

Binning Metagenomic Contigs Using Contig Embedding and Decomposed Tetranucleotide Frequency

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Overview of CedtBin
2.3. Contig Embedding
2.3.1. Preprocessing
2.3.2. Modified Mask Language Model
- Eighty percent of the time, a continuous k-mer centered on the k-mer is replaced using [MASK];
- Ten percent of the time, a continuous k-mer centered on the k-mer is randomly replaced with another k-mer;
- Ten percent of the time, the k-mer remains unchanged.
2.3.3. Pooling
2.4. Decomposed Tetranucleotide Frequency
2.4.1. Tetranucleotide Frequency Calculation
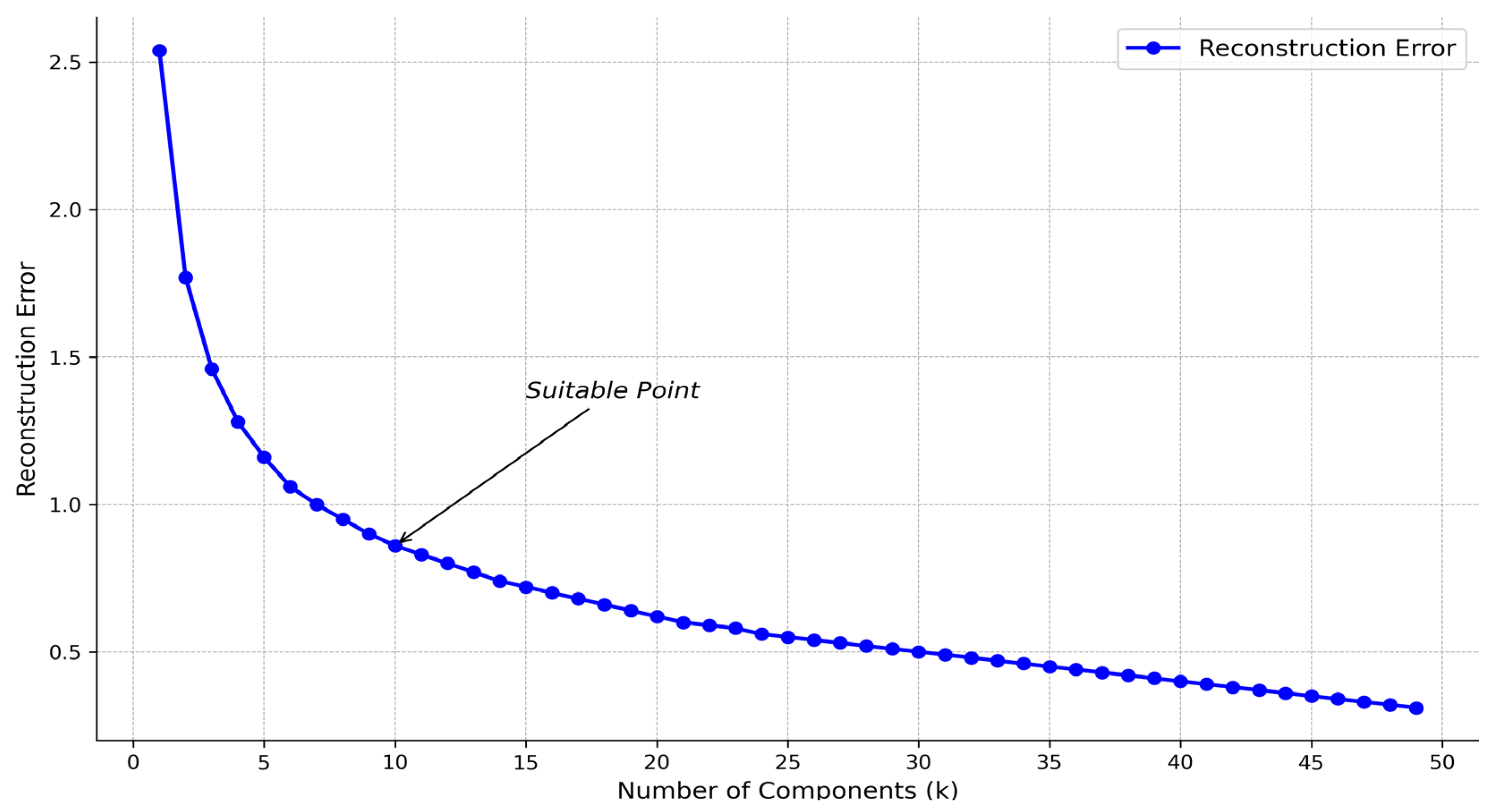
2.4.2. Non-Negative Matrix Factorization
2.4.3. Clustering
- i.
