
Chromosome-Level Genome Assembly of Protosalanx chinensis and Response to Air Exposure Stress



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA and RNA Sequencing
2.2. Sequencing QC and Genome Assembly
2.3. Identification of Repetitive Sequences
2.4. Genome Annotation
2.5. Phylogenetic and Gene Family Analysis
2.6. Transcriptome under Air Exposure Stress
3. Results
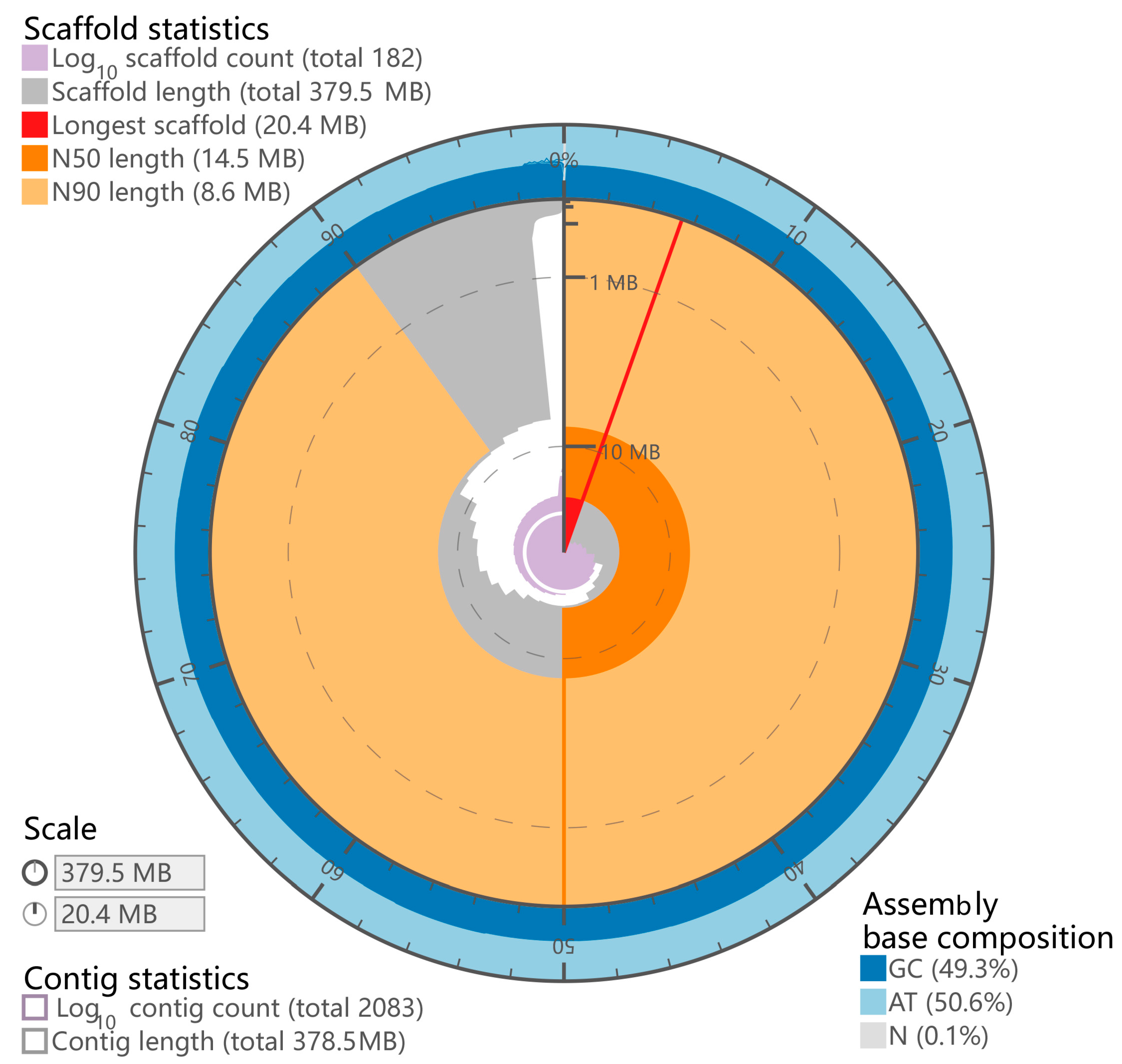
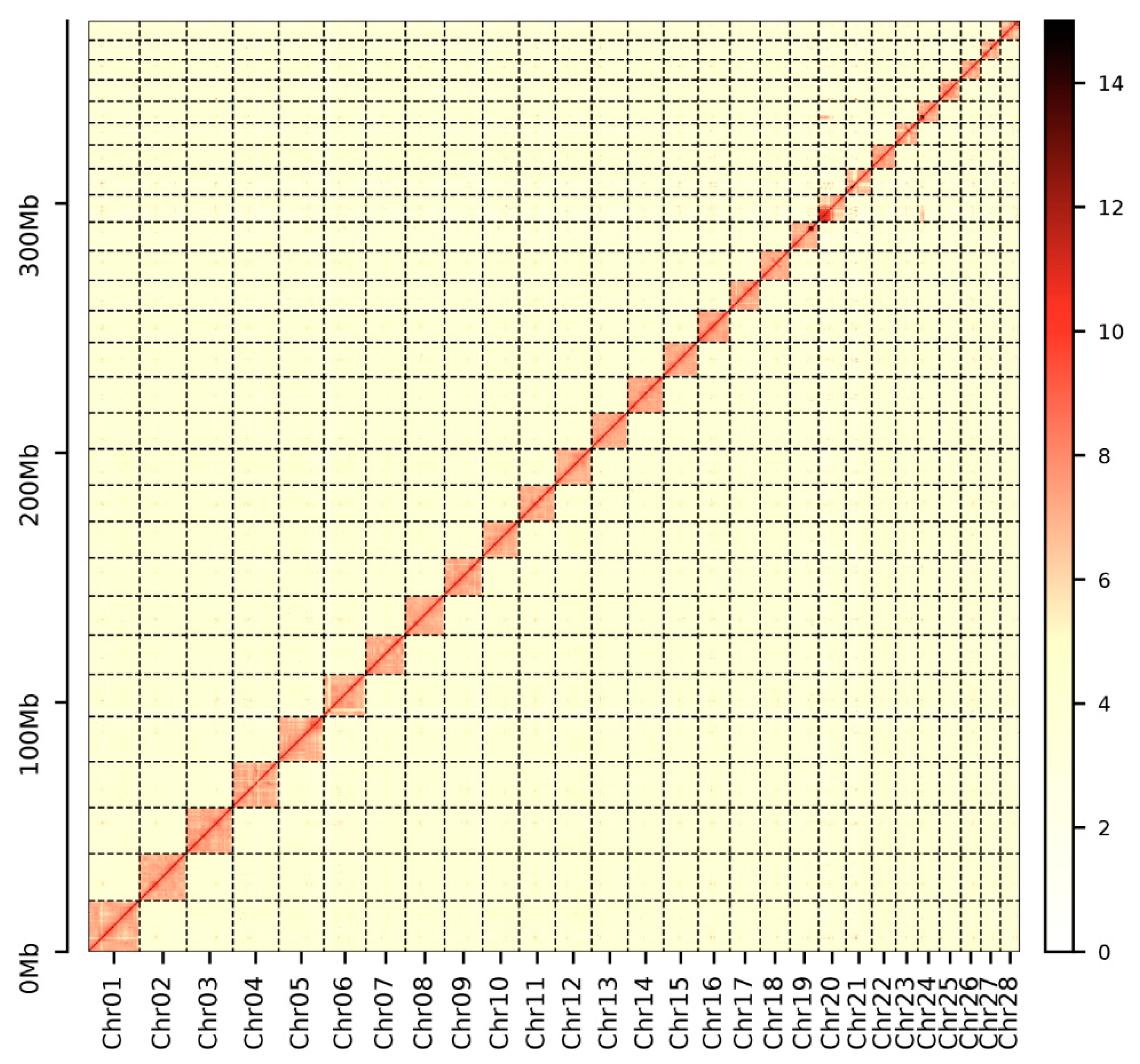
3.1. Chromosome-Scale Genome Assembly
3.2. Repetitive Sequences
3.3. Genome Annotation
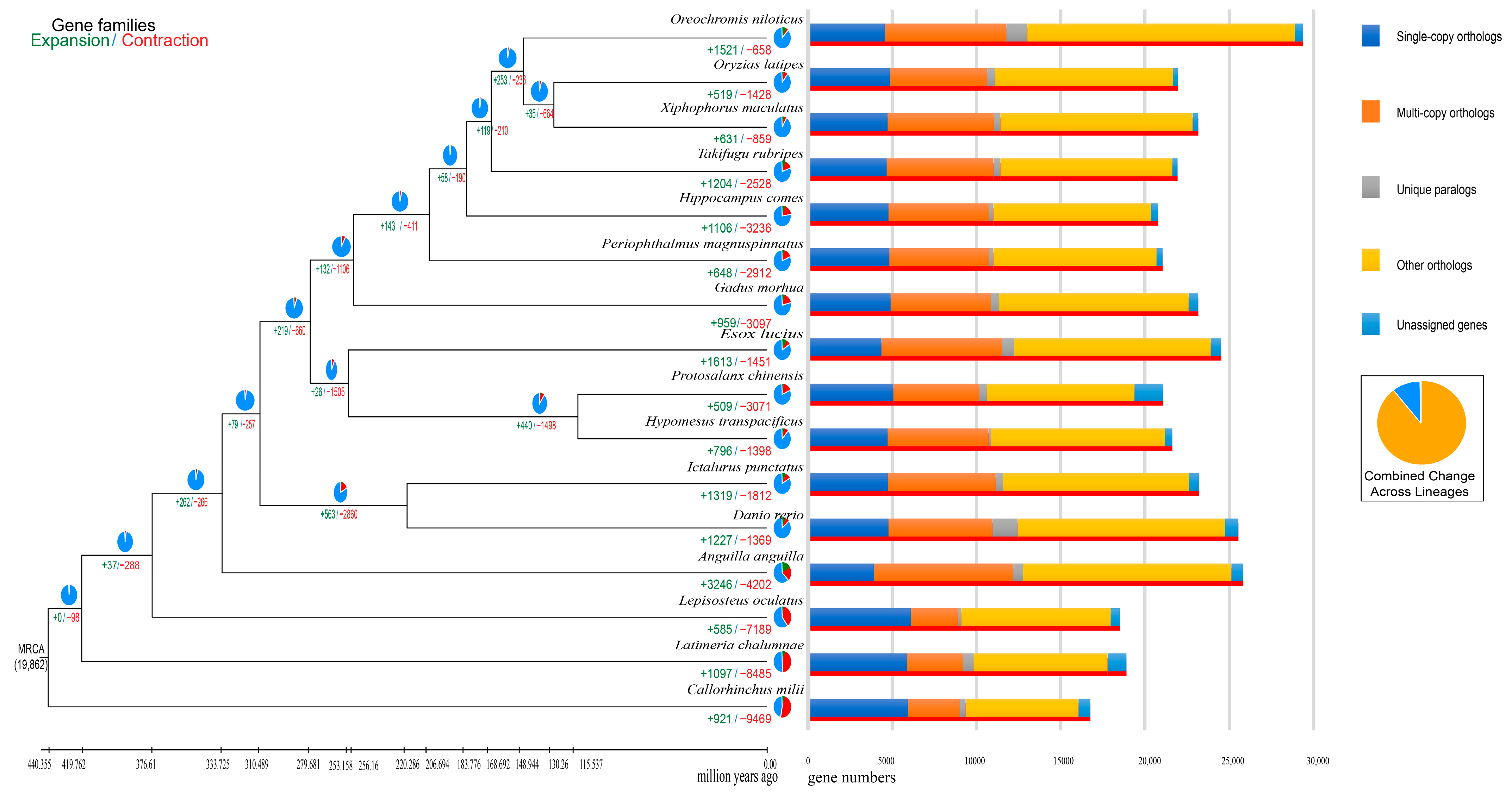
3.4. Phylogenetic and Gene Family Analysis
3.5. Gene Expression under Air Exposure Stress
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tang, F.; Gao, W.; Li, H.; Liu, W. Biology and fishery ecology of Protosalanx chinensis: A review. J. Fish. China 2020, 44, 2100–2111. [Google Scholar]
- Liu, K.; Xu, D.; Li, J.; Bian, C.; Duan, J.; Zhou, Y.; Zhang, M.; You, X.; You, Y.; Chen, J.; et al. Whole Genome Sequencing of Chinese Clearhead Icefish, Protosalanx hyalocranius. Gigascience 2017, 6, giw012. [Google Scholar] [CrossRef]
- Zhang, J.; Qi, J.; Shi, F.; Pan, H.; Liu, M.; Tian, R.; Geng, Y.; Li, H.; Qu, Y.; Chen, J.; et al. Insights into the Evolution of Neoteny from the Genome of the Asian Icefish Protosalanx chinensis. iScience 2020, 23, 101267. [Google Scholar] [CrossRef]
- Wang, Z.; Fu, C.; Lei, G. Biodiversity of Chinese Icefishes (Salangidae) and Their Conserving Strategies. Biodivers. Sci. 2002, 10, 416–424. [Google Scholar] [CrossRef]
- Zhang, Y.; Dong, S.; Wang, Q.; Sun, Z. The isozyme genetic structures in large icefish (Protosalanx hyalocranius) and Taihu Lake icefish (Neosalanx taihuensis). J. Dalian Fish. Coll. 2005, 20, 111–115. [Google Scholar]
