
Intercellular Adhesion Molecule 1: More than a Leukocyte Adhesion Molecule

Simple Summary
Abstract
1. Introduction
2. ICAM1 Gene
2.1. Gene Structure and Regulation of Transcription
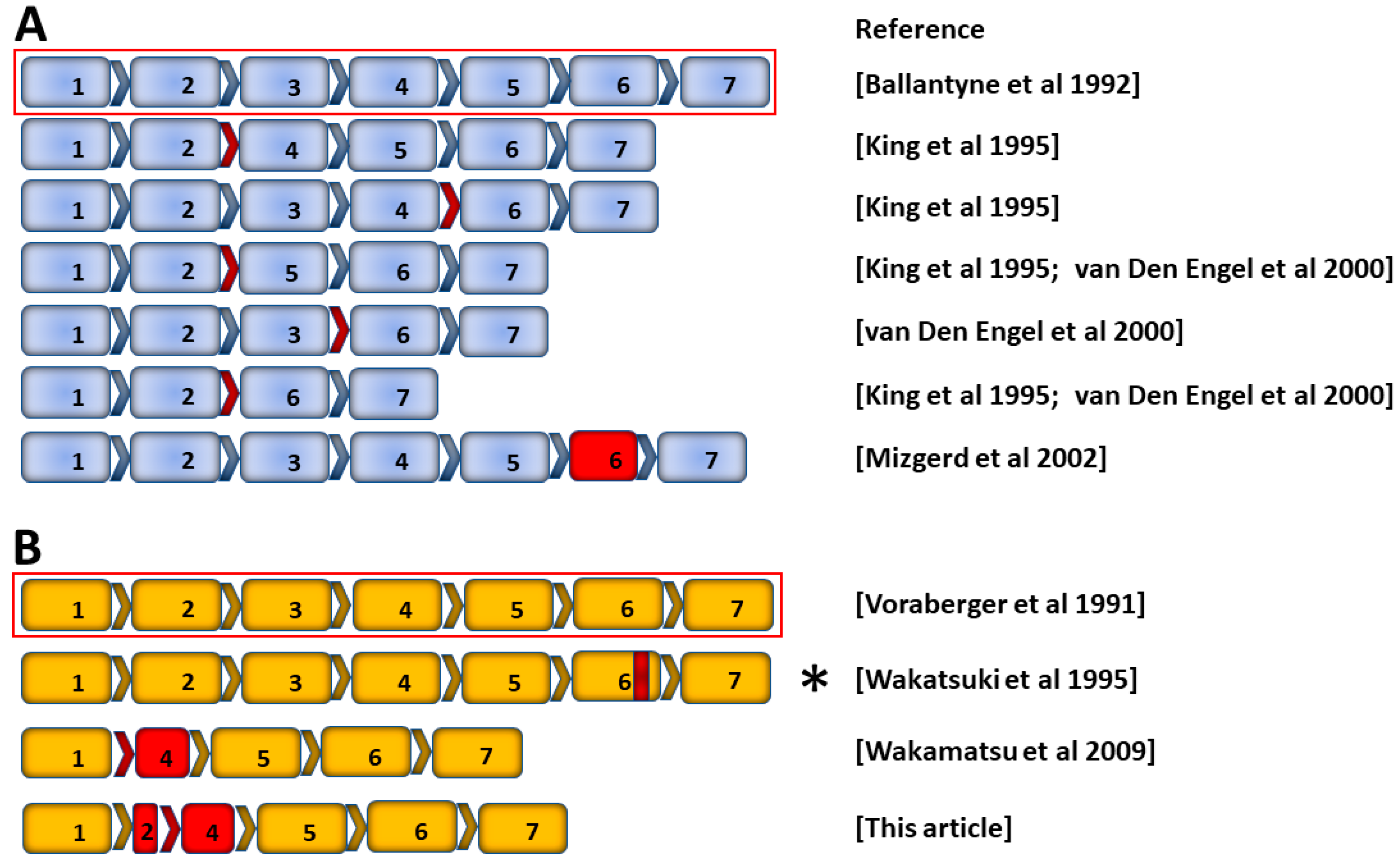
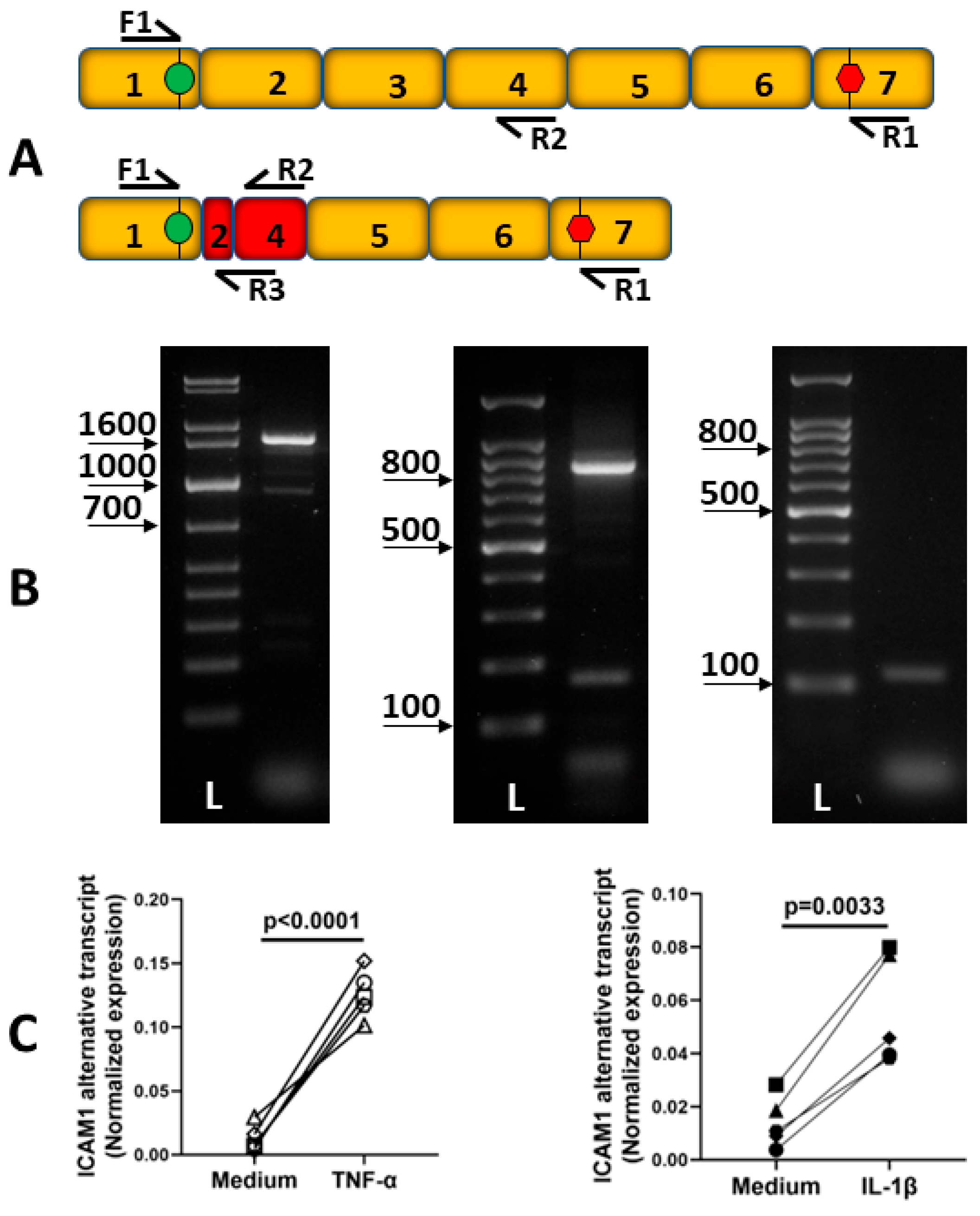
2.2. Alternative Transcripts
2.3. Post-Transcriptional Regulation
2.4. Icam1 Gene-Deficient Mouse Models
3. ICAM-1 Protein
3.1. Protein Structure
3.2. Post-Translational Modification
3.3. ICAM-1 Ligands
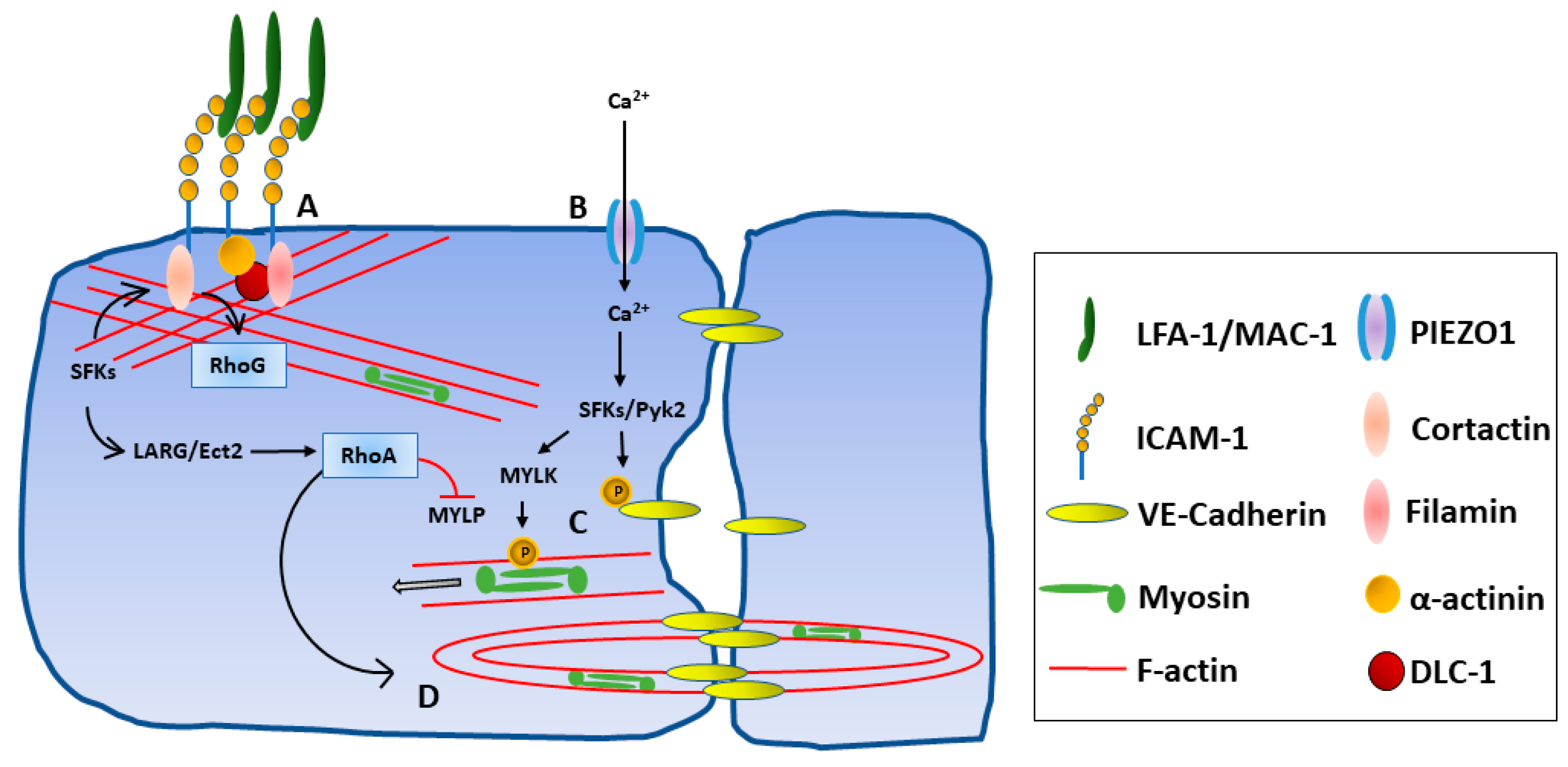
3.4. Intracellular Signaling
3.5. Soluble ICAM-1 Protein
4. Roles of ICAM-1 in Immune Function
4.1. Leukocyte Trafficking
4.2. Immune Synapse
4.3. Wound Healing
5. Involvement of ICAM-1 in Disease
5.1. Sepsis
5.2. Malaria
5.3. Rhinovirus Infections
5.4. Multiple Sclerosis
5.5. Inflammatory Bowel Disease
5.6. Uveitis
5.7. Cancer
5.8. Cardiovascular Diseases
6. Therapeutic Targeting of ICAM-1
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dustin, M.L.; Rothlein, R.; Bhan, A.K.; Dinarello, C.A.; Springer, T.A. Induction by IL 1 and interferon-gamma: Tissue distribution, biochemistry, and function of a natural adherence molecule (ICAM-1). J. Immunol. 1986, 137, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Roebuck, K.A.; Finnegan, A. Regulation of intercellular adhesion molecule-1 (CD54) gene expression. J. Leukoc. Biol. 1999, 66, 876–888. [Google Scholar] [CrossRef] [PubMed]
- Voraberger, G.; Schäfer, R.; Stratowa, C. Cloning of the human gene for intercellular adhesion molecule 1 and analysis of its 5’-regulatory region. Induction by cytokines and phorbol ester. J. Immunol. 1991, 147, 2777–2786. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qu, Y.; Niu, T.; Wang, H.; Liu, K. O-GlcNAc modification of Sp1 mediates hyperglycaemia-induced ICAM-1 up-regulation in endothelial cells. Biochem. Biophys. Res. Commun. 2017, 484, 79–84. [Google Scholar] [CrossRef]
- Liu, H.; Cheng, W.L.; Jiang, X.; Wang, P.X.; Fang, C.; Zhu, X.Y.; Huang, Z.; She, Z.G.; Li, H. Ablation of interferon regulatory factor 3 protects against atherosclerosis in apolipoprotein E-deficient mice. Hypertension 2017, 69, 510–520. [Google Scholar] [CrossRef]
- Roebuck, K.A.; Rahman, A.; Lakshminarayanan, V.; Janakidevi, K.; Malik, A.B. H2O2 and tumor necrosis factor-α activate intercellular adhesion molecule 1 (ICAM-1) gene transcription through distinct cis-regulatory elements within the ICAM-1 promoter. J. Biol. Chem. 1995, 270, 18966–18974. [Google Scholar] [CrossRef]
- Ledebur, H.C.; Parks, T.P. Transcriptional regulation of the intercellular adhesion molecule-1 gene by inflammatory cytokines in human endothelial cells. Essential roles of a variant NF-κB site and p65 homodimers. J. Biol. Chem. 1995, 270, 933–943. [Google Scholar] [CrossRef]
- Caldenhoven, E.; Coffer, P.; Yuan, J.; Van de Stolpe, A.; Horn, F.; Kruijer, W.; Van der Saag, P.T. Stimulation of the human intercellular adhesion molecule-1 promoter by interleukin-6 and interferon-gamma involves binding of distinct factors to a palindromic response element. J. Biol. Chem. 1994, 269, 21146–21154. [Google Scholar] [CrossRef]
- Morigi, M.; Angioletti, S.; Imberti, B.; Donadelli, R.; Micheletti, G.; Figliuzzi, M.; Remuzzi, A.; Zoja, C.; Remuzzi, G. Leukocyte-endothelial interaction is augmented by high glucose concentrations and hyperglycemia in a NF-kB-dependent fashion. J. Clin. Investig. 1998, 101, 1905–1915. [Google Scholar] [CrossRef]
- Morigi, M.; Zoja, C.; Figliuzzi, M.; Foppolo, M.; Micheletti, G.; Bontempelli, M.; Saronni, M.; Remuzzi, G.; Remuzzi, A. Fluid shear stress modulates surface expression of adhesion molecules by endothelial cells. Blood 1995, 85, 1696–1703. [Google Scholar] [CrossRef]
- Wertheimer, S.J.; Myers, C.L.; Wallace, R.W.; Parks, T.P. Intercellular adhesion molecule-1 gene expression in human endothelial cells. Differential regulation by tumor necrosis factor-alpha and phorbol myristate acetate. J. Biol. Chem. 1992, 267, 12030–12035. [Google Scholar] [CrossRef]
- Diamond, M.S.; Staunton, D.E.; de Fougerolles, A.R.; Stacker, S.A.; Garcia-Aguilar, J.; Hibbs, M.L.; Springer, T.A. ICAM-1 (CD54): A counter-receptor for Mac-1 (CD11b/CD18). J. Cell Biol. 1990, 111, 3129–3139. [Google Scholar] [CrossRef]
- Staunton, D.E.; Dustin, M.L.; Erickson, H.P.; Springer, T.A. The arrangement of the immunoglobulin-like domains of ICAM-1 and the binding sites for LFA-1 and rhinovirus. Cell 1990, 61, 243–254. [Google Scholar] [CrossRef]
- McCourt, P.A.; Ek, B.; Forsberg, N.; Gustafson, S. Intercellular adhesion molecule-1 is a cell surface receptor for hyaluronan. J. Biol. Chem. 1994, 269, 30081–30084. [Google Scholar] [CrossRef]
- Languino, L.R.; Plescia, J.; Duperray, A.; Brian, A.A.; Plow, E.F.; Geltosky, J.E.; Altieri, D.C. Fibrinogen mediates leukocyte adhesion to vascular endothelium through an ICAM-1-dependent pathway. Cell 1993, 73, 1423–1434. [Google Scholar] [CrossRef]
- Greve, J.M.; Davis, G.; Meyer, A.M.; Forte, C.P.; Yost, S.C.; Marlor, C.W.; Kamarck, M.E.; McClelland, A. The major human rhinovirus receptor is ICAM-1. Cell 1989, 56, 839–847. [Google Scholar] [CrossRef]
- Smith, J.D.; Craig, A.G.; Kriek, N.; Hudson-Taylor, D.; Kyes, S.; Fagan, T.; Pinches, R.; Baruch, D.I.; Newbold, C.I.; Miller, L.H. Identification of a Plasmodium falciparum intercellular adhesion molecule-1 binding domain: A parasite adhesion trait implicated in cerebral malaria. Proc. Natl. Acad. Sci. USA 2000, 97, 1766–1771. [Google Scholar] [CrossRef]
