
DT-13 Mediates Ligand-Dependent Activation of PPARγ Response Elements In Vitro



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
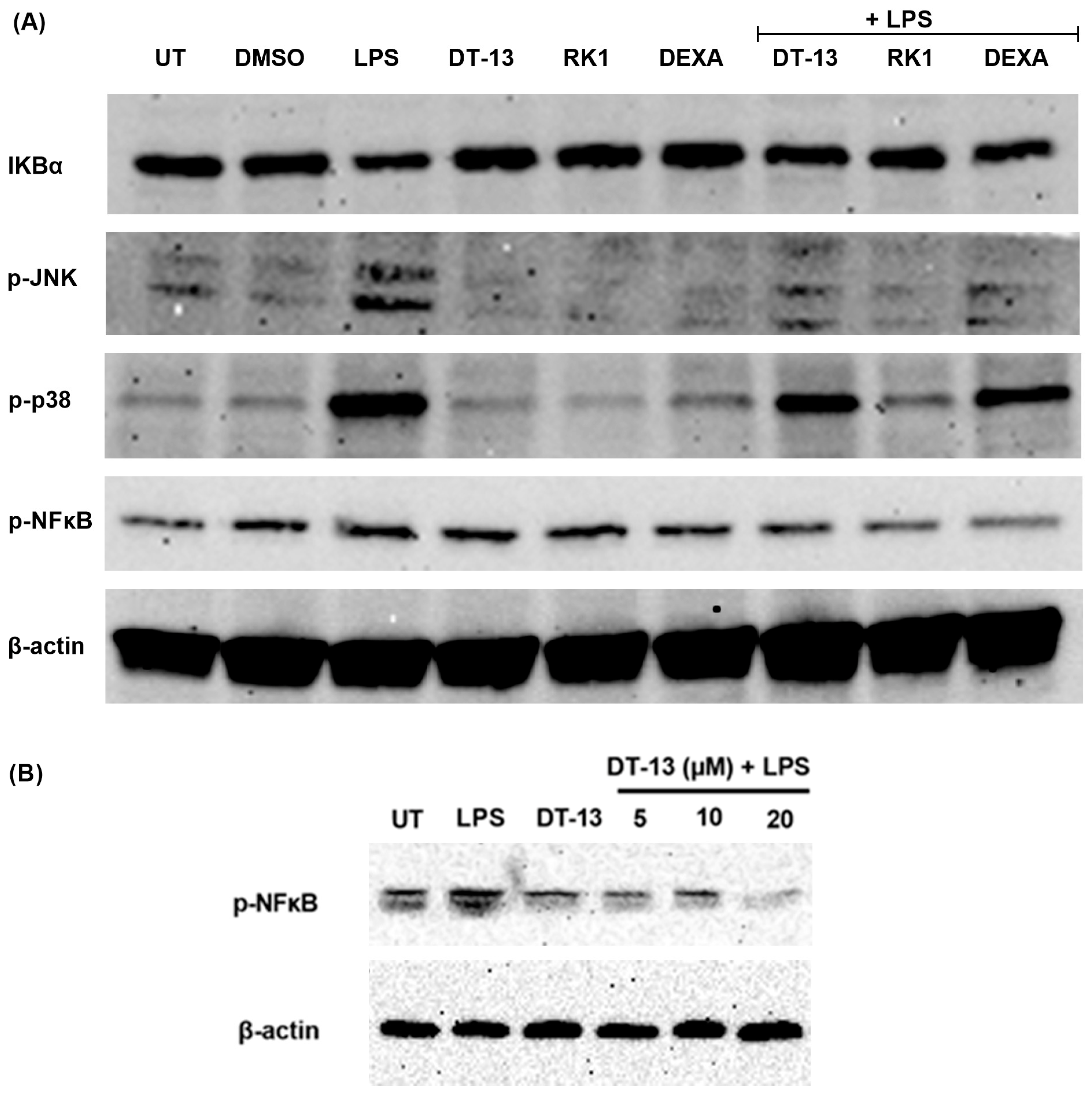
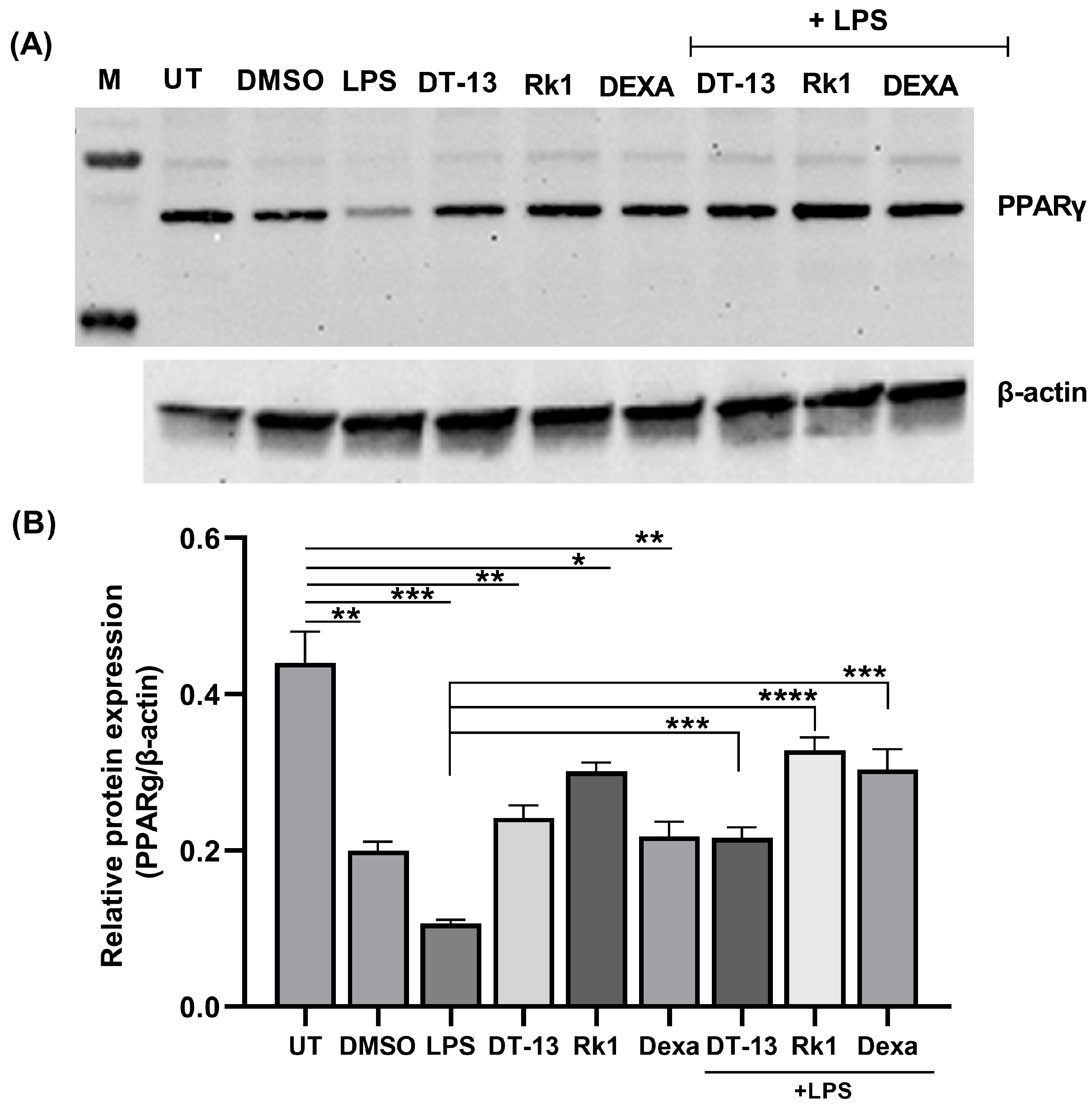
3.1. Saponins as an Inducer of PPARγ in LPS-Activated RAW Macrophages
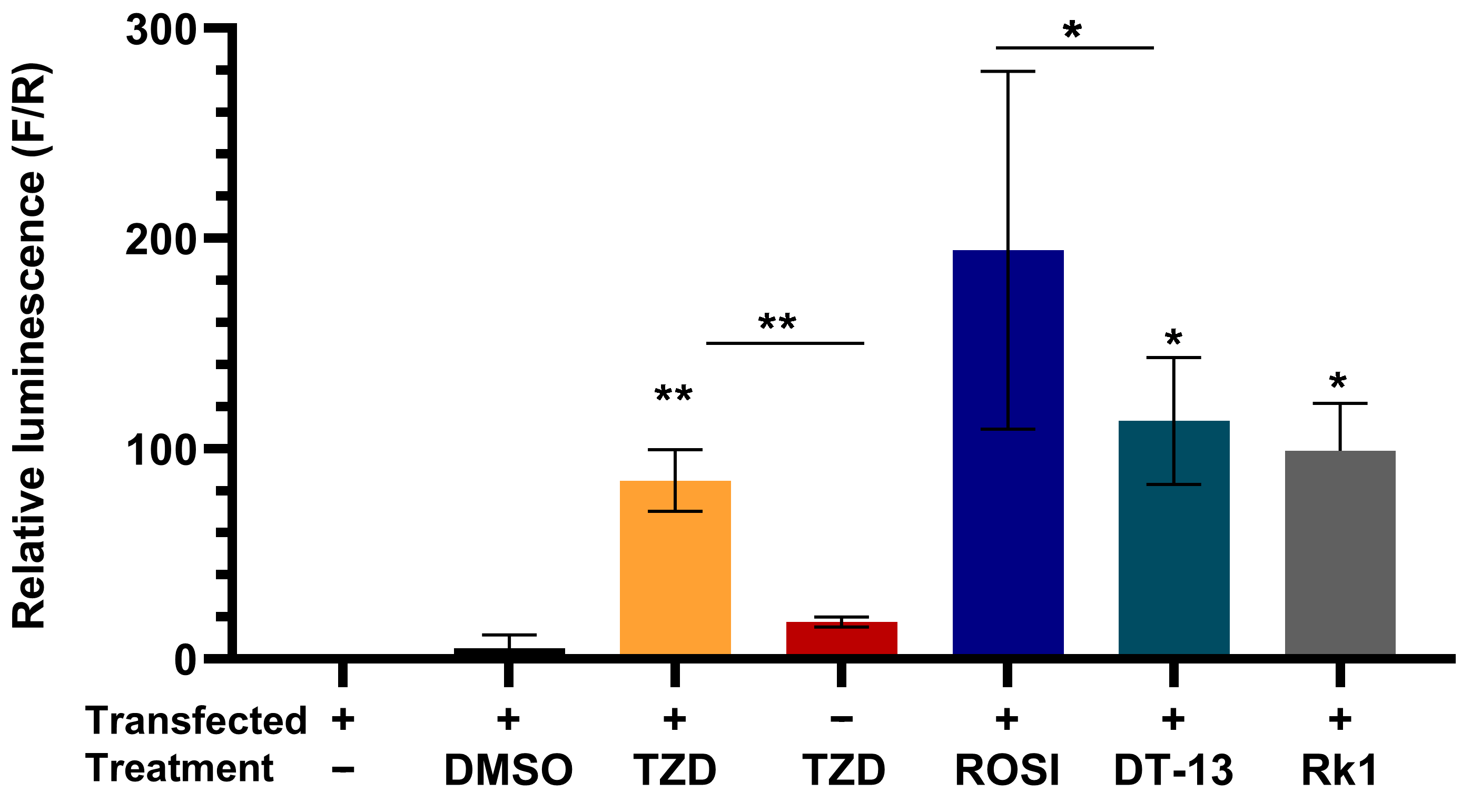
3.2. HEK293FT Cells as a Model System to Screen the Ligands of PPARγ
3.3. Saponins Activate PPARγ In Vitro
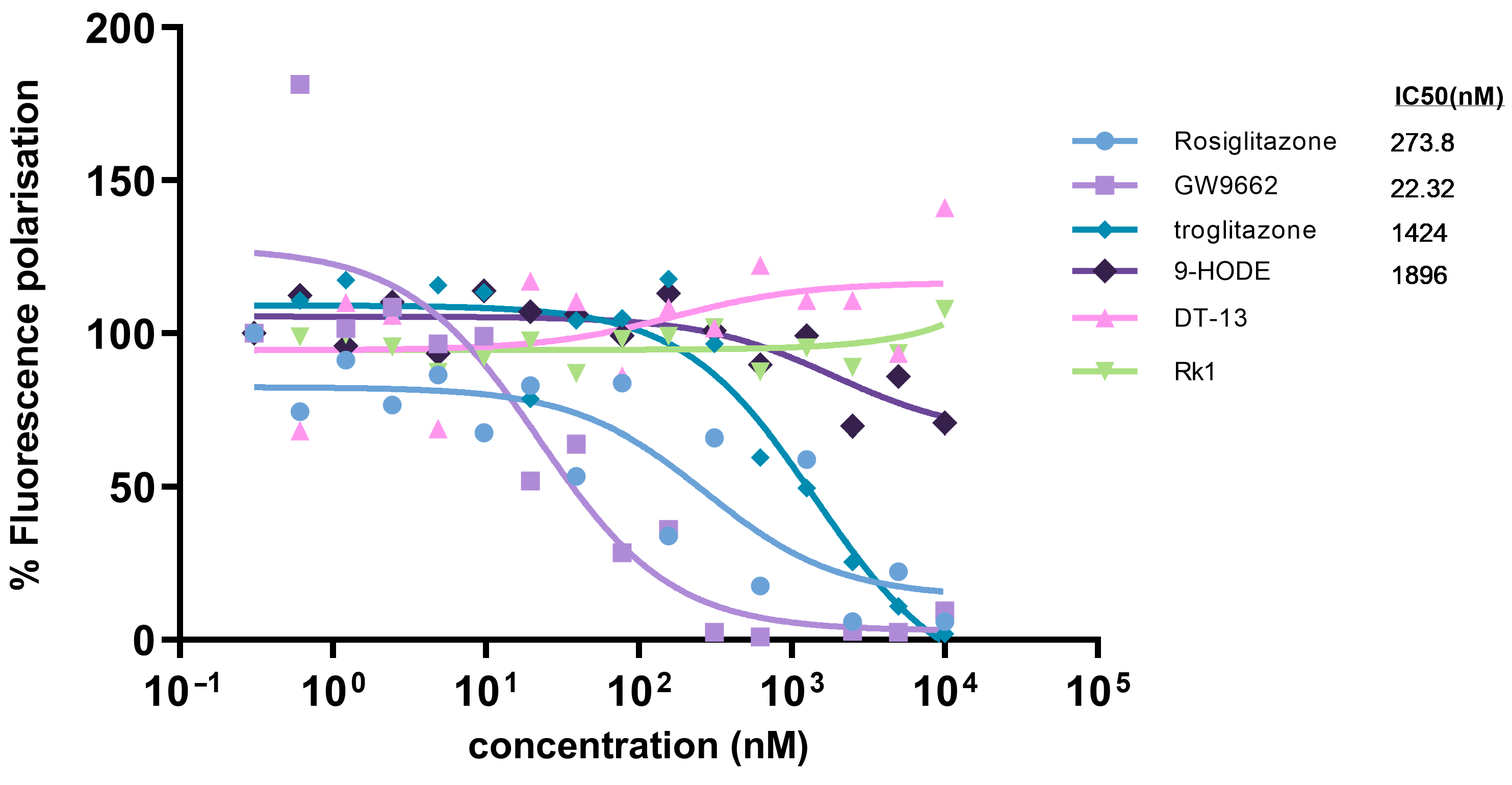
3.4. Comparative Analysis of Binding Efficiency of PPARγ Ligands and DT-13
3.5. In Silico Binding of DT-13 to PPARγ
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- King, T.C. 2—Inflammation, Inflammatory Mediators, and Immune-Mediated Disease. In Elsevier’s Integrated Pathology; King, T.C., Ed.; Mosby: Philadelphia, PA, USA, 2007; pp. 21–57. [Google Scholar]
- Elgazzar, A.H.; Elmonayeri, M. Inflammation. In The Pathophysiologic Basis of Nuclear Medicine; Elgazzar, A.H., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 69–98. [Google Scholar]
- Swanson, L.; Katkar, G.D.; Tam, J.; Pranadinata, R.F.; Chareddy, Y.; Coates, J.; Anandachar, M.S.; Castillo, V.; Olson, J.; Nizet, V.; et al. TLR4 signaling and macrophage inflammatory responses are dampened by GIV/Girdin. Proc. Natl. Acad. Sci. USA 2020, 117, 26895–26906. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Jang, W.Y.; Hwang, J.Y.; Cho, J.Y. Ginsenosides from Panax ginseng as Key Modulators of NF-κB Signaling Are Powerful Anti-Inflammatory and Anticancer Agents. Int. J. Mol. Sci. 2023, 24, 6119. [Google Scholar] [CrossRef]
- Zhu, J.; Luo, C.; Wang, P.; He, Q.; Zhou, J.; Peng, H. Saikosaponin A mediates the inflammatory response by inhibiting the MAPK and NF-kappaB pathways in LPS-stimulated RAW 264.7 cells. Exp. Ther. Med. 2013, 5, 1345–1350. [Google Scholar] [CrossRef]