- Jian, Y.; Xun, X.; HongBo, L. Bioaccumulation of elements in icefish Protosalanx hyalocranius from the Taihu Lake and Hongze Lake. Oceanol. Et Limnol. Sin. Hai Yang Yu Hu Chao 2009, 40, 201–207. [Google Scholar]
- Kang, B.; Deng, J.; Wang, Z.; Zhang, J. Transplantation of Icefish (Salangidae) in China: Glory or Disaster? Rev. Aquac. 2015, 7, 13–27. [Google Scholar] [CrossRef]
- Skrzynska, A.K.; Maiorano, E.; Bastaroli, M.; Naderi, F.; Míguez, J.M.; Martínez-Rodríguez, G.; Mancera, J.M.; Martos-Sitcha, J.A. Impact of Air Exposure on Vasotocinergic and Isotocinergic Systems in Gilthead Sea Bream (Sparus aurata): New Insights on Fish Stress Response. Front. Physiol. 2018, 9, 96. [Google Scholar] [CrossRef]
- Ikert, H.; Lynch, M.D.J.; Doxey, A.C.; Giesy, J.P.; Servos, M.R.; Katzenback, B.A.; Craig, P.M. High Throughput Sequencing of MicroRNA in Rainbow Trout Plasma, Mucus, and Surrounding Water Following Acute Stress. Front. Physiol. 2021, 11, 588313. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce Acceleration-Supported Software for Integrated Quality Control and Preprocessing of High-Throughput Sequencing Data. Gigascience 2018, 7, gix120. [Google Scholar] [CrossRef]
- Marçais, G.; Kingsford, C. A Fast, Lock-Free Approach for Efficient Parallel Counting of Occurrences of k-Mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast Reference-Free Genome Profiling from Short Reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-Resolved de Novo Assembly Using Phased Assembly Graphs with Hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Roach, M.J.; Schmidt, S.A.; Borneman, A.R. Purge Haplotigs: Allelic Contig Reassignment for Third-Gen Diploid Genome Assemblies. BMC Bioinform. 2018, 19, 460. [Google Scholar] [CrossRef] [PubMed]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef]