- Barragan, A.; Brossier, F.; Sibley, L.D. Transepithelial migration of Toxoplasma gondii involves an interaction of intercellular adhesion molecule 1 (ICAM-1) with the parasite adhesin MIC2. Cell Microbiol. 2005, 7, 561–568. [Google Scholar] [CrossRef]
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704. [Google Scholar] [CrossRef]
- Grakoui, A.; Bromley, S.K.; Sumen, C.; Davis, M.M.; Shaw, A.S.; Allen, P.M.; Dustin, M.L. The immunological synapse: A molecular machine controlling T cell activation. Science 1999, 285, 221–227. [Google Scholar] [CrossRef]
- Dressler, J.; Bachmann, L.; Muller, E. Enhanced expression of ICAM-1 (CD 54) in human skin wounds: Diagnostic value in legal medicine. Inflamm. Res. 1997, 46, 434–435. [Google Scholar] [CrossRef] [PubMed]
- Zonneveld, R.; Martinelli, R.; Shapiro, N.I.; Kuijpers, T.W.; Plötz, F.B.; Carman, C.V. Soluble adhesion molecules as markers for sepsis and the potential pathophysiological discrepancy in neonates, children and adults. Crit. Care 2014, 18, 204. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.E.; Siahaan, T.J. Targeting ICAM-1/LFA-1 interaction for controlling autoimmune diseases: Designing peptide and small molecule inhibitors. Peptides 2003, 24, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Reina, M.; Espel, E. Role of LFA-1 and ICAM-1 in Cancer. Cancers 2017, 9, 153. [Google Scholar] [CrossRef]
- Lawson, C.; Wolf, S. ICAM-1 signaling in endothelial cells. Pharmacol. Rep. 2009, 61, 22–32. [Google Scholar] [CrossRef]
- Kilgannon, P.; Turner, T.; Meyer, J.; Wisdom, W.; Gallatin, W.M. Mapping of the ICAM-5 (Telencephalin) gene, a neuronal member of the ICAM family, to a location between ICAM-1 and ICAM-3 on human chromosome 19p13.2. Genomics 1998, 54, 328–330. [Google Scholar] [CrossRef]
- Sansom, D.; Borrow, J.; Solomon, E.; Trowsdale, J. The human ICAM2 gene maps to 17q23–25. Genomics 1991, 11, 462–464. [Google Scholar] [CrossRef]
- Hou, J.; Baichwal, V.; Cao, Z. Regulatory elements and transcription factors controlling basal and cytokine-induced expression of the gene encoding intercellular adhesion molecule 1. Proc. Natl. Acad. Sci. USA 1994, 91, 11641–11645. [Google Scholar] [CrossRef]
- Lv, Y.; Kim, K.; Sheng, Y.; Cho, J.; Qian, Z.; Zhao, Y.Y.; Hu, G.; Pan, D.; Malik, A.B.; Hu, G. YAP controls endothelial activation and vascular inflammation through TRAF6. Circ. Res. 2018, 123, 43–56. [Google Scholar] [CrossRef]
- Kempe, S.; Kestler, H.; Lasar, A.; Wirth, T. NF-κB controls the global pro-inflammatory response in endothelial cells: Evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 2005, 33, 5308–5319. [Google Scholar] [CrossRef]
- Valenzuela, N.M. Late phase endothelial cell inflammation is characterized by interferon response genes and driven by JAK/STAT, not NFκB. Vascul. Pharmacol. 2022, 146, 107090. [Google Scholar] [CrossRef]
- Murphy, J.M.; Jeong, K.; Rodriguez, Y.A.R.; Kim, J.H.; Ahn, E.E.; Lim, S.S. FAK and Pyk2 activity promote TNF-α and IL-1β-mediated pro-inflammatory gene expression and vascular inflammation. Sci. Rep. 2019, 9, 7617. [Google Scholar] [CrossRef]
- Zhu, L.; Yang, H.; Chao, Y.; Gu, Y.; Zhang, J.; Wang, F.; Yu, W.; Ye, P.; Chu, P.; Kong, X.; et al. Akt phosphorylation regulated by IKKε in response to low shear stress leads to endothelial inflammation via activating IRF3. Cell. Signal. 2021, 80, 109900. [Google Scholar] [CrossRef]
- Look, D.C.; Pelletier, M.R.; Holtzman, M.J. Selective interaction of a subset of interferon-gamma response element-binding proteins with the intercellular adhesion molecule-1 (ICAM-1) gene promoter controls the pattern of expression on epithelial cells. J. Biol. Chem. 1994, 269, 8952–8958. [Google Scholar] [CrossRef]
- Walter, M.J.; Look, D.C.; Tidwell, R.M.; Roswit, W.T.; Holtzman, M.J. Targeted inhibition of interferon-gamma-dependent intercellular adhesion molecule-1 (ICAM-1) expression using dominant-negative Stat1. J. Biol. Chem. 1997, 272, 28582–28589. [Google Scholar] [CrossRef]
- Wung, B.S.; Ni, C.W.; Wang, D.L. ICAM-1 induction by TNFalpha and IL-6 is mediated by distinct pathways via Rac in endothelial cells. J. Biomed. Sci. 2005, 12, 91–101. [Google Scholar] [CrossRef]
- King, P.D.; Sandberg, E.T.; Selvakumar, A.; Fang, P.; Beaudet, A.L.; Dupont, B. Novel isoforms of murine intercellular adhesion molecule-1 generated by alternative RNA splicing. J. Immunol. 1995, 154, 6080–6093. [Google Scholar] [CrossRef]
- van Den Engel, N.K.; Heidenthal, E.; Vinke, A.; Kolb, H.; Martin, S. Circulating forms of intercellular adhesion molecule (ICAM)-1 in mice lacking membranous ICAM-1. Blood 2000, 95, 1350–1355. [Google Scholar] [CrossRef]
- Mizgerd, J.P.; Spieker, M.R.; Lupa, M.M. Exon truncation by alternative splicing of murine ICAM-1. Physiol. Genomics 2002, 12, 47–51. [Google Scholar] [CrossRef]
- Horley, K.J.; Carpenito, C.; Baker, B.; Takei, F. Molecular cloning of murine intercellular adhesion molecule (ICAM-1). EMBO J. 1989, 8, 2889–2896. [Google Scholar] [CrossRef]
- Ballantyne, C.M.; Sligh, J.E., Jr.; Dai, X.Y.; Beaudet, A.L. Characterization of the murine Icam-1 gene. Genomics 1992, 14, 1076–1080. [Google Scholar] [CrossRef] [PubMed]
- Human Gene: ICAM1. Available online: https://asia.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000090339;r=19:10271093-10286615;t=ENST00000264832 (accessed on 20 April 2023).