- Passos, F.R.S.; Araújo-Filho, H.G.; Monteiro, B.S.; Shanmugam, S.; Araújo, A.A.d.S.; Almeida, J.R.G.d.S.; Thangaraj, P.; Júnior, L.J.Q.; Quintans, J.d.S.S. Anti-inflammatory and modulatory effects of steroidal saponins and sapogenins on cytokines: A review of pre-clinical research. Phytomedicine 2022, 96, 153842. [Google Scholar] [CrossRef]
- Hassan, H.S.; Sule, M.I.; Musa, A.M.; Musa, K.Y.; Abubakar, M.S.; Hassan, A.S. Anti-inflammatory activity of crude saponin extracts from five Nigerian medicinal plants. Afr. J. Tradit. Complement. Altern. Med. 2012, 9, 250–255. [Google Scholar] [CrossRef]
- Lee, D.C.; Lau, A.S. Effects of Panax ginseng on tumor necrosis factor-alpha-mediated inflammation: A mini-review. Molecules 2011, 16, 2802–2816. [Google Scholar] [CrossRef]
- Khan, M.I.; Karima, G.; Khan, M.Z.; Shin, J.H.; Kim, J.D. Therapeutic Effects of Saponins for the Prevention and Treatment of Cancer by Ameliorating Inflammation and Angiogenesis and Inducing Antioxidant and Apoptotic Effects in Human Cells. Int. J. Mol. Sci. 2022, 23, 10665. [Google Scholar] [CrossRef]
- Zhang, J.D.; Xu, Z.; Cao, Y.B.; Chen, H.S.; Yan, L.; An, M.M.; Gao, P.H.; Wang, Y.; Jia, X.M.; Jiang, Y.Y. Antifungal activities and action mechanisms of compounds from Tribulus terrestris L. J. Ethnopharmacol. 2006, 103, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wang, H.; Zheng, M.; Xu, W.; Yang, Y.; Shi, F. Ginsenoside Rg3 suppresses the NLRP3 inflammasome activation through inhibition of its assembly. FASEB J. 2020, 34, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, M.; Han, Y.; Zhai, K.; Tang, Y.; Qin, X.; Cao, Z.; Yu, B.; Kou, J. The saponin DT-13 attenuates