- Dudchenko, O.; Batra, S.S.; Omer, A.D.; Nyquist, S.K.; Hoeger, M.; Durand, N.C.; Shamim, M.S.; Machol, I.; Lander, E.S.; Aiden, A.P.; et al. De Novo Assembly of the Aedes aegypti Genome Using Hi-C Yields Chromosome-Length Scaffolds. Science 2017, 356, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, H. LTR_FINDER: An Efficient Tool for the Prediction of Full-Length LTR Retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef] [PubMed]
- Chen, N. Using Repeat Masker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinform. 2004, 5, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem Repeats Finder: A Program to Analyze DNA Sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome Assembly from Long-Read RNA-Seq Alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated Eukaryotic Gene Structure Annotation Using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef]
- Keilwagen, J.; Hartung, F.; Grau, J. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-Seq Data. Gene Predict. Methods Protoc. 2019, 1962, 161–177. [Google Scholar]
- Stanke, M.; Waack, S. Gene Prediction with a Hidden Markov Model and a New Intron Submodel. Bioinformatics 2003, 19, ii215–ii225. [Google Scholar] [CrossRef]
- Korf, I. Gene Finding in Novel Genomes. BMC Bioinformatics 2004, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic Orthology Inference for Comparative Genomics. Genome Biol. 2019, 20, 1–14. [Google Scholar] [CrossRef]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for Large-Scale Multiple Sequence Alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A Toolkit for Detection and Evolutionary Analysis of Gene Synteny and Collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- De Bie, T.; Cristianini, N.; Demuth, J.P.; Hahn, M.W. CAFE: A Computational Tool for the Study of Gene Family Evolution. Bioinformatics 2006, 22, 1269–1271. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Trapnell, C.; Donaghey, J.; Rinn, J.L.; Pachter, L. Improving RNA-Seq Expression Estimates by Correcting for Fragment Bias. Genome Biol. 2011, 12, R22. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT Protein Sequence Data Bank and Its Supplement TrEMBL in 1999. Nucleic Acids Res. 1999, 27, 49–54. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Gui, J.-F.; Zhou, L.; Li, X.-Y. Rethinking Fish Biology and Biotechnologies in the Challenge Era for Burgeoning Genome Resources and Strengthening Food Security. Water Biol. Secur. 2022, 1, 100002. [Google Scholar] [CrossRef]
- Lu, G.; Luo, M. Genomes of Major Fishes in World Fisheries and Aquaculture: Status, Application and Perspective. Aquac. Fish. 2020, 5, 163–173. [Google Scholar] [CrossRef]
- Tang, F.-J.; Liu, W.; Wang, J.-L.; Li, Z.; Xie, S.-G. Diet Composition and Transition of Clearhead Icefish (Protosalanx hyalocranius) in Lake Xingkai; Kunming Institute of Zoology, Chinese Academy of Sciences: Kunming, China, 2013; Volume 34, pp. 493–498. [Google Scholar] [PubMed]
- Harris, R.M.; Hofmann, H.A. Seeing Is Believing: Dynamic Evolution of Gene Families. Proc. Natl. Acad. Sci. USA 2015, 112, 1252–1253. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Li, W.; Wu, B.; Chen, J.; Chen, X. Transcriptome Analysis Reveals New Insights into Immune Response to Hypoxia Challenge of Large Yellow Croaker (Larimichthys crocea). Fish Shellfish Immunol. 2020, 98, 738–747. [Google Scholar] [CrossRef]
- Lu, Y.-P.; Zheng, P.-H.; Zhang, X.-X.; Li, J.-T.; Zhang, Z.-L.; Xu, J.-R.; Meng, Y.-Q.; Li, J.-J.; Xian, J.-A.; Wang, A.-L. New Insights into the Regulation Mechanism of Red Claw Crayfish (Cherax quadricarinatus) Hepatopancreas under Air Exposure Using Transcriptome Analysis. Fish Shellfish Immunol. 2023, 132, 108505. [Google Scholar] [CrossRef]