- Wakamatsu, A.; Kimura, K.; Yamamoto, J.; Nishikawa, T.; Nomura, N.; Sugano, S.; Isogai, T. Identification and functional analyses of 11,769 full-length human cDNAs focused on alternative splicing. DNA Res. 2009, 16, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Wakatsuki, T.; Kimura, K.; Kimura, F.; Shinomiya, N.; Ohtsubo, M.; Ishizawa, M.; Yamamoto, M. A distinct mRNA encoding a soluble form of ICAM-1 molecule expressed in human tissues. Cell. Adhes. Commun. 1995, 3, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Ohh, M.; Smith, C.A.; Carpenito, C.; Takei, F. Regulation of intercellular adhesion molecule-1 gene expression involves multiple mRNA stabilization mechanisms: Effects of interferon-γ and phorbol myristate acetate. Blood 1994, 84, 2632–2639. [Google Scholar] [CrossRef]
- Shi, J.X.; Su, X.; Xu, J.; Zhang, W.Y.; Shi, Y. MK2 posttranscriptionally regulates TNF-α-induced expression of ICAM-1 and IL-8 via tristetraprolin in human pulmonary microvascular endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L793–L799. [Google Scholar] [CrossRef]
- Wu, T.; Shi, J.X.; Geng, S.; Zhou, W.; Shi, Y.; Su, X. The MK2/HuR signaling pathway regulates TNF-α-induced ICAM-1 expression by promoting the stabilization of ICAM-1 mRNA. BMC Pulm. Med. 2016, 16, 84. [Google Scholar] [CrossRef]
- Suárez, Y.; Wang, C.; Manes, T.D.; Pober, J.S. Cutting edge: TNF-induced microRNAs regulate TNF-induced expression of E-selectin and intercellular adhesion molecule-1 on human endothelial cells: Feedback control of inflammation. J. Immunol. 2010, 184, 21–25. [Google Scholar] [CrossRef]
- Ohta, M.; Kihara, T.; Toriuchi, K.; Aoki, H.; Iwaki, S.; Kakita, H.; Yamada, Y.; Aoyama, M. IL-6 promotes cell adhesion in human endothelial cells via microRNA-126-3p suppression. Exp. Cell. Res. 2020, 393, 112094. [Google Scholar] [CrossRef]
- Yao, M.Y.; Zhang, W.H.; Ma, W.T.; Liu, Q.H.; Xing, L.H.; Zhao, G.F. Long non-coding RNA MALAT1 exacerbates acute respiratory distress syndrome by upregulating ICAM-1 expression via microRNA-150-5p downregulation. Aging 2020, 12, 6570–6585. [Google Scholar] [CrossRef]
- Zhang, L.; Meng, W.; Chen, X.; Ning, Y.; Sun, M.; Wang, R. MiR-150-5p regulates the functions of type 2 innate lymphoid cells via the ICAM-1/p38 MAPK axis in allergic rhinitis. Mol. Cell. Biochem. 2022, 477, 1009–1022. [Google Scholar] [CrossRef]
- Tabet, F.; Vickers, K.C.; Torres, L.F.C.; Wiese, C.B.; Shoucri, B.M.; Lambert, G.; Catherinet, C.; Prado-Lourenco, L.; Levin, M.G.; Thacker, S.; et al. HDL-transferred microRNA-223 regulates ICAM-1 expression in endothelial cells. Nat. Commun. 2014, 5, 3292. [Google Scholar] [CrossRef]
- Guo, M.; Fan, S.; Chen, Q.; Jia, C.; Qiu, M.; Bu, Y.; Tang, W.H.; Zhang, Y. Platelet-derived microRNA-223 attenuates TNF-α induced monocytes adhesion to arterial endothelium by targeting ICAM-1 in Kawasaki disease. Front. Immunol. 2022, 13, 922868. [Google Scholar] [CrossRef]
- Szilágyi, B.; Fejes, Z.; Rusznyák, Á.; Fenyvesi, F.; Pócsi, M.; Halmi, S.; Griger, Z.; Kunapuli, S.P.; Kappelmayer, J.; Nagy, B., Jr. Platelet microparticles enriched in miR-223 reduce ICAM-1-dependent vascular inflammation in septic conditions. Front. Physiol. 2021, 12, 658524. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhao, C.; Wang, Y.; Li, X.; Xue, Y.; Ma, C. LncRNA NEAT1 promote inflammatory responses in coronary slow flow through regulating miR-148b-3p/ICAM-1 axis. J. Inflamm. Res. 2021, 14, 2445–2463. [Google Scholar] [CrossRef]
- Ding, S.; Liu, J.; Han, X.; Ding, W.; Liu, Z.; Zhu, Y.; Zhan, W.; Wan, Y.; Gai, S.; Hou, J.; et al. ICAM-1-related noncoding RNA accelerates atherosclerosis by amplifying NF-κB signaling. J. Mol. Cell. Cardiol. 2022, 170, 75–86. [Google Scholar] [CrossRef]
- Lumsden, A.L.; Ma, Y.; Ashander, L.M.; Stempel, A.J.; Keating, D.J.; Smith, J.R.; Appukuttan, B. ICAM-1-related long non-coding RNA: Promoter analysis and expression in human retinal endothelial cells. BMC Res. Notes 2018, 11, 285. [Google Scholar] [CrossRef]
- Guo, W.; Liu, S.; Cheng, Y.; Lu, L.; Shi, J.; Xu, G.; Li, N.; Cheng, K.; Wu, M.; Cheng, S.; et al. ICAM-1-related noncoding RNA in cancer stem cells maintains ICAM-1 expression in hepatocellular carcinoma. Clin. Cancer Res. 2016, 22, 2041–2050. [Google Scholar] [CrossRef]
- Xu, H.; Gonzalo, J.A.; St. Pierre, Y.; Williams, I.R.; Kupper, T.S.; Cotran, R.S.; Springer, T.A.; Gutierrez-Ramos, J.C. Leukocytosis and resistance to septic shock in intercellular adhesion molecule 1-deficient mice. J. Exp. Med. 1994, 180, 95–109. [Google Scholar] [CrossRef]
- Sligh, J.E., Jr.; Ballantyne, C.M.; Rich, S.S.; Hawkins, H.K.; Smith, C.W.; Bradley, A.; Beaudet, A.L. Inflammatory and immune responses are impaired in mice deficient in intercellular adhesion molecule 1. Proc. Natl. Acad. Sci. USA 1993, 90, 8529–8533. [Google Scholar] [CrossRef]
- Ramos, T.N.; Bullard, D.C.; Barnum, S.R. ICAM-1: Isoforms and phenotypes. J. Immunol. 2014, 192, 4469–4474. [Google Scholar] [CrossRef]
- Hayflick, J.S.; Kilgannon, P.; Gallatin, W.M. The intercellular adhesion molecule (ICAM) family of proteins. New members and novel functions. Immunol. Res. 1998, 17, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.C.U.; Jablonski-Westrich, D.; Haubold, U.; Gutierrez-Ramos, J.-C.; Springer, T.; Hamann, A. Overlapping and selective roles of endothelial intercellular adhesion molecule-1 (ICAM-1) and ICAM-2 in lymphocyte trafficking 1. J. Immunol. 2003, 171, 2588–2593. [Google Scholar] [CrossRef] [PubMed]
- Steiner, O.; Coisne, C.; Cecchelli, R.; Boscacci, R.; Deutsch, U.; Engelhardt, B.; Lyck, R. Differential roles for endothelial ICAM-1, ICAM-2, and VCAM-1 in shear-resistant T cell arrest, polarization, and directed crawling on blood–brain barrier endothelium. J. Immunol. 2010, 185, 4846–4855. [Google Scholar] [CrossRef] [PubMed]
- Jahromi, N.H.; Marchetti, L.; Moalli, F.; Duc, D.; Basso, C.; Tardent, H.; Kaba, E.; Deutsch, U.; Pot, C.; Sallusto, F.; et al. Intercellular adhesion molecule-1 (ICAM-1) and ICAM-2 differentially contribute to peripheral activation and CNS entry of autoaggressive Th1 and Th17 cells in experimental autoimmune encephalomyelitis. Front. Immunol. 2020, 10, 3056. [Google Scholar] [CrossRef]
- Yang, Y.; Jun, C.-D.; Liu, J.-H.; Zhang, R.; Joachimiak, A.; Springer, T.A.; Wang, J.-H. Structural basis for dimerization of ICAM-1 on the cell surface. Mol. Cell. 2004, 14, 269–276. [Google Scholar] [CrossRef]
- Miller, J.; Knorr, R.; Ferrone, M.; Houdei, R.; Carron, C.P.; Dustin, M.L. Intercellular adhesion molecule-1 dimerization and its consequences for adhesion mediated by lymphocyte function associated-1. J. Exp. Med. 1995, 182, 1231–1241. [Google Scholar] [CrossRef]
- Reilly, P.L.; Woska, J.R., Jr.; Jeanfavre, D.D.; McNally, E.; Rothlein, R.; Bormann, B.J. The native structure of intercellular adhesion molecule-1 (ICAM-1) is a dimer. Correlation with binding to LFA-1. J. Immunol. 1995, 155, 529–532. [Google Scholar] [CrossRef]
- Chen, X.; Kim, T.D.; Carman, C.V.; Mi, L.Z.; Song, G.; Springer, T.A. Structural plasticity in Ig superfamily domain 4 of ICAM-1 mediates cell surface dimerization. Proc. Natl. Acad. Sci. USA 2007, 104, 15358–15363. [Google Scholar] [CrossRef]
- Diamond, M.S.; Staunton, D.E.; Marlin, S.D.; Springer, T.A. Binding of the integrin Mac-1 (CD11b/CD18) to the third immunoglobulin-like domain of ICAM-1 (CD54) and its regulation by glycosylation. Cell 1991, 65, 961–971. [Google Scholar] [CrossRef]
- Jiménez, D.; Roda-Navarro, P.; Springer, T.A.; Casasnovas, J.M. Contribution of N-linked glycans to the conformation and function of intercellular adhesion molecules (ICAMs). J. Biol. Chem. 2005, 280, 5854–5861. [Google Scholar] [CrossRef]
- Scott, D.W.; Dunn, T.S.; Ballestas, M.E.; Litovsky, S.H.; Patel, R.P. Identification of a high-mannose ICAM-1 glycoform: Effects of ICAM-1 hypoglycosylation on monocyte adhesion and outside in signaling. Am. J. Physiol. Cell. Physiol. 2013, 305, C228–C237. [Google Scholar] [CrossRef]
- Regal-McDonald, K.; Xu, B.; Barnes, J.W.; Patel, R.P. High-mannose intercellular adhesion molecule-1 enhances CD16(+) monocyte adhesion to the endothelium. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H1028–H1038. [Google Scholar] [CrossRef]
- Springer, T.A. Traffic signals for lymphocyte recirculation and leukocyte emigration: The multistep paradigm. Cell 1994, 76, 301–314. [Google Scholar] [CrossRef]
- Lu, C.; Shimaoka, M.; Ferzly, M.; Oxvig, C.; Takagi, J.; Springer, T.A. An isolated, surface-expressed I domain of the integrin αLβ2 is sufficient for strong adhesive function when locked in the open conformation with a disulfide bond. Proc. Natl. Acad. Sci. USA 2001, 98, 2387–2392. [Google Scholar] [CrossRef]
- Wen, L.; Lyu, Q.; Ley, K.; Goult, B.T. Structural basis of β2 integrin inside-out activation. Cells 2022, 11, 3039. [Google Scholar] [CrossRef]
- Shimaoka, M.; Xiao, T.; Liu, J.H.; Yang, Y.; Dong, Y.; Jun, C.D.; McCormack, A.; Zhang, R.; Joachimiak, A.; Takagi, J.; et al. Structures of the alpha L I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell 2003, 112, 99–111. [Google Scholar] [CrossRef]
- Lee, J.-O.; Rieu, P.; Arnaout, M.A.; Liddington, R. Crystal structure of the A domain from the alpha subunit of integrin CR3 (CD11 b/CD18). Cell 1995, 80, 631–638. [Google Scholar] [CrossRef]
- Mao, D.; Lü, S.; Li, N.; Zhang, Y.; Long, M. Conformational stability analyses of alpha subunit I domain of LFA-1 and Mac-1. PLoS ONE 2011, 6, e24188. [Google Scholar] [CrossRef]
