsumi, S.; Ito, A. Calcium phosphate clusters. Biomaterials 2001, 22, 2921–2929. [Google Scholar] [CrossRef]
- Posner, A.S.; Beebe, R.A. The surface chemistry of bone mineral and related calcium phosphates. Semin. Arthritis Rheum. 1975, 4, 267–291. [Google Scholar] [CrossRef]
- Posner, A.S.; Betts, F.; Blumenthal, N.C. Role of ATP and Mg in the stabilization of biological and synthetic amorphous calcium phosphates. Calcif. Tissue Res. 1977, 22, 208–212. [Google Scholar] [CrossRef]
- Blumenthal, N.C.; Betts, F.; Posner, A.S. Stabilization of amorphous calcium phosphate by Mg and ATP. Calcif. Tissue Res. 1977, 23, 245–250. [Google Scholar] [CrossRef]
- Urry, D.W. Neutral sites for calcium ion binding to elastin and collagen: A charge neutralization theory for calcification and its relationship to atherosclerosis. Proc. Natl. Acad. Sci. USA 1971, 68, 810–814. [Google Scholar] [CrossRef]
- Khavandgar, Z.; Roman, H.; Li, J.; Lee, S.; Vali, H.; Brinckmann, J.; Davis, E.C.; Murshed, M. Elastin haploinsufficiency impedes the progression of arterial calcification in MGP-deficient mice. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2014, 29, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Triffitt, J.T.; Gebauer, U.; Ashton, B.A.; Owen, M.E.; Reynolds, J.J. Origin of plasma alpha2HS-glycoprotein and its accumulation in bone. Nature 1976, 262, 226–227. [Google Scholar] [CrossRef] [PubMed]
- Schinke, T.; Amendt, C.; Trindl, A.; Pöschke, O.; Müller-Esterl, W.; Jahnen-Dechent, W. The serum protein alpha2-HS glycoprotein/fetuin inhibits apatite formation in vitro and in mineralizing calvaria cells. A possible role in mineralization and calcium homeostasis. J. Biol. Chem. 1996, 271, 20789–20796. [Google Scholar] [CrossRef]
- Kocyigit, I.; Unal, A.; Elcik, D.; Korkar, H.; Sen, A.; Yasan, M.; Eroglu, E.; Sipahioglu, M.H.; Tokgoz, B.; Oymak, O. Association Between Cardiac Valvular Calcification and Serum Fetuin-A Levels in Renal Transplant Recipients. Transplant. Proc. 2015, 47, 1398–1401. [Google Scholar] [CrossRef] [PubMed]
- Muzasti, R.A.; Loesnihari, R. High Fetuin-A Level as a Protective Factor to Abdominal Aortic Calcification in Indonesian Regular Hemodialysis Patients. Open Access Maced. J. Med. Sci. 2019, 7, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Demiryurek, B.E.; Gundogdu, A.A. Serum Fetuin-A Levels in Patients with Bilateral Basal Ganglia Calcification. Neurosci. Lett. 2018, 666, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Mutluay, R.; Konca Değertekin, C.; Işıktaş Sayılar, E.; Derici, Ü.; Gültekin, S.; Gönen, S.; Arınsoy, S.T.; Sindel, M.Ş. Serum fetuin-A is associated with the components of MIAC(malnutrition, inflammation, atherosclerosis, calcification) syndrome in different stages of chronic kidney disease. Turk. J. Med. Sci. 2019, 49, 327–335. [Google Scholar] [CrossRef]
- Koca, N.; Ersoy, A.; Şensoy, B.; Kırhan, E.; Güllülü, S.; Ersoy, C.; Dirican, M.; Sarandöl, E. The association between cardiac valvular calcification and fetuin-A levels in kidney transplant recipients. Clin. Exp. Nephrol. 2019, 23, 1250–1256. [Google Scholar] [CrossRef]
- Ulutas, O.; Taskapan, M.C.; Dogan, A.; Baysal, T.; Taskapan, H. Vascular calcification is not related to serum fetuin-A and osteopontin levels in hemodialysis patients. Int. Urol. Nephrol. 2018, 50, 137–142. [Google Scholar] [CrossRef]
- Bjørklund, G.; Svanberg, E.; Dadar, M.; Card, D.J.; Chirumbolo, S.; Harrington, D.J.; Aaseth, J. The Role of Matrix Gla Protein (MGP) in Vascular Calcification. Curr. Med. Chem. 2020, 27, 1647–1660. [Google Scholar] [CrossRef] [PubMed]