- Wu, L.; Tang, D.; Shen, C.; Bai, Y.; Jiang, K.; Yu, Q.; Wang, Z. Comparative Transcriptome Analysis of the Gills of Cardisoma armatum Provides Novel Insights into the Terrestrial Adaptive Related Mechanism of Air Exposure Stress. Genomics 2021, 113, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.N.; Allen, J.I.; McVeigh, A.; Shaw, J. Lysosomal and Autophagic Reactions as Predictive Indicators of Environmental Impact in Aquatic Animals. Autophagy 2006, 2, 217–220. [Google Scholar] [CrossRef]
- McGeachy, M.J.; Cua, D.J.; Gaffen, S.L. The IL-17 Family of Cytokines in Health and Disease. Immunity 2019, 50, 892–906. [Google Scholar] [CrossRef]
- Xue, T.; Liu, Y.; Cao, M.; Zhang, X.; Fu, Q.; Yang, N.; Li, C. Genome-Wide Identification of Interleukin-17 (IL-17)/Interleukin-17 Receptor (IL- 17R) in Turbot (Scophthalmus maximus) and Expression Pattern Analysis after Vibrio anguillarum Infection. Dev. Comp. Immunol. 2021, 121, 104070. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, R.; Wang, X.; Zhu, H.; Tian, Z. Transcriptome Analysis Reveals Molecular Mechanisms Responsive to Acute Cold Stress in the Tropical Stenothermal Fish Tiger Barb (Puntius tetrazona). BMC Genom. 2020, 21, 737. [Google Scholar] [CrossRef]
- Brijs, J.; Sandblom, E.; Axelsson, M.; Sundell, K.; Sundh, H.; Huyben, D.; Broström, R.; Kiessling, A.; Berg, C.; Gräns, A. The Final Countdown: Continuous Physiological Welfare Evaluation of Farmed Fish during Common Aquaculture Practices before and during Harvest. Aquaculture 2018, 495, 903–911. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, W.; Li, C. Heat and Hypoxia Exposure Mediates Circadian Rhythms Response via Methylation Modification in Apostichopus Japonicas. Front. Mar. Sci. 2021, 8, 721465. [Google Scholar] [CrossRef]