- Create an Annoy index. Add all data points to the index and then build a random projection tree.
- ii.
- Calculate k-distance. For each data point, use the Annoy algorithm to find its k nearest neighbors and record the distance of the kth nearest neighbor. Store the k-distance values of all points in a list and sort them.
- iii.
- Draw a k-distance graph. Use Matplotlib to draw a k-distance graph and select the inflection point from the graph as the Eps of the DBSCAN algorithm. For example, in Figure 2, it can be found that there is a sharp increase between the distance 0.6 and 0.9, so Eps can take the inflection point position here as 0.7.
- iv.
- Grid search. After determining Eps, use the grid search method to find it in the preset MinPts value and select the optimal MinPts according to the clustering results.
3. Results
3.1. Experimental Instructions
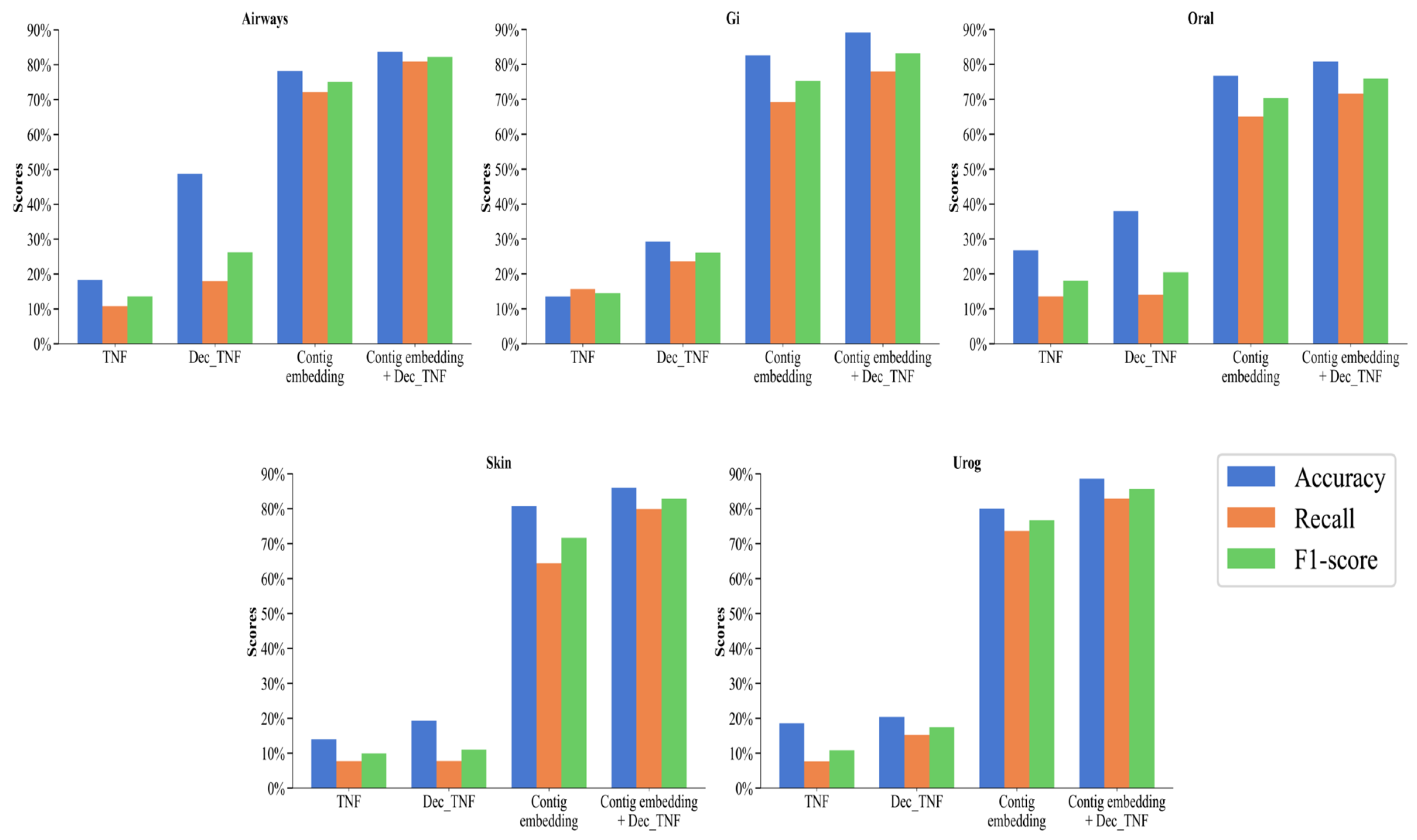
3.2. Results and Analysis of Different Features
3.2.1. Evaluation Metrics
3.2.2. Binning Performance of Different Features
3.3. Results and Analysis of Different Binning Methods
3.3.1. Simulated Dataset CAMI2-HMP Binning Results
3.3.2. Real Dataset MetaHIT Binning Results
3.4. Memory Usage and Runtime
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Riesenfeld, C.S.; Schloss, P.D.; Handelsman, J. Metagenomics: Genomic analysis of microbial communities. Annu. Rev. Genet. 2004, 38, 525–552. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; Boushey, H.A. The microbiome in asthma. J. Allergy Clin. Immunol. 2015, 135, 25–30. [Google Scholar] [CrossRef]
- Huang, Y.J.; Marsland, B.J.; Bunyavanich, S.; O’Mahony, L.; Leung, D.Y.; Muraro, A.; Fleisher, T.A. The microbiome in allergic disease: Current understanding and future opportunities—2017 PRACTALL document of the American Academy of Allergy, Asthma & Immunology and the European Academy of Allergy and Clinical Immunology. J. Allergy Clin. Immunol. 2017, 139, 1099–1110. [Google Scholar]
- Severance, E.G.; Yolken, R.H.; Eaton, W.W. Autoimmune diseases, gastrointestinal disorders and the microbiome in schizophrenia: More than a gut feeling. Schizophr. Res. 2016, 176, 23–35. [Google Scholar] [CrossRef]
- Teeling, H.; Waldmann, J.; Lombardot, T.; Bauer, M.; Glöckner, F.O. TETRA: A web-service and a stand-alone program for the analysis and comparison of tetranucleotide usage patterns in DNA sequences. BMC Bioinform. 2004, 5, 163. [Google Scholar] [CrossRef] [PubMed]