tumor necrosis factor-α-induced vascular inflammation associated with Src/NF-kB/MAPK pathway modulation. Int. J. Biol. Sci. 2015, 11, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Tong, F.; Xu, M.; Shu, Y.; Li, Z. DT-13 inhibits the proliferation of pancreatic cancer by inducing apoptosis via AMPK-mTOR signaling. Biochem. Biophys. Res. Commun. 2024, 695, 149451. [Google Scholar] [CrossRef]
- Tao, J.; Wang, H.; Zhou, H.; Li, S. The saponin monomer of dwarf lilyturf tuber, DT-13, reduces L-type calcium currents during hypoxia in adult rat ventricular myocytes. Life Sci. 2005, 77, 3021–3030. [Google Scholar] [CrossRef]
- Raina, S.; Hübner, E.; Samuel, E.; Nagel, G.; Fuchs, H. DT-13 attenuates inflammation by inhibiting NLRP3-inflammasome related genes in RAW264.7 macrophages. Biochem. Biophys. Res. Commun. 2024, 708, 149763. [Google Scholar] [CrossRef]
- Huang, W.; Glass, C.K. Nuclear receptors and inflammation control: Molecular mechanisms and pathophysiological relevance. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1542–1549. [Google Scholar] [CrossRef]
- Glass, C.K.; Ogawa, S. Combinatorial roles of nuclear receptors in inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 44–55. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat. Rev. Immunol. 2010, 10, 365–376. [Google Scholar] [CrossRef]
- Mukohda, M.; Lu, K.T.; Guo, D.F.; Wu, J.; Keen, H.L.; Liu, X.; Ketsawatsomkron, P.; Stump, M.; Rahmouni, K.; Quelle, F.W.; et al. Hypertension-Causing Mutation in Peroxisome Proliferator-Activated Receptor γ Impairs Nuclear Export of Nuclear Factor-κB p65 in Vascular Smooth Muscle. Hypertension 2017, 70, 174–182. [Google Scholar] [CrossRef]