- Price, P.A.; Urist, M.R.; Otawara, Y. Matrix Gla protein, a new gamma-carboxyglutamic acid-containing protein which is associated with the organic matrix of bone. Biochem. Biophys. Res. Commun. 1983, 117, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Ducy, P.; McKee, M.D.; Pinero, G.J.; Loyer, E.; Behringer, R.R.; Karsenty, G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997, 386, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Munroe, P.B.; Olgunturk, R.O.; Fryns, J.P.; Van Maldergem, L.; Ziereisen, F.; Yuksel, B.; Gardiner, R.M.; Chung, E. Mutations in the gene encoding the human matrix Gla protein cause Keutel syndrome. Nat. Genet. 1999, 21, 142–144. [Google Scholar] [CrossRef] [PubMed]
- Roumeliotis, S.; Dounousi, E.; Eleftheriadis, T.; Liakopoulos, V. Association of the Inactive Circulating Matrix Gla Protein with Vitamin K Intake, Calcification, Mortality, and Cardiovascular Disease: A Review. Int. J. Mol. Sci. 2019, 20, 628. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Fusaro, M.; Ciceri, P.; Gasperoni, L.; Cianciolo, G. The Role of Vitamin K in Vascular Calcification. Adv. Chronic Kidney Dis. 2019, 26, 437–444. [Google Scholar] [CrossRef]
- Franzén, A.; Heinegård, D. Isolation and characterization of two sialoproteins present only in bone calcified matrix. Biochem. J. 1985, 232, 715–724. [Google Scholar] [CrossRef]
- Jono, S.; Peinado, C.; Giachelli, C.M. Phosphorylation of osteopontin is required for inhibition of vascular smooth muscle cell calcification. J. Biol. Chem. 2000, 275, 20197–20203. [Google Scholar] [CrossRef]
- Ikeda, T.; Shirasawa, T.; Esaki, Y.; Yoshiki, S.; Hirokawa, K. Osteopontin mRNA is expressed by smooth muscle-derived foam cells in human atherosclerotic lesions of the aorta. J. Clin. Investig. 1993, 92, 2814–2820. [Google Scholar] [CrossRef]
- O’Brien, E.R.; Garvin, M.R.; Stewart, D.K.; Hinohara, T.; Simpson, J.B.; Schwartz, S.M.; Giachelli, C.M. Osteopontin is synthesized by macrophage, smooth muscle, and endothelial cells in primary and restenotic human coronary atherosclerotic plaques. Arterioscler. Thromb. J. Vasc. Biol. 1994, 14, 1648–1656. [Google Scholar] [CrossRef]
- Shanahan, C.M.; Cary, N.R.; Metcalfe, J.C.; Weissberg, P.L. High expression of genes for calcification-regulating proteins in human atherosclerotic plaques. J. Clin. Investig. 1994, 93, 2393–2402. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, C.M.; Weissberg, P.L.; Metcalfe, J.C. Isolation of gene markers of differentiated and proliferating vascular smooth muscle cells. Circ. Res. 1993, 73, 193–204. [Google Scholar] [CrossRef]
- Proudfoot, D.; Skepper, J.N.; Shanahan, C.M.; Weissberg, P.L. Calcification of human vascular cells in vitro is correlated with high levels of matrix Gla protein and low levels of osteopontin expression. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Lomashvili, K.A.; Cobbs, S.; Hennigar, R.A.; Hardcastle, K.I.; O’Neill, W.C. Phosphate-induced vascular calcification: Role of pyrophosphate and osteopontin. J. Am. Soc. Nephrol. JASN 2004, 15, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- Lomashvili, K.A.; Garg, P.; Narisawa, S.; Millan, J.L.; O’Neill, W.C. Upregulation of alkaline phosphatase and pyrophosphate hydrolysis: Potential mechanism for uremic vascular calcification. Kidney Int. 2008, 73, 1024–1030. [Google Scholar] [CrossRef]
- Lomashvili, K.A.; Narisawa, S.; Millán, J.L.; O’Neill, W.C. Vascular calcification is dependent on plasma levels of pyrophosphate. Kidney Int. 2014, 85, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Lomashvili, K.A.; Khawandi, W.; O’Neill, W.C. Reduced plasma pyrophosphate levels in hemodialysis patients. J. Am. Soc. Nephrol. JASN 2005, 16, 2495–2500. [Google Scholar] [CrossRef]
- Pomozi, V.; Brampton, C.; van de Wetering, K.; Zoll, J.; Calio, B.; Pham, K.; Owens, J.B.; Marh, J.; Moisyadi, S.; Váradi, A.; et al. Pyrophosphate Supplementation Prevents Chronic and Acute Calcification in ABCC6-Deficient Mice. Am. J. Pathol. 2017, 187, 1258–1272. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Rivera-Torres, J.; Osorio, F.G.; Acín-Pérez, R.; Enriquez, J.A.; López-Otín, C.; Andrés, V. Defective extracellular pyrophosphate metabolism promotes vascular calcification in a mouse model of Hutchinson-Gilford progeria syndrome that is ameliorated on pyrophosphate treatment. Circulation 2013, 127, 2442–2451. [Google Scholar] [CrossRef]