- Chatterji, S.; Yamazaki, I.; Bai, Z.; Eisen, J.A. CompostBin: A DNA composition-based algorithm for binning environmental shotgun reads. In Proceedings of the Research in Computational Molecular Biology: 12th Annual International Conference, RECOMB 2008, Singapore, 30 March–2 April 2008; Proceedings 12. pp. 17–28. [Google Scholar]
- Wu, Y.-W.; Ye, Y. A novel abundance-based algorithm for binning metagenomic sequences using l-tuples. J. Comput. Biol. 2011, 18, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. MetaCluster-TA: Taxonomic annotation for metagenomic data based on assembly-assisted binning. BMC Genom. 2014, 15 (Suppl. S1), S12. [Google Scholar] [CrossRef]
- Xing, X.; Liu, J.S.; Zhong, W. MetaGen: Reference-free learning with multiple metagenomic samples. Genome Biol. 2017, 18, 187. [Google Scholar] [CrossRef]
- Nielsen, H.B.; Almeida, M.; Juncker, A.S.; Rasmussen, S.; Li, J.; Sunagawa, S.; Plichta, D.R.; Gautier, L.; Pedersen, A.G.; Le Chatelier, E. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 2014, 32, 822–828. [Google Scholar] [CrossRef]
- Alneberg, J.; Bjarnason, B.S.; De Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Lahti, L.; Loman, N.J.; Andersson, A.F.; Quince, C. Binning metagenomic contigs by coverage and composition. Nat. Methods 2014, 11, 1144–1146. [Google Scholar] [CrossRef]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2016, 32, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Jiang, Y.; Wang, J.; Zhang, H.; Luo, H. BMC3C: Binning metagenomic contigs using codon usage, sequence composition and read coverage. Bioinformatics 2018, 34, 4172–4179. [Google Scholar] [CrossRef] [PubMed]
- Nissen, J.N.; Johansen, J.; Allesøe, R.L.; Sønderby, C.K.; Armenteros, J.J.A.; Grønbech, C.H.; Jensen, L.J.; Nielsen, H.B.; Petersen, T.N.; Winther, O. Improved metagenome binning and assembly using deep variational autoencoders. Nat. Biotechnol. 2021, 39, 555–560. [Google Scholar] [CrossRef]
- Zhang, P.; Jiang, Z.; Wang, Y.; Li, Y. CLMB: Deep contrastive learning for robust metagenomic binning. In Proceedings of the International Conference on Research in Computational Molecular Biology, San Diego, CA, USA, 22–25 May 2022; pp. 326–348. [Google Scholar]