- Kosiakova, H.; Berdyshev, A.; Dosenko, V.; Drevytska, T.; Herasymenko, O.; Hula, N. The involvement of peroxisome proliferator-activated receptor gamma (PPARγ) in anti-inflammatory activity of N-stearoylethanolamine. Heliyon 2022, 8, e11336. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Glass, C.K. Nuclear receptors versus inflammation: Mechanisms of transrepression. Trends Endocrinol. Metab. 2006, 17, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Bensinger, S.J.; Tontonoz, P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature 2008, 454, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhong, S.; Li, T.; Zhang, J. Saponins as modulators of nuclear receptors. Crit. Rev. Food Sci. Nutr. 2020, 60, 94–107. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Waltenberger, B.; Pferschy-Wenzig, E.-M.; Blunder, M.; Liu, X.; Malainer, C.; Blazevic, T.; Schwaiger, S.; Rollinger, J.M.; Heiss, E.H.; et al. Natural product agonists of peroxisome proliferator-activated receptor gamma (PPARγ): A review. Biochem. Pharmacol. 2014, 92, 73–89. [Google Scholar] [CrossRef]
- Ma, J.J.; Zhang, T.; Fang, N.; Zou, Y.; Gong, Q.H.; Yu, L.M.; Chen, D.X. Establishment of a cell-based drug screening model for identifying agonists of human peroxisome proliferator-activated receptor gamma (PPARγ). J. Pharm. Pharmacol. 2012, 64, 719–726. [Google Scholar] [CrossRef]
- Vasaturo, M.; Fiengo, L.; De Tommasi, N.; Sabatino, L.; Ziccardi, P.; Colantuoni, V.; Bruno, M.; Cerchia, C.; Novellino, E.; Lupo, A.; et al. A compound-based proteomic approach discloses 15-ketoatractyligenin methyl ester as a new PPARγ partial agonist with anti-proliferative ability. Sci. Rep. 2017, 7, 41273. [Google Scholar] [CrossRef]
- Wurm, F.M. Production of recombinant protein therapeutics in cultivated mammalian cells. Nat. Biotechnol. 2004, 22, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