- Sans, E.; Delachanal, E.; Duperray, A. Analysis of the roles of ICAM-1 in neutrophil transmigration using a reconstituted mammalian cell expression model: Implication of ICAM-1 cytoplasmic domain and Rho-dependent signaling pathway. J. Immunol. 2001, 166, 544–551. [Google Scholar] [CrossRef]
- Sulimai, N.; Brown, J.; Lominadze, D. The role of nuclear factor-kappa B in fibrinogen-induced inflammatory responses in cultured primary neurons. Biomolecules 2022, 12, 1741. [Google Scholar] [CrossRef]
- Etienne, S.; Adamson, P.; Greenwood, J.; Strosberg, A.D.; Cazaubon, S.; Couraud, P.O. ICAM-1 signaling pathways associated with Rho activation in microvascular brain endothelial cells. J. Immunol. 1998, 161, 5755–5761. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, O.; Yanez-Mo, M.; Serrador, J.M.; Montoya, M.C.; Vicente-Manzanares, M.; Tejedor, R.; Furthmayr, H.; Sanchez-Madrid, F. Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J. Cell Biol. 2002, 157, 1233–1245. [Google Scholar] [CrossRef] [PubMed]
- Celli, L.; Ryckewaert, J.-J.; Delachanal, E.; Duperray, A. Evidence of a functional role for interaction between ICAM-1 and nonmuscle α-actinins in leukocyte diapedesis1. J. Immunol. 2006, 177, 4113–4121. [Google Scholar] [CrossRef] [PubMed]
- Kanters, E.; van Rijssel, J.; Hensbergen, P.J.; Hondius, D.; Mul, F.P.; Deelder, A.M.; Sonnenberg, A.; van Buul, J.D.; Hordijk, P.L. Filamin B mediates ICAM-1-driven leukocyte transendothelial migration. J. Biol. Chem. 2008, 283, 31830–31839. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, L.; van der Stoel, M.; Rianna, C.; van Stalborch, A.M.; de Ligt, A.; Hoogenboezem, M.; Tol, S.; van Rijssel, J.; Szulcek, R.; Bogaard, H.J.; et al. Stiffness-induced endothelial DLC-1 expression forces leukocyte spreading through stabilization of the ICAM-1 adhesome. Cell Rep. 2018, 24, 3115–3124. [Google Scholar] [CrossRef]
- Lessey-Morillon, E.C.; Osborne, L.D.; Monaghan-Benson, E.; Guilluy, C.; O’Brien, E.T.; Superfine, R.; Burridge, K. The RhoA guanine nucleotide exchange factor, LARG, mediates ICAM-1-dependent mechanotransduction in endothelial cells to stimulate transendothelial migration. J. Immunol. 2014, 192, 3390–3398. [Google Scholar] [CrossRef]
- Heemskerk, N.; Schimmel, L.; Oort, C.; van Rijssel, J.; Yin, T.; Ma, B.; van Unen, J.; Pitter, B.; Huveneers, S.; Goedhart, J.; et al. F-actin-rich contractile endothelial pores prevent vascular leakage during leukocyte diapedesis through local RhoA signalling. Nat. Commun. 2016, 7, 10493. [Google Scholar] [CrossRef]
- Lyck, R.; Reiss, Y.; Gerwin, N.; Greenwood, J.; Adamson, P.; Engelhardt, B. T-cell interaction with ICAM-1/ICAM-2 double-deficient brain endothelium in vitro: The cytoplasmic tail of endothelial ICAM-1 is necessary for transendothelial migration of T cells. Blood 2003, 102, 3675–3683. [Google Scholar] [CrossRef]
- Sano, H.; Nakagawa, N.; Chiba, R.; Kurasawa, K.; Saito, Y.; Iwamoto, I. Cross-linking of intercellular adhesion molecule-1 induces interleukin-8 and RANTES production through the activation of MAP kinases in human vascular endothelial cells. Biochem. Biophys. Res. Commun. 1998, 250, 694–698. [Google Scholar] [CrossRef]
- Dragoni, S.; Hudson, N.; Kenny, B.A.; Burgoyne, T.; McKenzie, J.A.; Gill, Y.; Blaber, R.; Futter, C.E.; Adamson, P.; Greenwood, J.; et al. Endothelial MAPKs direct ICAM-1 signaling to divergent inflammatory functions. J. Immunol. 2017, 198, 4074–4085. [Google Scholar] [CrossRef]
- Giorelli, M.; De Blasi, A.; Defazio, G.; Avolio, C.; Iacovelli, L.; Livrea, P.; Trojano, M. Differential regulation of membrane bound and soluble ICAM 1 in human endothelium and blood mononuclear cells: Effects of interferon beta-1a. Cell. Commun. Adhes. 2002, 9, 259–272. [Google Scholar] [CrossRef][Green Version]
- Champagne, B.; Tremblay, P.; Cantin, A.; St. Pierre, Y. Proteolytic cleavage of ICAM-1 by human neutrophil elastase1. J. Immunol. 1998, 161, 6398–6405. [Google Scholar] [CrossRef]
- Tsakadze, N.L.; Sithu, S.D.; Sen, U.; English, W.R.; Murphy, G.; D’Souza, S.E. Tumor necrosis factor-alpha-converting enzyme (TACE/ADAM-17) mediates the ectodomain cleavage of intercellular adhesion molecule-1 (ICAM-1). J. Biol. Chem. 2006, 281, 3157–3164. [Google Scholar] [CrossRef]
- Leeuwenberg, J.F.; Smeets, E.F.; Neefjes, J.J.; Shaffer, M.A.; Cinek, T.; Jeunhomme, T.M.; Ahern, T.J.; Buurman, W.A. E-selectin and intercellular adhesion molecule-1 are released by activated human endothelial cells in vitro. Immunology 1992, 77, 543–549. [Google Scholar]
- Rothlein, R.; Mainolfi, E.A.; Czajkowski, M.; Marlin, S.D. A form of circulating ICAM-1 in human serum. J. Immunol. 1991, 147, 3788–3793. [Google Scholar] [CrossRef]
- Sessler, C.N.; Windsor, A.C.; Schwartz, M.; Watson, L.; Fisher, B.J.; Sugerman, H.J.; Fowler, A.A., 3rd. Circulating ICAM-1 is increased in septic shock. Am. J. Respir. Crit. Care Med. 1995, 151, 1420–1427. [Google Scholar] [CrossRef]
- Styles, J.N.; Converse, R.R.; Griffin, S.M.; Wade, T.J.; Klein, E.; Nylander-French, L.A.; Stewart, J.R.; Sams, E.; Hudgens, E.; Egorov, A.I. Human cytomegalovirus infections are associated with elevated biomarkers of vascular injury. Front. Cell. Infect. Microbiol. 2020, 10, 334. [Google Scholar] [CrossRef]
- Egorov, A.I.; Converse, R.R.; Griffin, S.M.; Styles, J.N.; Sams, E.; Hudgens, E.; Wade, T.J. Latent Toxoplasma gondii infections are associated with elevated biomarkers of inflammation and vascular injury. BMC Infect. Dis. 2021, 21, 188. [Google Scholar] [CrossRef]
- Kaur, S.; Hussain, S.; Kolhe, K.; Kumar, G.; Tripathi, D.M.; Tomar, A.; Kale, P.; Narayanan, A.; Bihari, C.; Bajpai, M.; et al. Elevated plasma ICAM1 levels predict 28-day mortality in cirrhotic patients with COVID-19 or bacterial sepsis. JHEP Rep. 2021, 3, 100303. [Google Scholar] [CrossRef]
- Sprenger, A.; Schardt, C.; Rotsch, M.; Zehrer, M.; Wolf, M.; Havemann, K.; Heymanns, J. Soluble intercellular adhesion molecule-1 in patients with lung cancer and benign lung diseases. J. Cancer Res. Clin. Oncol. 1997, 123, 632–638. [Google Scholar] [CrossRef]
- Schellerer, V.S.; Langheinrich, M.C.; Zver, V.; Grützmann, R.; Stürzl, M.; Gefeller, O.; Naschberger, E.; Merkel, S. Soluble intercellular adhesion molecule-1 is a prognostic marker in colorectal carcinoma. Int. J. Colorectal Dis. 2019, 34, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Köstler, W.J.; Tomek, S.; Brodowicz, T.; Budinsky, A.C.; Flamm, M.; Hejna, M.; Krainer, M.; Wiltschke, C.; Zielinski, C.C. Soluble ICAM-1 in breast cancer: Clinical significance and biological implications. Cancer Immunol. Immunother. 2001, 50, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.D.; Bielinski, S.J.; Suarez-Lopez, J.R.; Reiner, A.P.; Bailey, K.; Thyagarajan, B.; Carr, J.J.; Duprez, D.A.; Jacobs, D.R., Jr. Circulating soluble intercellular adhesion molecule 1 and subclinical atherosclerosis: The Coronary Artery Risk Development in Young Adults Study. Clin. Chem. 2012, 58, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Verity, D.H.; Wallace, G.R.; Seed, P.T.; Kanawati, C.A.; Ayesh, I.; Holland-Gladwish, J.; Stanford, M.R. Soluble adhesion molecules in Behçet’s disease. Ocul. Immunol. Inflamm. 1998, 6, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Sari, R.A.; Taysi, S.; Erdem, F.; Yilmaz, O.; Keleş, S.; Kiziltunç, A.; Odabaş, A.R.; Cetinkaya, R. Correlation of serum levels of soluble intercellular adhesion molecule-1 with disease activity in systemic lupus erythematosus. Rheumatol. Int. 2002, 21, 149–152. [Google Scholar] [CrossRef]
- Furukawa, S.; Imai, K.; Matsubara, T.; Yone, K.; Yachi, A.; Okumura, K.; Yabuta, K. Increased levels of circulating intercellular adhesion molecule 1 in kawasaki disease. Arthritis Rheum. 1992, 35, 672–677. [Google Scholar] [CrossRef]
- Cush, J.J.; Rothlein, R.; Lindsley, H.B.; Mainolfi, E.A.; Lipsky, P.E. Increased levels of circulating intercellular adhesion molecule 1 in the sera of patients with rheumatoid arthritis. Arthritis Rheum. 1993, 36, 1098–1102. [Google Scholar] [CrossRef]
- Robledo, O.; Papaioannou, A.; Ochietti, B.; Beauchemin, C.; Legault, D.; Cantin, A.; King, P.D.; Daniel, C.; Alakhov, V.Y.; Potworowski, E.F.; et al. ICAM-1 isoforms: Specific activity and sensitivity to cleavage by leukocyte elastase and cathepsin G. Eur. J. Immunol. 2003, 33, 1351–1360. [Google Scholar] [CrossRef]
- Fiore, E.; Fusco, C.; Romero, P.; Stamenkovic, I. Matrix metalloproteinase 9 (MMP-9/gelatinase B) proteolytically cleaves ICAM-1 and participates in tumor cell resistance to natural killer cell-mediated cytotoxicity. Oncogene 2002, 21, 5213–5223. [Google Scholar] [CrossRef]
- Morsing, S.K.H.; Rademakers, T.; Brouns, S.L.N.; Stalborch, A.D.V.; Donners, M.; van Buul, J.D. ADAM10-mediated cleavage of ICAM-1 is involved in neutrophil transendothelial migration. Cells 2021, 10, 232. [Google Scholar] [CrossRef]
- Zarbock, A.; Ley, K.; McEver, R.P.; Hidalgo, A. Leukocyte ligands for endothelial selectins: Specialized glycoconjugates that mediate rolling and signaling under flow. Blood 2011, 118, 6743–6751. [Google Scholar] [CrossRef]
- Wójciak-Stothard, B.; Williams, L.; Ridley, A.J. Monocyte adhesion and spreading on human endothelial cells is dependent on Rho-regulated receptor clustering. J. Cell. Biol. 1999, 145, 1293–1307. [Google Scholar] [CrossRef]
- Sumagin, R.; Prizant, H.; Lomakina, E.; Waugh, R.E.; Sarelius, I.H. LFA-1 and Mac-1 define characteristically different intralumenal crawling and emigration patterns for monocytes and neutrophils in situ. J. Immunol. 2010, 185, 7057–7066. [Google Scholar] [CrossRef]