- Líndez, P.P.; Johansen, J.; Kutuzova, S.; Sigurdsson, A.I.; Nissen, J.N.; Rasmussen, S. Adversarial and variational autoencoders improve metagenomic binning. Commun. Biol. 2023, 6, 1073. [Google Scholar] [CrossRef]
- Pan, S.; Zhu, C.; Zhao, X.-M.; Coelho, L.P. A deep siamese neural network improves metagenome-assembled genomes in microbiome datasets across different environments. Nat. Commun. 2022, 13, 2326. [Google Scholar] [CrossRef]
- Lamurias, A.; Sereika, M.; Albertsen, M.; Hose, K.; Nielsen, T.D. Metagenomic binning with assembly graph embeddings. Bioinformatics 2022, 38, 4481–4487. [Google Scholar] [CrossRef]
- Sczyrba, A.; Hofmann, P.; Belmann, P.; Koslicki, D.; Janssen, S.; Dröge, J.; Gregor, I.; Majda, S.; Fiedler, J.; Dahms, E. Critical assessment of metagenome interpretation—A benchmark of metagenomics software. Nat. Methods 2017, 14, 1063–1071. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Devlin, J.; Chang, M.-W.; Lee, K.; Toutanova, K. Bert: Pre-training of deep bidirectional transformers for language understanding. arXiv 2018, arXiv:1810.04805. [Google Scholar]
- Ji, Y.; Zhou, Z.; Liu, H.; Davuluri, R.V. DNABERT: Pre-trained Bidirectional Encoder Representations from Transformers model for DNA-language in genome. Bioinformatics 2021, 37, 2112–2120. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Seung, H.S. Algorithms for non-negative matrix factorization. Adv. Neural Inf. Process. Syst. 2000, 13, 535–541. [Google Scholar]
- Ester, M.; Kriegel, H.-P.; Sander, J.; Xu, X. A density-based algorithm for discovering clusters in large spatial databases with noise. In Proceedings of the Kdd, Portland, OR, USA, 2–4 August 1996; pp. 226–231. [Google Scholar]
- Annoy: Approximate Nearest Neighbors in C++/Python. Available online: https://github.com/spotify/annoy (accessed on 26 July 2024).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Datasets | Samples | Genus | Species | Strains | Contigs | N50 |
|---|---|---|---|---|---|---|
| Airways | 10 | 139 | 311 | 639 | 187,685 | 28,290 |
| Gi | 10 | 63 | 141 | 250 | 81,602 | 701,026 |
| Oral | 10 | 107 | 249 | 673 | 201,563 | 25,427 |
| Skin | 10 | 106 | 247 | 531 | 173,927 | 66,557 |
| Urog | 9 | 47 | 118 | 234 | 57,762 | 877,598 |
| MetaHIT | 264 | 70 | 219 | 291 | 179,534 | 8461 |
| RECALL | ||||||||
|---|---|---|---|---|---|---|---|---|
| Dataset | Binner | 0.50 | 0.60 | 0.70 | 0.8 | 0.9 | 0.95 | 0.99 |