- Dalton, A.C.; Barton, W.A. Over-expression of secreted proteins from mammalian cell lines. Protein Sci. 2014, 23, 517–525. [Google Scholar] [CrossRef]
- Andréll, J.; Tate, C.G. Overexpression of membrane proteins in mammalian cells for structural studies. Mol. Membr. Biol. 2013, 30, 52–63. [Google Scholar] [CrossRef]
- Fuchs, H.; Niesler, N.; Trautner, A.; Sama, S.; Jerz, G.; Panjideh, H.; Weng, A. Glycosylated Triterpenoids as Endosomal Escape Enhancers in Targeted Tumor Therapies. Biomedicines 2017, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.S.; Wells, R.A. Cross-Talk between PPARs and the Partners of RXR: A Molecular Perspective. PPAR Res. 2009, 2009, 925309. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Rachez, C.; Freedman, L.P. Discrete roles for peroxisome proliferator-activated receptor gamma and retinoid X receptor in recruiting nuclear receptor coactivators. Mol. Cell. Biol. 2000, 20, 8008–8017. [Google Scholar] [CrossRef] [PubMed]
- Willems, S.; Gellrich, L.; Chaikuad, A.; Kluge, S.; Werz, O.; Heering, J.; Knapp, S.; Lorkowski, S.; Schubert-Zsilavecz, M.; Merk, D. Endogenous vitamin E metabolites mediate allosteric PPARγ activation with unprecedented co-regulatory interactions. Cell Chem. Biol. 2021, 28, 1489–1500.e8. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Orhan, I.E.; Banach, M.; Rollinger, J.M.; Barreca, D.; Weckwerth, W.; Bauer, R.; Bayer, E.A.; et al. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Nasim, N.; Sandeep, I.S.; Mohanty, S. Plant-derived natural products for drug discovery: Current approaches and prospects. Nucleus 2022, 65, 399–411. [Google Scholar] [CrossRef]