- Phillipson, M.; Heit, B.; Colarusso, P.; Liu, L.; Ballantyne, C.M.; Kubes, P. Intraluminal crawling of neutrophils to emigration sites: A molecularly distinct process from adhesion in the recruitment cascade. J. Exp. Med. 2006, 203, 2569–2575. [Google Scholar] [CrossRef]
- Carman, C.V.; Springer, T.A. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J. Cell. Biol. 2004, 167, 377–388. [Google Scholar] [CrossRef]
- Heemskerk, N.; van Rijssel, J.; van Buul, J.D. Rho-GTPase signaling in leukocyte extravasation: An endothelial point of view. Cell. Adh. Migr. 2014, 8, 67–75. [Google Scholar] [CrossRef]
- Allingham, M.J.; van Buul, J.D.; Burridge, K. ICAM-1-mediated, Src- and Pyk2-dependent vascular endothelial cadherin tyrosine phosphorylation is required for leukocyte transendothelial migration. J. Immunol. 2007, 179, 4053–4064. [Google Scholar] [CrossRef]
- Grönloh, M.L.B.; Arts, J.J.G.; Martínez, S.P.; van der Veen, A.A.; Kempers, L.; van Steen, A.C.I.; Roelofs, J.; Nolte, M.A.; Goedhart, J.; van Buul, J.D. Endothelial transmigration hotspots limit vascular leakage through heterogeneous expression of ICAM-1. EMBO Rep. 2023, 24, e55483. [Google Scholar] [CrossRef]
- Dias, M.C.; Quesada, A.O.; Soldati, S.; Bösch, F.; Gruber, I.; Hildbrand, T.; Sönmez, D.; Khire, T.; Witz, G.; McGrath, J.L.; et al. Brain endothelial tricellular junctions as novel sites for T cell diapedesis across the blood-brain barrier. J. Cell. Sci. 2021, 134, jcs253880. [Google Scholar] [CrossRef]
- Sumagin, R.; Sarelius, I.H. Intercellular adhesion molecule-1 enrichment near tricellular endothelial junctions is preferentially associated with leukocyte transmigration and signals for reorganization of these junctions to accommodate leukocyte passage. J. Immunol. 2010, 184, 5242–5252. [Google Scholar] [CrossRef]
- Abadier, M.; Jahromi, N.H.; Alves, L.C.; Boscacci, R.; Vestweber, D.; Barnum, S.; Deutsch, U.; Engelhardt, B.; Lyck, R. Cell surface levels of endothelial ICAM-1 influence the transcellular or paracellular T-cell diapedesis across the blood–brain barrier. Eur. J. Immunol. 2015, 45, 1043–1058. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.T.; Chang, H.Y.; Haraldsen, G.; Jahnsen, F.L.; Troyanskaya, O.G.; Chang, D.S.; Wang, Z.; Rockson, S.G.; van de Rijn, M.; Botstein, D.; et al. Endothelial cell diversity revealed by global expression profiling. Proc. Natl. Acad. Sci. USA 2003, 100, 10623–10628. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.R.; Choi, D.; Chipps, T.J.; Pan, Y.; Zamora, D.O.; Davies, M.H.; Babra, B.; Powers, M.R.; Planck, S.R.; Rosenbaum, J.T. Unique gene expression profiles of donor-matched human retinal and choroidal vascular endothelial cells. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2676–2684. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.R.; David, L.L.; Appukuttan, B.; Wilmarth, P.A. Angiogenic and immunologic proteins identified by deep proteomic profiling of human retinal and choroidal vascular endothelial cells: Potential targets for new biologic drugs. Am. J. Ophthalmol. 2018, 193, 197–229. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, A.S.; Schewitz-Bowers, L.P.; Wei, L.; Lee, R.W.; Smith, J.R. Intercellular adhesion molecule 1 mediates migration of Th1 and Th17 cells across human retinal vascular endothelium. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6917–6925. [Google Scholar] [CrossRef]
- Bharadwaj, A.S.; Stempel, A.J.; Olivas, A.; Franzese, S.E.; Ashander, L.M.; Ma, Y.; Lie, S.; Appukuttan, B.; Smith, J.R. Molecular signals involved in human B cell migration into the retina: In vitro investigation of ICAM-1, VCAM-1, and CXCL13. Ocul. Immunol. Inflamm. 2017, 25, 811–819. [Google Scholar] [CrossRef]
- Sumagin, R.; Sarelius, I.H. TNF-alpha activation of arterioles and venules alters distribution and levels of ICAM-1 and affects leukocyte-endothelial cell interactions. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2116–H2125. [Google Scholar] [CrossRef]
- Huang, A.J.; Manning, J.E.; Bandak, T.M.; Ratau, M.C.; Hanser, K.R.; Silverstein, S.C. Endothelial cell cytosolic free calcium regulates neutrophil migration across monolayers of endothelial cells. J. Cell. Biol. 1993, 120, 1371–1380. [Google Scholar] [CrossRef]
- Etienne-Manneville, S.; Manneville, J.B.; Adamson, P.; Wilbourn, B.; Greenwood, J.; Couraud, P.O. ICAM-1-coupled cytoskeletal rearrangements and transendothelial lymphocyte migration involve intracellular calcium signaling in brain endothelial cell lines. J. Immunol. 2000, 165, 3375–3383. [Google Scholar] [CrossRef]
- Wang, S.; Wang, B.; Shi, Y.; Möller, T.; Stegmeyer, R.I.; Strilic, B.; Li, T.; Yuan, Z.; Wang, C.; Wettschureck, N.; et al. Mechanosensation by endothelial PIEZO1 is required for leukocyte diapedesis. Blood 2022, 140, 171–183. [Google Scholar] [CrossRef]
- Rodríguez-Fernández, J.L.; Criado-García, O. The actin cytoskeleton at the immunological synapse of dendritic cells. Front. Cell. Dev. Biol. 2021, 9, 679500. [Google Scholar] [CrossRef]
- Zaretsky, I.; Atrakchi, O.; Mazor, R.D.; Stoler-Barak, L.; Biram, A.; Feigelson, S.W.; Gitlin, A.D.; Engelhardt, B.; Shulman, Z. ICAMs support B cell interactions with T follicular helper cells and promote clonal selection. J. Exp. Med. 2017, 214, 3435–3448. [Google Scholar] [CrossRef]
- Scholer, A.; Hugues, S.; Boissonnas, A.; Fetler, L.; Amigorena, S. Intercellular adhesion molecule-1-dependent stable interactions between T Cells and dendritic cells determine CD8+ T cell memory. Immunity 2008, 28, 258–270. [Google Scholar] [CrossRef]
- Kozlovski, S.; Atrakchi, O.; Feigelson, S.W.; Shulman, Z.; Alon, R. Stable contacts of naïve CD4 T cells with migratory dendritic cells are ICAM-1-dependent but dispensable for proliferation in vivo. Cell. Adh Migr. 2019, 13, 314–320. [Google Scholar] [CrossRef]
- Feigelson, S.W.; Solomon, A.; Biram, A.; Hatzav, M.; Lichtenstein, M.; Regev, O.; Kozlovski, S.; Varol, D.; Curato, C.; Leshkowitz, D.; et al. ICAMs are not obligatory for functional immune synapses between naive CD4 T cells and lymph node DCs. Cell Rep. 2018, 22, 849–859. [Google Scholar] [CrossRef]
- Nagaoka, T.; Kaburagi, Y.; Hamaguchi, Y.; Hasegawa, M.; Takehara, K.; Steeber, D.A.; Tedder, T.F.; Sato, S. Delayed wound healing in the absence of intercellular adhesion molecule-1 or L-selectin expression. Am. J. Pathol. 2000, 157, 237–247. [Google Scholar] [CrossRef]
- Gay, A.N.; Mushin, O.P.; Lazar, D.A.; Naik-Mathuria, B.J.; Yu, L.; Gobin, A.; Smith, C.W.; Olutoye, O.O. Wound healing characteristics of ICAM-1 null mice devoid of all isoforms of ICAM-1. J. Surg. Res. 2011, 171, e1–e7. [Google Scholar] [CrossRef][Green Version]
- Sumagin, R.; Brazil, J.C.; Nava, P.; Nishio, H.; Alam, A.; Luissint, A.C.; Weber, D.A.; Neish, A.S.; Nusrat, A.; Parkos, C.A. Neutrophil interactions with epithelial-expressed ICAM-1 enhances intestinal mucosal wound healing. Mucosal Immunol. 2016, 9, 1151–1162. [Google Scholar] [CrossRef]
- Dalal, P.J.; Sumagin, R. Emerging functions of ICAM-1 in macrophage efferocytosis and wound healing. J. Cell. Immunol. 2020, 2, 250–253. [Google Scholar] [CrossRef]
- Wiesolek, H.L.; Bui, T.M.; Lee, J.J.; Dalal, P.; Finkielsztein, A.; Batra, A.; Thorp, E.B.; Sumagin, R. Intercellular adhesion molecule 1 functions as an efferocytosis receptor in inflammatory macrophages. Am. J. Pathol. 2020, 190, 874–885. [Google Scholar] [CrossRef]
- Davies, S.P.; Reynolds, G.M.; Wilkinson, A.L.; Li, X.; Rose, R.; Leekha, M.; Liu, Y.S.; Gandhi, R.; Buckroyd, E.; Grove, J.; et al. Hepatocytes delete regulatory T cells by enclysis, a CD4+ T cell engulfment process. Cell. Rep. 2019, 29, 1610–1620.e1614. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, F.; Pape, H.C.; Harwood, P.; Müller, K.; Hoevel, P.; Pütz, C.; Siemann, A.; Krettek, C.; van Griensven, M. Role of adhesion molecule ICAM in the pathogenesis of polymicrobial sepsis. Exp. Toxicol. Pathol. 2005, 56, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Ode, Y.; Aziz, M.; Wang, P. CIRP increases ICAM-1(+) phenotype of neutrophils exhibiting elevated iNOS and NETs in sepsis. J. Leukoc. Biol. 2018, 103, 693–707. [Google Scholar] [CrossRef] [PubMed]
- Bohatschek, M.; Werner, A.; Raivich, G. Systemic LPS injection leads to granulocyte influx into normal and injured brain: Effects of ICAM-1 deficiency. Exp. Neurol. 2001, 172, 137–152. [Google Scholar] [CrossRef]
- Berendt, A.R.; Simmons, D.L.; Tansey, J.; Newbold, C.I.; Marsh, K. Intercellular adhesion molecule-1 is an endothelial cell adhesion receptor for Plasmodium falciparum. Nature 1989, 341, 57–59. [Google Scholar] [CrossRef]
- Staunton, D.E.; Merluzzi, V.J.; Rothlein, R.; Barton, R.; Marlin, S.D.; Springer, T.A. A cell adhesion molecule, ICAM-1, is the major surface receptor for rhinoviruses. Cell 1989, 56, 849–853. [Google Scholar] [CrossRef]
- Terajima, M.; Yamaya, M.; Sekizawa, K.; Okinaga, S.; Suzuki, T.; Yamada, N.; Nakayama, K.; Ohrui, T.; Oshima, T.; Numazaki, Y.; et al. Rhinovirus infection of primary cultures of human tracheal epithelium: Role of ICAM-1 and IL-1beta. Am. J. Physiol. 1997, 273, L749–L759. [Google Scholar] [CrossRef]
- Bullard, D.C.; Hu, X.; Schoeb, T.R.; Collins, R.G.; Beaudet, A.L.; Barnum, S.R. Intercellular adhesion molecule-1 expression is required on multiple cell types for the development of experimental autoimmune encephalomyelitis. J. Immunol. 2007, 178, 851–857. [Google Scholar] [CrossRef]