- De Oliveira, R.B.; Louvet, L.; Riser, B.L.; Barreto, F.C.; Benchitrit, J.; Rezg, R.; Poirot, S.; Jorgetti, V.; Drüeke, T.B.; Massy, Z.A. Peritoneal delivery of sodium pyrophosphate blocks the progression of pre-existing vascular calcification in uremic apolipoprotein-E knockout mice. Calcif. Tissue Int. 2015, 97, 179–192. [Google Scholar] [CrossRef]
- O’Neill, W.C.; Lomashvili, K.A.; Malluche, H.H.; Faugere, M.-C.; Riser, B.L. Treatment with pyrophosphate inhibits uremic vascular calcification. Kidney Int. 2011, 79, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Riser, B.L.; Barreto, F.C.; Rezg, R.; Valaitis, P.W.; Cook, C.S.; White, J.A.; Gass, J.H.; Maizel, J.; Louvet, L.; Drueke, T.B.; et al. Daily peritoneal administration of sodium pyrophosphate in a dialysis solution prevents the development of vascular calcification in a mouse model of uraemia. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2011, 26, 3349–3357. [Google Scholar] [CrossRef] [PubMed]
- Rutsch, F.; Ruf, N.; Vaingankar, S.; Toliat, M.R.; Suk, A.; Höhne, W.; Schauer, G.; Lehmann, M.; Roscioli, T.; Schnabel, D.; et al. Mutations in ENPP1 are associated with “idiopathic” infantile arterial calcification. Nat. Genet. 2003, 34, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.; Polewski, M.; van Etten, D.; Terkeltaub, R. Chondrogenesis mediated by PPi depletion promotes spontaneous aortic calcification in NPP1-/- mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 686–691. [Google Scholar] [CrossRef]
- Albright, R.A.; Stabach, P.; Cao, W.; Kavanagh, D.; Mullen, I.; Braddock, A.A.; Covo, M.S.; Tehan, M.; Yang, G.; Cheng, Z.; et al. ENPP1-Fc prevents mortality and vascular calcifications in rodent model of generalized arterial calcification of infancy. Nat. Commun. 2015, 6, 10006. [Google Scholar] [CrossRef] [PubMed]
- St Hilaire, C.; Ziegler, S.G.; Markello, T.C.; Brusco, A.; Groden, C.; Gill, F.; Carlson-Donohoe, H.; Lederman, R.J.; Chen, M.Y.; Yang, D.; et al. NT5E mutations and arterial calcifications. N. Engl. J. Med. 2011, 364, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Narisawa, S.; Yadav, M.C.; Millán, J.L. In vivo overexpression of tissue-nonspecific alkaline phosphatase increases skeletal mineralization and affects the phosphorylation status of osteopontin. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2013, 28, 1587–1598. [Google Scholar] [CrossRef]
- Tani, T.; Fujiwara, M.; Orimo, H.; Shimizu, A.; Narisawa, S.; Pinkerton, A.B.; Millán, J.L.; Tsuruoka, S. Inhibition of tissue-nonspecific alkaline phosphatase protects against medial arterial calcification and improves survival probability in the CKD-MBD mouse model. J. Pathol. 2020, 250, 30–41. [Google Scholar] [CrossRef]
- Li, Q.; Huang, J.; Pinkerton, A.B.; Millan, J.L.; van Zelst, B.D.; Levine, M.A.; Sundberg, J.P.; Uitto, J. Inhibition of Tissue-Nonspecific Alkaline Phosphatase Attenuates Ectopic Mineralization in the Abcc6-/- Mouse Model of PXE but Not in the Enpp1 Mutant Mouse Models of GACI. J. Investig. Dermatol. 2019, 139, 360–368. [Google Scholar] [CrossRef]
- Ziegler, S.G.; Ferreira, C.R.; MacFarlane, E.G.; Riddle, R.C.; Tomlinson, R.E.; Chew, E.Y.; Martin, L.; Ma, C.-T.; Sergienko, E.; Pinkerton, A.B.; et al. Ectopic calcification in pseudoxanthoma elasticum responds to inhibition of tissue-nonspecific alkaline phosphatase. Sci. Transl. Med. 2017, 9, eaal1669. [Google Scholar] [CrossRef]
- Forster, I.C.; Hernando, N.; Biber, J.; Murer, H. Phosphate transporters of the SLC20 and SLC34 families. Mol. Aspects Med. 2013, 34, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Biber, J.; Hernando, N.; Forster, I.; Murer, H. Regulation of phosphate transport in proximal tubules. Pflugers Arch. 2009, 458, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Jensen, N.; Schrøder, H.D.; Hejbøl, E.K.; Füchtbauer, E.-M.; de Oliveira, J.R.M.; Pedersen, L. Loss of function of Slc20a2 associated with familial idiopathic Basal Ganglia calcification in humans causes brain calcifications in mice. J. Mol. Neurosci. MN 2013, 51, 994–999. [Google Scholar] [CrossRef]