- Chaachouay, N.; Zidane, L. Plant-Derived Natural Products: A Source for Drug Discovery and Development. Drugs Drug Candidates 2024, 3, 184–207. [Google Scholar] [CrossRef]
- Al Sharif, M.; Alov, P.; Diukendjieva, A.; Vitcheva, V.; Simeonova, R.; Krasteva, I.; Shkondrov, A.; Tsakovska, I.; Pajeva, I. Molecular determinants of PPARγ partial agonism and related in silico/in vivo studies of natural saponins as potential type 2 diabetes modulators. Food Chem. Toxicol. 2018, 112, 47–59. [Google Scholar] [CrossRef]
- Montanari, R.; Capelli, D.; Tava, A.; Galli, A.; Laghezza, A.; Tortorella, P.; Loiodice, F.; Pochetti, G. Screening of saponins and sapogenins from Medicago species as potential PPARγ agonists and X-ray structure of the complex PPARγ/caulophyllogenin. Sci. Rep. 2016, 6, 27658. [Google Scholar] [CrossRef]
- Winoto, A.; Littman, D.R. Nuclear Hormone Receptors in T Lymphocytes. Cell 2002, 109 (Suppl. S1), S57–S66. [Google Scholar] [CrossRef]
- Simonin, M.A.; Bordji, K.; Boyault, S.; Bianchi, A.; Gouze, E.; Bécuwe, P.; Dauça, M.; Netter, P.; Terlain, B. PPAR-γ ligands modulate effects of LPS in stimulated rat synovial fibroblasts. Am. J. Physiol. Cell Physiol. 2002, 282, C125–C133. [Google Scholar] [CrossRef] [PubMed]
- Fajas, L.; Auboeuf, D.; Raspé, E.; Schoonjans, K.; Lefebvre, A.-M.; Saladin, R.; Najib, J.; Laville, M.; Fruchart, J.-C.; Deeb, S.; et al. The Organization, Promoter Analysis, and Expression of the Human PPARγ Gene. J. Biol. Chem. 1997, 272, 18779–18789. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Wang, Z.; Jie, W.; Fu, X.; Li, B.; Xu, H.; Liu, Y.; Li, M.; Kim, E.; Yang, Y.; et al. The kinase inhibitor BX795 suppresses the inflammatory response via multiple kinases. Biochem. Pharmacol. 2020, 174, 113797. [Google Scholar] [CrossRef]