- Taniguchi, T.; Tsukada, H.; Nakamura, H.; Kodama, M.; Fukuda, K.; Saito, T.; Miyasaka, M.; Seino, Y. Effects of the anti-ICAM-1 monoclonal antibody on dextran sodium sulphate-induced colitis in rats. J. Gastroenterol. Hepatol. 1998, 13, 945–949. [Google Scholar] [CrossRef]
- Sans, M.; Panés, J.; Ardite, E.; Elizalde, J.I.; Arce, Y.; Elena, M.; Palacín, A.; Fernández-Checa, J.C.; Anderson, D.C.; Lobb, R.; et al. VCAM-1 and ICAM-1 mediate leukocyte-endothelial cell adhesion in rat experimental colitis. Gastroenterology 1999, 116, 874–883. [Google Scholar] [CrossRef]
- Rosette, C.; Roth, R.B.; Oeth, P.; Braun, A.; Kammerer, S.; Ekblom, J.; Denissenko, M.F. Role of ICAM1 in invasion of human breast cancer cells. Carcinogenesis 2005, 26, 943–950. [Google Scholar] [CrossRef]
- Taftaf, R.; Liu, X.; Singh, S.; Jia, Y.; Dashzeveg, N.K.; Hoffmann, A.D.; El-Shennawy, L.; Ramos, E.K.; Adorno-Cruz, V.; Schuster, E.J.; et al. ICAM1 initiates CTC cluster formation and trans-endothelial migration in lung metastasis of breast cancer. Nat. Commun. 2021, 12, 4867. [Google Scholar] [CrossRef]
- Strell, C.; Lang, K.; Niggemann, B.; Zaenker, K.S.; Entschladen, F. Surface molecules regulating rolling and adhesion to endothelium of neutrophil granulocytes and MDA-MB-468 breast carcinoma cells and their interaction. Cell. Mol. Life Sci. 2007, 64, 3306–3316. [Google Scholar] [CrossRef]
- Regev, O.; Kizner, M.; Roncato, F.; Dadiani, M.; Saini, M.; Castro-Giner, F.; Yajuk, O.; Kozlovski, S.; Levi, N.; Addadi, Y.; et al. ICAM-1 on breast cancer cells suppresses lung metastasis but is dispensable for tumor growth and killing by cytotoxic T cells. Front. Immunol. 2022, 13, 849701. [Google Scholar] [CrossRef]
- Yang, M.; Liu, J.; Piao, C.; Shao, J.; Du, J. ICAM-1 suppresses tumor metastasis by inhibiting macrophage M2 polarization through blockade of efferocytosis. Cell Death Dis. 2015, 6, e1780. [Google Scholar] [CrossRef]
- Lim, E.J.; Kang, J.H.; Kim, Y.J.; Kim, S.; Lee, S.J. ICAM-1 promotes cancer progression by regulating SRC activity as an adapter protein in colorectal cancer. Cell Death Dis. 2022, 13, 417. [Google Scholar] [CrossRef]
- O’Brien, K.D.; McDonald, T.O.; Chait, A.; Allen, M.D.; Alpers, C.E. Neovascular expression of E-selectin, intercellular adhesion molecule-1, and vascular cell adhesion molecule-1 in human atherosclerosis and their relation to intimal leukocyte content. Circulation 1996, 93, 672–682. [Google Scholar] [CrossRef]
- Kevil, C.G.; Patel, R.P.; Bullard, D.C. Essential role of ICAM-1 in mediating monocyte adhesion to aortic endothelial cells. Am. J. Physiol. Cell. Physiol. 2001, 281, C1442–C1447. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Moldawer, L.L.; Opal, S.M.; Reinhart, K.; Turnbull, I.R.; Vincent, J.L. Sepsis and septic shock. Nat. Rev. Dis. Primers 2016, 2, 16045. [Google Scholar] [CrossRef]
- Aird, W.C. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 2003, 101, 3765–3777. [Google Scholar] [CrossRef]
- Murao, A.; Arif, A.; Brenner, M.; Denning, N.L.; Jin, H.; Takizawa, S.; Nicastro, B.; Wang, P.; Aziz, M. Extracellular CIRP and TREM-1 axis promotes ICAM-1-Rho-mediated NETosis in sepsis. FASEB J. 2020, 34, 9771–9786. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hong, L.J.; Huang, J.Y.; Jiang, Q.; Tao, R.R.; Tan, C.; Lu, N.N.; Wang, C.K.; Ahmed, M.M.; Lu, Y.M.; et al. P2RX7 sensitizes Mac-1/ICAM-1-dependent leukocyte-endothelial adhesion and promotes neurovascular injury during septic encephalopathy. Cell Res. 2015, 25, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Mándi, Y.; Farkas, G.; Ocsovszky, I.; Nagy, Z. Inhibition of tumor necrosis factor production and ICAM-1 expression by pentoxifylline: Beneficial effects in sepsis syndrome. Res. Exp. Med. 1995, 195, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Moxon, C.A.; Gibbins, M.P.; McGuinness, D.; Milner, D.A., Jr.; Marti, M. New insights into malaria pathogenesis. Annu. Rev. Pathol. 2020, 15, 315–343. [Google Scholar] [CrossRef]
- Venugopal, K.; Hentzschel, F.; Valkiūnas, G.; Marti, M. Plasmodium asexual growth and sexual development in the haematopoietic niche of the host. Nat. Rev. Microbiol. 2020, 18, 177–189. [Google Scholar] [CrossRef]
- Turner, G.D.; Morrison, H.; Jones, M.; Davis, T.M.; Looareesuwan, S.; Buley, I.D.; Gatter, K.C.; Newbold, C.I.; Pukritayakamee, S.; Nagachinta, B.; et al. An immunohistochemical study of the pathology of fatal malaria. Evidence for widespread endothelial activation and a potential role for intercellular adhesion molecule-1 in cerebral sequestration. Am. J. Pathol. 1994, 145, 1057–1069. [Google Scholar]
- Ockenhouse, C.F.; Betageri, R.; Springer, T.A.; Staunton, D.E. Plasmodium falciparum-infected erythrocytes bind ICAM-1 at a site distinct from LFA-1, Mac-1, and human rhinovirus. Cell 1992, 68, 63–69. [Google Scholar] [CrossRef]
- Sinha, S.; Qidwai, T.; Kanchan, K.; Anand, P.; Jha, G.N.; Pati, S.S.; Mohanty, S.; Mishra, S.K.; Tyagi, P.K.; Sharma, S.K.; et al. Variations in host genes encoding adhesion molecules and susceptibility to falciparum malaria in India. Malar. J. 2008, 7, 250. [Google Scholar] [CrossRef]
- Amodu, O.K.; Gbadegesin, R.A.; Ralph, S.A.; Adeyemo, A.A.; Brenchley, P.E.; Ayoola, O.O.; Orimadegun, A.E.; Akinsola, A.K.; Olumese, P.E.; Omotade, O.O. Plasmodium falciparum malaria in south-west Nigerian children: Is the polymorphism of ICAM-1 and E-selectin genes contributing to the clinical severity of malaria? Acta Trop. 2005, 95, 248–255. [Google Scholar] [CrossRef]
- Fernandez-Reyes, D.; Craig, A.G.; Kyes, S.A.; Peshu, N.; Snow, R.W.; Berendt, A.R.; Marsh, K.; Newbold, C.I. A High frequency African coding polymorphism in the N-terminal domain of ICAM-1 predisposing to cerebral malaria in Kenya. Hum. Mol. Genet. 1997, 6, 1357–1360. [Google Scholar] [CrossRef]
- Mwanziva, C.; Mpina, M.; Balthazary, S.; Mkali, H.; Mbugi, E.; Mosha, F.; Chilongola, J. Child hospitalization due to severe malaria is associated with the ICAM-1Kilifi allele but not adherence patterns of Plasmodium falciparum infected red blood cells to ICAM-1. Acta Trop. 2010, 116, 45–50. [Google Scholar] [CrossRef]
- Kun, J.F.; Klabunde, J.; Lell, B.; Luckner, D.; Alpers, M.; May, J.; Meyer, C.; Kremsner, P.G. Association of the ICAM-1Kilifi mutation with protection against severe malaria in Lambaréné, Gabon. Am. J. Trop. Med. Hyg. 1999, 61, 776–779. [Google Scholar] [CrossRef][Green Version]
- Blankson, S.O.; Dadjé, D.S.; Traikia, N.; Alao, M.J.; Ayivi, S.; Amoussou, A.; Deloron, P.; Ndam, N.T.; Milet, J.; Basco, L.K.; et al. ICAM-1 (Kilifi) variant is not associated with cerebral and severe malaria pathogenesis in Beninese children. Malar. J. 2022, 21, 115. [Google Scholar] [CrossRef]
- Fry, A.E.; Auburn, S.; Diakite, M.; Green, A.; Richardson, A.; Wilson, J.; Jallow, M.; Sisay-Joof, F.; Pinder, M.; Griffiths, M.J.; et al. Variation in the ICAM1 gene is not associated with severe malaria phenotypes. Genes. Immun. 2008, 9, 462–469. [Google Scholar] [CrossRef]
- Bellamy, R.; Kwiatkowski, D.; Hill, A.V.S. Absence of an association between intercellular adhesion molecule 1, complement receptor 1 and interleukin 1 receptor antagonist gene polymorphisms and severe malaria in a West African population. Trans. R Soc. Trop. Med. Hyg. 1998, 92, 312–316. [Google Scholar] [CrossRef]
- Shukla, S.D.; Shastri, M.D.; Vanka, S.K.; Jha, N.K.; Dureja, H.; Gupta, G.; Chellappan, D.K.; Oliver, B.G.; Dua, K.; Walters, E.H. Targeting intercellular adhesion molecule-1 (ICAM-1) to reduce rhinovirus-induced acute exacerbations in chronic respiratory diseases. Inflammopharmacology 2022, 30, 725–735. [Google Scholar] [CrossRef]
- Tomassini, J.E.; Graham, D.; DeWitt, C.M.; Lineberger, D.W.; Rodkey, J.A.; Colonno, R.J. cDNA cloning reveals that the major group rhinovirus receptor on HeLa cells is intercellular adhesion molecule 1. Proc. Natl. Acad. Sci. USA 1989, 86, 4907–4911. [Google Scholar] [CrossRef]
- Winther, B.; Arruda, E.; Witek, T.J.; Marlin, S.D.; Tsianco, M.M.; Innes, D.J.; Hayden, F.G. Expression of ICAM-1 in nasal epithelium and levels of soluble ICAM-1 in nasal lavage fluid during human experimental rhinovirus infection. Arch. Otolaryngol. Head Neck Surg. 2002, 128, 131–136. [Google Scholar] [CrossRef]
- Altman, L.C.; Ayars, G.H.; Baker, C.; Luchtel, D.L. Cytokines and eosinophil-derived cationic proteins upregulate intercellular adhesion molecule-1 on human nasal epithelial cells. J. Allergy Clin. Immunol. 1993, 92, 527–536. [Google Scholar] [CrossRef]
- Turner, R.B.; Wecker, M.T.; Pohl, G.; Witek, T.J.; McNally, E.; St. George, R.; Winther, B.; Hayden, F.G. Efficacy of tremacamra, a soluble intercellular adhesion molecule 1, for experimental rhinovirus infection: A randomized clinical trial. JAMA 1999, 281, 1797–1804. [Google Scholar] [CrossRef]
- Traub, S.; Nikonova, A.; Carruthers, A.; Dunmore, R.; Vousden, K.A.; Gogsadze, L.; Hao, W.; Zhu, Q.; Bernard, K.; Zhu, J.; et al. An anti-human ICAM-1 antibody inhibits rhinovirus-induced exacerbations of lung inflammation. PLoS Pathog. 2013, 9, e1003520. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Nave, K.A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Dobson, R.; Giovannoni, G. Multiple sclerosis—A review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef]
- Samoilova, E.B.; Horton, J.L.; Chen, Y. Experimental autoimmune encephalomyelitis in intercellular adhesion molecule-1-deficient mice. Cell. Immunol. 1998, 190, 83–89. [Google Scholar] [CrossRef]