| Airways | MetaBAT2 | 82 | 73 | 68 | 57 | 38 | 31 | 21 |
| VAMB | 126 | 123 | 118 | 110 | 77 | 58 | 42 | |
| CedtBin | 142 | 135 | 131 | 119 | 85 | 66 | 48 | |
| Gi | MetaBAT2 | 99 | 96 | 94 | 89 | 80 | 68 | 59 |
| VAMB | 114 | 112 | 110 | 108 | 92 | 86 | 70 | |
| CedtBin | 133 | 130 | 128 | 115 | 93 | 86 | 71 | |
| Oral | MetaBAT2 | 86 | 84 | 81 | 79 | 76 | 61 | 45 |
| VAMB | 180 | 170 | 164 | 156 | 130 | 110 | 82 | |
| CedtBin | 183 | 168 | 163 | 156 | 133 | 113 | 84 | |
| Skin | MetaBAT2 | 105 | 98 | 93 | 77 | 68 | 57 | 48 |
| VAMB | 140 | 139 | 136 | 119 | 94 | 79 | 62 | |
| CedtBin | 154 | 150 | 143 | 125 | 100 | 85 | 73 | |
| Urog | MetaBAT2 | 75 | 73 | 71 | 69 | 67 | 63 | 45 |
| VAMB | 118 | 113 | 106 | 99 | 87 | 74 | 51 | |
| CedtBin | 132 | 122 | 115 | 105 | 95 | 83 | 58 | |
| RECALL | ||||||||
|---|---|---|---|---|---|---|---|---|
| Dataset | Binner | 0.50 | 0.60 | 0.70 | 0.8 | 0.9 | 0.95 | 0.99 |
| Airways | MetaBAT2 | 78 | 71 | 64 | 55 | 39 | 30 | 19 |
| VAMB | 103 | 100 | 96 | 90 | 61 | 43 | 28 | |
| CedtBin | 105 | 104 | 99 | 92 | 65 | 47 | 32 | |
| Gi | MetaBAT2 | 92 | 88 | 87 | 83 | 76 | 67 | 57 |
| VAMB | 81 | 80 | 80 | 80 | 71 | 68 | 57 | |
| CedtBin | 111 | 105 | 104 | 100 | 85 | 81 | 68 | |
| Oral | MetaBAT2 | 85 | 84 | 79 | 76 | 68 | 57 | 40 |
| VAMB | 129 | 124 | 121 | 117 | 98 | 81 | 59 | |
| CedtBin | 130 | 124 | 119 | 116 | 97 | 82 | 60 | |
| Skin | MetaBAT2 | 101 | 95 | 91 | 75 | 65 | 53 | 44 |
| VAMB | 108 | 107 | 104 | 90 | 66 | 58 | 47 | |
| CedtBin | 115 | 111 | 109 | 101 | 79 | 63 | 55 | |
| Urog | MetaBAT2 | 65 | 62 | 61 | 61 | 58 | 55 | 40 |
| VAMB | 71 | 71 | 68 | 66 | 59 | 55 | 40 | |
| CedtBin | 82 | 79 | 73 | 69 | 65 | 60 | 52 | |
| RECALL | ||||||||
|---|---|---|---|---|---|---|---|---|
| Dataset | Binner | 0.50 | 0.60 | 0.70 | 0.8 | 0.9 | 0.95 | 0.99 |
| Airways | MetaBAT2 | 49 | 44 | 43 | 35 | 24 | 17 | 9 |
| VAMB | 55 | 53 | 51 | 49 | 30 | 17 | 10 | |
| CedtBin | 57 | 56 | 53 | 49 | 34 | 23 | 13 | |
| Gi | MetaBAT2 | 56 | 54 | 52 | 45 | 39 | 32 | 28 |
| VAMB | 45 | 44 | 44 | 44 | 37 | 35 | 32 | |
| CedtBin | 55 | 55 | 52 | 48 | 43 | 40 | 35 | |
| Oral | MetaBAT2 | 57 | 54 | 53 | 48 | 44 | 38 | 27 |
| VAMB | 65 | 63 | 62 | 61 | 53 | 46 | 36 | |
| CedtBin | 63 | 62 | 60 | 60 | 55 | 47 | 35 | |
| Skin | MetaBAT2 | 65 | 63 | 62 | 53 | 48 | 39 | 34 |
| VAMB | 55 | 55 | 54 | 51 | 39 | 34 | 28 | |