- Jerônimo, R.; Moraes, M.N.; de Assis, L.V.M.; Ramos, B.C.; Rocha, T.; Castrucci, A.M.d.L. Thermal Stress in Danio Rerio: A Link between Temperature, Light, Thermo-TRP Channels, and Clock Genes. J. Therm. Biol. 2017, 68, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, Y.; Wang, Z.; Xu, K.; Xiao, X.; Mu, W. Comparison of Effects in Sustained and Diel-Cycling Hypoxia on Hypoxia Tolerance, Histology, Physiology and Expression of Clock Genes in High Latitude Fish Phoxinus lagowskii. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2021, 260, 111020. [Google Scholar] [CrossRef]
- Peng, L.-B.; Wang, D.; Han, T.; Wen, Z.; Cheng, X.; Zhu, Q.-L.; Zheng, J.-L.; Wang, P. Histological, Antioxidant, Apoptotic and Transcriptomic Responses under Cold Stress and the Mitigation of Blue Wavelength Light of Zebrafish Eyes. Aquac. Rep. 2022, 26, 101291. [Google Scholar] [CrossRef]
- Mao, Y.; Zhang, G. A Complete, Telomere-to-Telomere Human Genome Sequence Presents New Opportunities for Evolutionary Genomics. Nat. Methods 2022, 19, 635–638. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Zhang, X.; Tang, X.; Zhou, Y.; Ding, Y.; Liu, H. Chromosome-Level Genome Assembly of Protosalanx chinensis and Response to Air Exposure Stress. Biology 2023, 12, 1266. https://doi.org/10.3390/biology12091266
Zhou Y, Zhang X, Tang X, Zhou Y, Ding Y, Liu H. Chromosome-Level Genome Assembly of Protosalanx chinensis and Response to Air Exposure Stress. Biology. 2023; 12(9):1266. https://doi.org/10.3390/biology12091266
Chicago/Turabian StyleZhou, Yanfeng, Xizhao Zhang, Xuemei Tang, Yifan Zhou, Yuting Ding, and Hong Liu. 2023. "Chromosome-Level Genome Assembly of Protosalanx chinensis and Response to Air Exposure Stress" Biology 12, no. 9: 1266. https://doi.org/10.3390/biology12091266
APA StyleZhou, Y., Zhang, X., Tang, X., Zhou, Y., Ding, Y., & Liu, H. (2023). Chromosome-Level Genome Assembly of Protosalanx chinensis and Response to Air Exposure Stress. Biology, 12(9), 1266. https://doi.org/10.3390/biology12091266