- Farhat-Younis, L.; Na, M.; Zarfin, A.; Khateeb, A.; Santana-Magal, N.; Richter, A.; Gutwillig, A.; Rasoulouniriana, D.; Gleiberman, A.; Beck, L.; et al. Expression of modified FcγRI enables myeloid cells to elicit robust tumor-specific cytotoxicity. eLife 2024, 12, RP91999. [Google Scholar] [CrossRef] [PubMed]
- Boyle, J.G.; Logan, P.J.; Ewart, M.A.; Reihill, J.A.; Ritchie, S.A.; Connell, J.M.; Cleland, S.J.; Salt, I.P. Rosiglitazone stimulates nitric oxide synthesis in human aortic endothelial cells via AMP-activated protein kinase. J. Biol. Chem. 2008, 283, 11210–11217. [Google Scholar] [CrossRef]
- Hall, M.D.; Yasgar, A.; Peryea, T.; Braisted, J.C.; Jadhav, A.; Simeonov, A.; Coussens, N.P. Fluorescence polarization assays in high-throughput screening and drug discovery: A review. Methods Appl. Fluoresc. 2016, 4, 022001. [Google Scholar] [CrossRef]
- Fang, M.; Webster, T.F.; Ferguson, P.L.; Stapleton, H.M. Characterizing the peroxisome proliferator-activated receptor (PPARγ) ligand binding potential of several major flame retardants, their metabolites, and chemical mixtures in house dust. Environ. Health Perspect. 2015, 123, 166–172. [Google Scholar] [CrossRef]
- Xiong, J.; Guo, J.; Huang, L.; Meng, B.; Ping, Q. Self-micelle formation and the incorporation of lipid in the formulation affect the intestinal absorption of Panax notoginseng. Int. J. Pharm. 2008, 360, 191–196. [Google Scholar] [CrossRef]
- Cho, M.C.; Lee, K.; Paik, S.G.; Yoon, D.Y. Peroxisome Proliferators-Activated Receptor (PPAR) Modulators and Metabolic Disorders. PPAR Res. 2008, 2008, 679137. [Google Scholar] [CrossRef]
- Zhou, J.P.; Yang, X.N.; Song, Y.; Zhou, F.; Liu, J.J.; Hu, Y.Q.; Chen, L.G. Rosiglitazone alleviates lipopolysaccharide-induced inflammation in RAW264.7 cells via inhibition of NF-kappaB and in a PPARγ-dependent manner. Exp. Ther. Med. 2021, 22, 743. [Google Scholar] [CrossRef]