| CedtBin | 68 | 65 | 64 | 62 | 53 | 45 | 39 | |
| Urog | MetaBAT2 | 33 | 31 | 31 | 30 | 27 | 25 | 21 |
| VAMB | 31 | 31 | 31 | 29 | 25 | 24 | 19 | |
| CedtBin | 35 | 33 | 32 | 30 | 29 | 29 | 23 | |
| RECALL | ||||||||
|---|---|---|---|---|---|---|---|---|
| Dataset | Binner | 0.50 | 0.60 | 0.70 | 0.8 | 0.9 | 0.95 | 0.99 |
| MetaHIT | MetaBAT2 | 80 | 77 | 57 | 25 | 3 | 0 | 0 |
| VAMB | 104 | 101 | 90 | 62 | 23 | 8 | 0 | |
| CedtBin | 113 | 112 | 95 | 66 | 25 | 9 | 0 | |
| Dataset | TNF | Dec_TNF | CedtBin | DBSCAN of CedtBin | Annoy-DBSCAN of CedtBin | |
|---|---|---|---|---|---|---|
| Airways | Time | 1 min 31 s | 2 min 18 s | 55 min 55 s | 5.00 s | 38.73 s |
| Memory | 55,506.55 MiB | 5246.63 MiB | 8680.34 MiB | 628.23 MiB | 725.45 MiB | |
| Gi | Time | 1 min 4 s | 1 min 12 s | 45 min 54 s | 3.36 s | 22.14 s |
| Memory | 9959.82 MiB | 3167.38 MiB | 5813.71 MiB | 206.89 MiB | 316.33 MiB | |
| Oral | Time | 1 min 33 s | 3 min 2 s | 51 min 38 s | 5.17 s | 35.56 s |
| Memory | 32,263.70 MiB | 4292.60 MiB | 6917.91 MiB | 516.80 MiB | 617.94 MiB | |
| Skin | Time | 1 min 38 s | 2 min 15 s | 51 min 38 s | 5.49 s | 33.35 s |
| Memory | 41,357.69 MiB | 5744.48 MiB | 8236.53 MiB | 556.55 MiB | 671.12 MiB | |
| Urog | Time | 42.91 s | 45.81 s | 28 min 38 s | 0.80 s | 8.13 s |
| Memory | 13,101.41 MiB | 3002.84 MiB | 4082.00 MiB | 80.91 MiB | 109.49 MiB | |
| MetaHIT | Time | 50.54 s | 1 min 41 s | 46 min 37 s | 4.75 s | 32.72 s |
| Memory | 49,518.02 MiB | 4245.40 MiB | 6813.13 MiB | 389.90 MiB | 490.63 MiB |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, L.; Shi, J.; Huang, B. Binning Metagenomic Contigs Using Contig Embedding and Decomposed Tetranucleotide Frequency. Biology 2024, 13, 755. https://doi.org/10.3390/biology13100755
Fu L, Shi J, Huang B. Binning Metagenomic Contigs Using Contig Embedding and Decomposed Tetranucleotide Frequency. Biology. 2024; 13(10):755. https://doi.org/10.3390/biology13100755
Chicago/Turabian StyleFu, Long, Jiabin Shi, and Baohua Huang. 2024. "Binning Metagenomic Contigs Using Contig Embedding and Decomposed Tetranucleotide Frequency" Biology 13, no. 10: 755. https://doi.org/10.3390/biology13100755
APA StyleFu, L., Shi, J., & Huang, B. (2024). Binning Metagenomic Contigs Using Contig Embedding and Decomposed Tetranucleotide Frequency. Biology, 13(10), 755. https://doi.org/10.3390/biology13100755