- Jansen, R.S.; Küçükosmanoglu, A.; de Haas, M.; Sapthu, S.; Otero, J.A.; Hegman, I.E.M.; Bergen, A.A.B.; Gorgels, T.G.M.F.; Borst, P.; van de Wetering, K. ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc. Natl. Acad. Sci. USA 2013, 110, 20206–20211. [Google Scholar] [CrossRef] [PubMed]
- Le Saux, O.; Urban, Z.; Tschuch, C.; Csiszar, K.; Bacchelli, B.; Quaglino, D.; Pasquali-Ronchetti, I.; Pope, F.M.; Richards, A.; Terry, S.; et al. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat. Genet. 2000, 25, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Bergen, A.A.; Plomp, A.S.; Schuurman, E.J.; Terry, S.; Breuning, M.; Dauwerse, H.; Swart, J.; Kool, M.; van Soest, S.; Baas, F.; et al. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat. Genet. 2000, 25, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Colin, S.; Chinetti-Gbaguidi, G.; Staels, B. Macrophage phenotypes in atherosclerosis. Immunol. Rev. 2014, 262, 153–166. [Google Scholar] [CrossRef]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Hamczyk, M.R.; Andrés, V. Alternatively activated macrophages exhibit an anticalcifying activity dependent on extracellular ATP/pyrophosphate metabolism. Am. J. Physiol. Cell Physiol. 2016, 310, C788–C799. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Hamczyk, M.R.; Andrés, V. Novel phosphate-activated macrophages prevent ectopic calcification by increasing extracellular ATP and pyrophosphate. PLoS ONE 2017, 12, e0174998. [Google Scholar] [CrossRef]
- Kronenberg, F.; Mora, S.; Stroes, E.S.G.; Ference, B.A.; Arsenault, B.J.; Berglund, L.; Dweck, M.R.; Koschinsky, M.; Lambert, G.; Mach, F.; et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: A European Atherosclerosis Society consensus statement. Eur. Heart J. 2022, 43, 3925–3946. [Google Scholar] [CrossRef]
- Tserensonom, M.; Yagi, S.; Ise, T.; Kawabata, Y.; Kadota, M.; Hara, T.; Kusunos, K.; Yamaguchi, K.; Yamada, H.; Soeki, T.; et al. Lipoprotein(a) is a risk factor of aortic valve calcification in patients with a risk of atherosclerosis. J. Med. Investig. JMI 2023, 70, 450–456. [Google Scholar] [CrossRef]
- Sun, H.; Unoki, H.; Wang, X.; Liang, J.; Ichikawa, T.; Arai, Y.; Shiomi, M.; Marcovina, S.M.; Watanabe, T.; Fan, J. Lipoprotein(a) enhances advanced atherosclerosis and vascular calcification in WHHL transgenic rabbits expressing human apolipoprotein(a). J. Biol. Chem. 2002, 277, 47486–47492. [Google Scholar] [CrossRef]
- Duarte Lau, F.; Giugliano, R.P. Lipoprotein(a) and its Significance in Cardiovascular Disease: A Review. JAMA Cardiol. 2022, 7, 760–769. [Google Scholar] [CrossRef]
- Villa-Bellosta, R. ATP-based therapy prevents vascular calcification and extends longevity in a mouse model of Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2019, 116, 23698–23704. [Google Scholar] [CrossRef]
- Villa-Bellosta, R. Dietary magnesium supplementation improves lifespan in a mouse model of progeria. EMBO Mol. Med. 2020, 12, e12423. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villa-Bellosta, R. Vascular Calcification: A Passive Process That Requires Active Inhibition. Biology 2024, 13, 111. https://doi.org/10.3390/biology13020111
Villa-Bellosta R. Vascular Calcification: A Passive Process That Requires Active Inhibition. Biology. 2024; 13(2):111. https://doi.org/10.3390/biology13020111
Chicago/Turabian StyleVilla-Bellosta, Ricardo. 2024. "Vascular Calcification: A Passive Process That Requires Active Inhibition" Biology 13, no. 2: 111. https://doi.org/10.3390/biology13020111
APA StyleVilla-Bellosta, R. (2024). Vascular Calcification: A Passive Process That Requires Active Inhibition. Biology, 13(2), 111. https://doi.org/10.3390/biology13020111