- Hu, X.; Barnum, S.R.; Wohler, J.E.; Schoeb, T.R.; Bullard, D.C. Differential ICAM-1 isoform expression regulates the development and progression of experimental autoimmune encephalomyelitis. Mol. Immunol. 2010, 47, 1692–1700. [Google Scholar] [CrossRef]
- Bullard, D.C.; Hu, X.; Crawford, D.; McDonald, K.; Ramos, T.N.; Barnum, S.R. Expression of a single ICAM-1 isoform on T cells is sufficient for development of experimental autoimmune encephalomyelitis. Eur. J. Immunol. 2014, 44, 1194–1199. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q. A comprehensive review and update on the pathogenesis of inflammatory bowel disease. J. Immunol. Res. 2019, 2019, 7247238. [Google Scholar] [CrossRef]
- Nielsen, O.H.; Langholz, E.; Hendel, J.; Brynskov, J. Circulating soluble intercellular adhesion molecule-1 (sICAM-1) in active inflammatory bowel disease. Dig. Dis. Sci. 1994, 39, 1918–1923. [Google Scholar] [CrossRef]
- Jones, S.C.; Banks, R.E.; Haidar, A.; Gearing, A.J.; Hemingway, I.K.; Ibbotson, S.H.; Dixon, M.F.; Axon, A.T. Adhesion molecules in inflammatory bowel disease. Gut 1995, 36, 724–730. [Google Scholar] [CrossRef]
- Bendjelloul, F.; Malý, P.; Mandys, V.; Jirkovská, M.; Prokesová, L.; Tucková, L.; Tlaskalová-Hogenová, H. Intercellular adhesion molecule-1 (ICAM-1) deficiency protects mice against severe forms of experimentally induced colitis. Clin. Exp. Immunol. 2000, 119, 57–63. [Google Scholar] [CrossRef]
- Bendjelloul, F.; Rossmann, P.; Malý, P.; Mandys, V.; Jirkovská, M.; Prokesová, L.; Tucková, L.; Tlaskalová-Hogenová, H. Detection of ICAM-1 in experimentally induced colitis of ICAM-1-deficient and wild-type mice: An immunohistochemical study. Histochem. J. 2000, 32, 703–709. [Google Scholar] [CrossRef]
- Burns, R.C.; Rivera-Nieves, J.; Moskaluk, C.A.; Matsumoto, S.; Cominelli, F.; Ley, K. Antibody blockade of ICAM-1 and VCAM-1 ameliorates inflammation in the SAMP-1/Yit adoptive transfer model of Crohn’s disease in mice. Gastroenterology 2001, 121, 1428–1436. [Google Scholar] [CrossRef]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef]
- de Smet, M.D.; Taylor, S.R.; Bodaghi, B.; Miserocchi, E.; Murray, P.I.; Pleyer, U.; Zierhut, M.; Barisani-Asenbauer, T.; LeHoang, P.; Lightman, S. Understanding uveitis: The impact of research on visual outcomes. Prog. Retin. Eye Res. 2011, 30, 452–470. [Google Scholar] [CrossRef]
- Smith, J.R.; Lai, T.Y.Y. Managing uveitis during the COVID-19 pandemic. Ophthalmology 2020, 127, e65–e67. [Google Scholar] [CrossRef]
- Arocker-Mettinger, E.; Steurer-Georgiew, L.; Steurer, M.; Huber-Spitzy, V.; Hoelzl, E.; Grabner, G.; Kuchar, A. Circulating ICAM-1 levels in serum of uveitis patients. Curr. Eye Res. 1992, 11, 161–166. [Google Scholar] [CrossRef]
- Klok, A.M.; Luyendijk, L.; Zaal, M.J.; Rothova, A.; Kijlstra, A. Soluble ICAM-1 serum levels in patients with intermediate uveitis. Br. J. Ophthalmol. 1999, 83, 847–851. [Google Scholar] [CrossRef]
- Martin, C.M.; Lacomba, M.S.; Molina, C.I.; Chamond, R.R.; Galera, J.M.; Estevez, E.C. Levels of soluble ICAM-1 and soluble IL-2R in the serum and aqueous humor of uveitis patients. Curr. Eye Res. 2000, 20, 287–292. [Google Scholar] [CrossRef]
- Whitcup, S.M.; Vistica, B.P.; Magone, M.T.; George, R.K. Elevated serum levels of soluble ICAM-1 in uveitis patients predict underlying systemic disease. Br. J. Ophthalmol. 1999, 83, 252–253. [Google Scholar] [CrossRef][Green Version]
- Webster, L.; Stanbury, R.M.; Chignell, A.H.; Limb, G.A. Vitreous intercellular adhesion molecule 1 in uveitis complicated by retinal detachment. Br. J. Ophthalmol. 1998, 82, 438–443. [Google Scholar] [CrossRef]
- Haydinger, C.D.; Ferreira, L.B.; Williams, K.A.; Smith, J.R. Mechanisms of macular edema. Front. Med. 2023, 10, 1128811. [Google Scholar] [CrossRef] [PubMed]
- Whitcup, S.M.; Chan, C.C.; Li, Q.; Nussenblatt, R.B. Expression of cell adhesion molecules in posterior uveitis. Arch. Ophthalmol. 1992, 110, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Ryan, F.J.; Ma, Y.; Ashander, L.M.; Kvopka, M.; Appukuttan, B.; Lynn, D.J.; Smith, J.R. Transcriptomic responses of human retinal vascular endothelial cells to inflammatory cytokines. Transl. Vis. Sci. Technol. 2022, 11, 27. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, A.S.; Appukuttan, B.; Wilmarth, P.A.; Pan, Y.; Stempel, A.J.; Chipps, T.J.; Benedetti, E.E.; Zamora, D.O.; Choi, D.; David, L.L.; et al. Role of the retinal vascular endothelial cell in ocular disease. Prog. Retin. Eye Res. 2013, 32, 102–180. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, J.; Wang, Y.; Calder, V.L. Lymphocyte adhesion and transendothelial migration in the central nervous system: The role of LFA-1, ICAM-1, VLA-4 and VCAM-1. off. Immunology 1995, 86, 408–415. [Google Scholar]
- Devine, L.; Lightman, S.L.; Greenwood, J. Role of LFA-1, ICAM-1, VLA-4 and VCAM-1 in lymphocyte migration across retinal pigment epithelial monolayers in vitro. Immunology 1996, 88, 456–462. [Google Scholar] [CrossRef]
- Dewispelaere, R.; Lipski, D.; Foucart, V.; Bruyns, C.; Frère, A.; Caspers, L.; Willermain, F. ICAM-1 and VCAM-1 are differentially expressed on blood-retinal barrier cells during experimental autoimmune uveitis. Exp. Eye Res. 2015, 137, 94–102. [Google Scholar] [CrossRef]
- Uchio, E.; Kijima, M.; Tanaka, S.; Ohno, S. Suppression of experimental uveitis with monoclonal antibodies to ICAM-1 and LFA-1. Investig. Ophthalmol. Vis. Sci. 1994, 35, 2626–2631. [Google Scholar]
- Whitcup, S.M.; Hikita, N.; Shirao, M.; Miyasaka, M.; Tamatani, T.; Mochizuki, M.; Nussenblatt, R.B.; Chan, C.C. Monoclonal antibodies against CD54 (ICAM-1) and CD11a (LFA-1) prevent and inhibit endotoxin-induced uveitis. Exp. Eye Res. 1995, 60, 597–601. [Google Scholar] [CrossRef]
- Becker, M.D.; Garman, K.; Whitcup, S.M.; Planck, S.R.; Rosenbaum, J.T. Inhibition of leukocyte sticking and infiltration, but not rolling, by antibodies to ICAM-1 and LFA-1 in murine endotoxin-induced uveitis. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2563–2566. [Google Scholar]
- Ferreira, L.B.; Furtado, J.M.; Charng, J.; Franchina, M.; Matthews, J.M.; Molan, A.A.L.; Hunter, M.; Mackey, D.A.; Smith, J.R. Prevalence of toxoplasmic retinochoroiditis in an Australian adult population: A community-based study. Ophthalmol. Retina 2022, 6, 963–968. [Google Scholar] [CrossRef]
- Smith, J.R.; Ashander, L.M.; Arruda, S.L.; Cordeiro, C.A.; Lie, S.; Rochet, E.; Belfort, R., Jr.; Furtado, J.M. Pathogenesis of ocular toxoplasmosis. Prog. Retin. Eye Res. 2021, 81, 100882. [Google Scholar] [CrossRef]
- Gao, N.; Wang, C.; Yu, Y.; Xie, L.; Xing, Y.; Zhang, Y.; Wang, Y.; Wu, J.; Cai, Y. LFA-1/ ICAM-1 promotes NK cell cytotoxicity associated with the pathogenesis of ocular toxoplasmosis in murine model. PLoS Negl. Trop. Dis. 2022, 16, e0010848. [Google Scholar] [CrossRef]
- Adamis, A.P. Is diabetic retinopathy an inflammatory disease? Br. J. Ophthalmol. 2002, 86, 363–365. [Google Scholar] [CrossRef]
- Miyamoto, K.; Khosrof, S.; Bursell, S.E.; Rohan, R.; Murata, T.; Clermont, A.C.; Aiello, L.P.; Ogura, Y.; Adamis, A.P. Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc. Natl. Acad. Sci. USA 1999, 96, 10836–10841. [Google Scholar] [CrossRef]
- Barouch, F.C.; Miyamoto, K.; Allport, J.R.; Fujita, K.; Bursell, S.E.; Aiello, L.P.; Luscinskas, F.W.; Adamis, A.P. Integrin-mediated neutrophil adhesion and retinal leukostasis in diabetes. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1153–1158. [Google Scholar]
- van der Wijk, A.E.; Hughes, J.M.; Klaassen, I.; Van Noorden, C.J.F.; Schlingemann, R.O. Is leukostasis a crucial step or epiphenomenon in the pathogenesis of diabetic retinopathy? J. Leukoc. Biol. 2017, 102, 993–1001. [Google Scholar] [CrossRef]
- Kammerer, S.; Roth, R.B.; Reneland, R.; Marnellos, G.; Hoyal, C.R.; Markward, N.J.; Ebner, F.; Kiechle, M.; Schwarz-Boeger, U.; Griffiths, L.R.; et al. Large-scale association study identifies ICAM gene region as breast and prostate cancer susceptibility locus. Cancer Res. 2004, 64, 8906–8910. [Google Scholar] [CrossRef]
- Usami, Y.; Ishida, K.; Sato, S.; Kishino, M.; Kiryu, M.; Ogawa, Y.; Okura, M.; Fukuda, Y.; Toyosawa, S. Intercellular adhesion molecule-1 (ICAM-1) expression correlates with oral cancer progression and induces macrophage/cancer cell adhesion. Int. J. Cancer 2013, 133, 568–578. [Google Scholar] [CrossRef]
- Tsai, S.T.; Wang, P.J.; Liou, N.J.; Lin, P.S.; Chen, C.H.; Chang, W.C. ICAM1 is a potential cancer stem cell marker of esophageal squamous cell carcinoma. PLoS ONE 2015, 10, e0142834. [Google Scholar] [CrossRef]
- Buitrago, D.; Keutgen, X.M.; Crowley, M.; Filicori, F.; Aldailami, H.; Hoda, R.; Liu, Y.F.; Hoda, R.S.; Scognamiglio, T.; Jin, M.; et al. Intercellular adhesion molecule-1 (ICAM-1) is upregulated in aggressive papillary thyroid carcinoma. Ann. Surg. Oncol. 2012, 19, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Kotteas, E.A.; Boulas, P.; Gkiozos, I.; Tsagkouli, S.; Tsoukalas, G.; Syrigos, K.N. The intercellular cell adhesion molecule-1 (ICAM-1) in lung cancer: Implications for disease progression and prognosis. Anticancer Res. 2014, 34, 4665–4672. [Google Scholar] [PubMed]
- Qiu, Z.; Wang, Y.; Zhang, Z.; Qin, R.; Peng, Y.; Tang, W.; Xi, Y.; Tian, G.; Zhang, Y. Roles of intercellular cell adhesion molecule-1 (ICAM-1) in colorectal cancer: Expression, functions, prognosis, tumorigenesis, polymorphisms and therapeutic implications. Front. Oncol. 2022, 12, 1052672. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Wang, Z.; Kuang, Y.; Wu, Z.; Zhao, S.; Zhang, Z.; Li, H.; Zheng, M.; Zhang, N.; Long, C.; et al. Intercellular adhesion molecule-1 as target for CAR-T-cell therapy of triple-negative breast cancer. Front. Immunol. 2020, 11, 573823. [Google Scholar] [CrossRef]