- Hughes, T.S.; Giri, P.K.; de Vera, I.M.; Marciano, D.P.; Kuruvilla, D.S.; Shin, Y.; Blayo, A.L.; Kamenecka, T.M.; Burris, T.P.; Griffin, P.R.; et al. An alternate binding site for PPARγ ligands. Nat. Commun. 2014, 5, 3571. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Brust, R.; Mosure, S.A.; Bass, J.; Munoz-Tello, P.; Lin, H.; Hughes, T.S.; Tang, M.; Ge, Q.; Kamenekca, T.M.; et al. Cooperative cobinding of synthetic and natural ligands to the nuclear receptor PPARγ. eLife 2018, 7, e43320. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, Y.; Li, W.; Li, H.; Yang, L.; Wang, J.; Mahdy, H.A.; Mehany, A.B.M.; Jaiash, D.A.; Santali, E.Y.; et al. Screening of Some Sulfonamide and Sulfonylurea Derivatives as Anti-Alzheimer’s Agents Targeting BACE1 and PPARγ. J. Chem. 2020, 2020, 1631243. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raina, S.; Samuel, E.; Fuchs, H. DT-13 Mediates Ligand-Dependent Activation of PPARγ Response Elements In Vitro. Biology 2024, 13, 1015. https://doi.org/10.3390/biology13121015
Raina S, Samuel E, Fuchs H. DT-13 Mediates Ligand-Dependent Activation of PPARγ Response Elements In Vitro. Biology. 2024; 13(12):1015. https://doi.org/10.3390/biology13121015
Chicago/Turabian StyleRaina, Shikha, Esther Samuel, and Hendrik Fuchs. 2024. "DT-13 Mediates Ligand-Dependent Activation of PPARγ Response Elements In Vitro" Biology 13, no. 12: 1015. https://doi.org/10.3390/biology13121015
APA StyleRaina, S., Samuel, E., & Fuchs, H. (2024). DT-13 Mediates Ligand-Dependent Activation of PPARγ Response Elements In Vitro. Biology, 13(12), 1015. https://doi.org/10.3390/biology13121015