- Guo, P.; Huang, J.; Wang, L.; Jia, D.; Yang, J.; Dillon, D.A.; Zurakowski, D.; Mao, H.; Moses, M.A.; Auguste, D.T. ICAM-1 as a molecular target for triple negative breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 14710–14715. [Google Scholar] [CrossRef]
- Figenschau, S.L.; Knutsen, E.; Urbarova, I.; Fenton, C.; Elston, B.; Perander, M.; Mortensen, E.S.; Fenton, K.A. ICAM1 expression is induced by proinflammatory cytokines and associated with TLS formation in aggressive breast cancer subtypes. Sci. Rep. 2018, 8, 11720. [Google Scholar] [CrossRef]
- Maeda, K.; Kang, S.M.; Sawada, T.; Nishiguchi, Y.; Yashiro, M.; Ogawa, Y.; Ohira, M.; Ishikawa, T.; Hirakawa, Y.S.C.K. Expression of intercellular adhesion molecule-1 and prognosis in colorectal cancer. Oncol. Rep. 2002, 9, 511–514. [Google Scholar] [CrossRef]
- Gu, W.; Yao, L.; Li, L.; Zhang, J.; Place, A.T.; Minshall, R.D.; Liu, G. ICAM-1 regulates macrophage polarization by suppressing MCP-1 expression via miR-124 upregulation. Oncotarget 2017, 8, 111882–111901. [Google Scholar] [CrossRef]
- Jung, M.; Yang, Y.; McCloskey, J.E.; Zaman, M.; Vedvyas, Y.; Zhang, X.; Stefanova, D.; Gray, K.D.; Min, I.M.; Zarnegar, R.; et al. Chimeric antigen receptor T cell therapy targeting ICAM-1 in gastric cancer. Mol. Ther. Oncolytics 2020, 18, 587–601. [Google Scholar] [CrossRef]
- Min, I.M.; Shevlin, E.; Vedvyas, Y.; Zaman, M.; Wyrwas, B.; Scognamiglio, T.; Moore, M.D.; Wang, W.; Park, S.; Park, S.; et al. CAR T therapy targeting ICAM-1 eliminates advanced human thyroid tumors. Clin. Cancer Res. 2017, 23, 7569–7583. [Google Scholar] [CrossRef]
- Kantari-Mimoun, C.; Barrin, S.; Vimeux, L.; Haghiri, S.; Gervais, C.; Joaquina, S.; Mittelstaet, J.; Mockel-Tenbrinck, N.; Kinkhabwala, A.; Damotte, D.; et al. CAR T-cell entry into tumor islets is a two-step process dependent on IFNγ and ICAM-1. Cancer Immunol. Res. 2021, 9, 1425–1438. [Google Scholar] [CrossRef]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: Update from the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef]
- Kuriakose, D.; Xiao, Z. Pathophysiology and treatment of stroke: Present status and future perspectives. Int. J. Mol. Sci. 2020, 21, 7609. [Google Scholar] [CrossRef]
- Frąk, W.; Wojtasińska, A.; Lisińska, W.; Młynarska, E.; Franczyk, B.; Rysz, J. Pathophysiology of cardiovascular diseases: New insights into molecular mechanisms of atherosclerosis, arterial hypertension, and coronary artery disease. Biomedicines 2022, 10, 1938. [Google Scholar] [CrossRef]
- Malakar, A.K.; Choudhury, D.; Halder, B.; Paul, P.; Uddin, A.; Chakraborty, S. A review on coronary artery disease, its risk factors, and therapeutics. J. Cell. Physiol. 2019, 234, 16812–16823. [Google Scholar] [CrossRef]
- Golledge, J. Update on the pathophysiology and medical treatment of peripheral artery disease. Nat. Rev. Cardiol. 2022, 19, 456–474. [Google Scholar] [CrossRef]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef]
- Poston, R.N.; Haskard, D.O.; Coucher, J.R.; Gall, N.P.; Johnson-Tidey, R.R. Expression of intercellular adhesion molecule-1 in atherosclerotic plaques. Am. J. Pathol. 1992, 140, 665–673. [Google Scholar]
- Davies, M.J.; Gordon, J.L.; Gearing, A.J.; Pigott, R.; Woolf, N.; Katz, D.; Kyriakopoulos, A. The expression of the adhesion molecules ICAM-1, VCAM-1, PECAM, and E-selectin in human atherosclerosis. J. Pathol. 1993, 171, 223–229. [Google Scholar] [CrossRef]
- van der Wal, A.C.; Das, P.K.; Tigges, A.J.; Becker, A.E. Adhesion molecules on the endothelium and mononuclear cells in human atherosclerotic lesions. Am. J. Pathol. 1992, 141, 1427–1433. [Google Scholar]
- Nakashima, Y.; Raines, E.W.; Plump, A.S.; Breslow, J.L.; Ross, R. Upregulation of VCAM-1 and ICAM-1 at atherosclerosis-prone sites on the endothelium in the ApoE-deficient mouse. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 842–851. [Google Scholar] [CrossRef]
- Watanabe, T.; Fan, J. Atherosclerosis and inflammation mononuclear cell recruitment and adhesion molecules with reference to the implication of ICAM-1/LFA-1 pathway in atherogenesis. Int. J. Cardiol. 1998, 66 (Suppl. S1), S45–S53. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.J.; Ballantyne, C.M.; Sharrett, A.R.; Smith, L.C.; Davis, C.E.; Gotto, A.M., Jr.; Boerwinkle, E. Circulating adhesion molecules VCAM-1, ICAM-1, and E-selectin in carotid atherosclerosis and incident coronary heart disease cases: The Atherosclerosis Risk In Communities (ARIC) study. Circulation 1997, 96, 4219–4225. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, K.; Matsumoto, M.; Sasaki, T.; Hashimoto, H.; Kuwabara, K.; Ohtsuki, T.; Hori, M. Involvement of ICAM-1 in the progression of atherosclerosis in APOE-knockout mice. Atherosclerosis 2002, 160, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Fotis, L.; Agrogiannis, G.; Vlachos, I.S.; Pantopoulou, A.; Margoni, A.; Kostaki, M.; Verikokos, C.; Tzivras, D.; Mikhailidis, D.P.; Perrea, D. Intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 at the early stages of atherosclerosis in a rat model. In Vivo 2012, 26, 243–250. [Google Scholar]
- Motawi, T.; Shaker, O.; Taha, N.; Raheem, M.A. Genetic variations in E-selectin and ICAM-1: Relation to atherosclerosis. Med. Sci. Monit. 2012, 18, Cr381–Cr389. [Google Scholar] [CrossRef]
- Li, D.; Qu, C.; Dong, P. The ICAM-1 K469E polymorphism is associated with the risk of coronary artery disease: A meta-analysis. Coron. Artery Dis. 2014, 25, 665–670. [Google Scholar] [CrossRef]
- Liu, A.; Wan, A.; Feng, A.; Rui, R.; Zhou, B. ICAM-1 gene rs5498 polymorphism decreases the risk of coronary artery disease. Medicine 2018, 97, e12523. [Google Scholar] [CrossRef]
- Bourdillon, M.C.; Poston, R.N.; Covacho, C.; Chignier, E.; Bricca, G.; McGregor, J.L. ICAM-1 deficiency reduces atherosclerotic lesions in double-knockout mice (ApoE(−/−)/ICAM-1(−/−)) fed a fat or a chow diet. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2630–2635. [Google Scholar] [CrossRef]
- Collins, R.G.; Velji, R.; Guevara, N.V.; Hicks, M.J.; Chan, L.; Beaudet, A.L. P-Selectin or intercellular adhesion molecule (ICAM)-1 deficiency substantially protects against atherosclerosis in apolipoprotein E-deficient mice. J. Exp. Med. 2000, 191, 189–194. [Google Scholar] [CrossRef]
- Nageh, M.F.; Sandberg, E.T.; Marotti, K.R.; Lin, A.H.; Melchior, E.P.; Bullard, D.C.; Beaudet, A.L. Deficiency of inflammatory cell adhesion molecules protects against atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1517–1520. [Google Scholar] [CrossRef]
- Cybulsky, M.I.; Iiyama, K.; Li, H.; Zhu, S.; Chen, M.; Iiyama, M.; Davis, V.; Gutierrez-Ramos, J.C.; Connelly, P.W.; Milstone, D.S. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J. Clin. Investig. 2001, 107, 1255–1262. [Google Scholar] [CrossRef]
- Marzolla, V.; Armani, A.; Mammi, C.; Moss, M.E.; Pagliarini, V.; Pontecorvo, L.; Antelmi, A.; Fabbri, A.; Rosano, G.; Jaffe, I.Z.; et al. Essential role of ICAM-1 in aldosterone-induced atherosclerosis. Int. J. Cardiol. 2017, 232, 233–242. [Google Scholar] [CrossRef]
- Rautou, P.E.; Leroyer, A.S.; Ramkhelawon, B.; Devue, C.; Duflaut, D.; Vion, A.C.; Nalbone, G.; Castier, Y.; Leseche, G.; Lehoux, S.; et al. Microparticles from human atherosclerotic plaques promote endothelial ICAM-1-dependent monocyte adhesion and transendothelial migration. Circ. Res. 2011, 108, 335–343. [Google Scholar] [CrossRef]
- Shirani, A.; Stüve, O. Natalizumab for multiple sclerosis: A case in point for the impact of translational neuroimmunology. J. Immunol. 2017, 198, 1381–1386. [Google Scholar] [CrossRef]
- Moral, M.E.G.; Siahaan, T.J. Conjugates of cell adhesion peptides for therapeutics and diagnostics against cancer and autoimmune diseases. Curr. Top. Med. Chem. 2017, 17, 3425–3443. [Google Scholar] [CrossRef][Green Version]
- Biswas, S.; Bryant, R.V.; Travis, S. Interfering with leukocyte trafficking in Crohn’s disease. Best. Pract. Res. Clin. Gastroenterol. 2019, 38–39, 101617. [Google Scholar] [CrossRef]
- Di Fusco, D.; Dinallo, V.; Marafini, I.; Figliuzzi, M.M.; Romano, B.; Monteleone, G. Antisense oligonucleotide: Basic concepts and therapeutic application in inflammatory bowel disease. Front. Pharmacol. 2019, 10, 305. [Google Scholar] [CrossRef]
- Atlantic Healthcare Announces Results from Phase 3 Trial of Alicaforsen Enema for Orphan-Designated Pouchitis. 2019. Available online: www.atlantichc.com/atlantic-healthcare-announces-results-from-phase-3-trial-of-alicaforsen-enema-for-orphan-designated-pouchitis/ (accessed on 3 April 2023).
- Reinisch, W.; Hung, K.; Hassan-Zahraee, M.; Cataldi, F. Targeting endothelial ligands: ICAM-1/alicaforsen, MAdCAM-1. J. Crohns Colitis 2018, 12, S669–S677. [Google Scholar] [CrossRef]
- Li, S.; Chen, L.; Wang, G.; Xu, L.; Hou, S.; Chen, Z.; Xu, X.; Wang, X.; Liu, F.; Du, Y.Z. Anti-ICAM-1 antibody-modified nanostructured lipid carriers: A pulmonary vascular endothelium-targeted device for acute lung injury therapy. J. Nanobiotechnol. 2018, 16, 105. [Google Scholar] [CrossRef]
- Jiang, S.; Li, S.; Hu, J.; Xu, X.; Wang, X.; Kang, X.; Qi, J.; Lu, X.; Wu, J.; Du, Y.; et al. Combined delivery of angiopoietin-1 gene and simvastatin mediated by anti-intercellular adhesion molecule-1 antibody-conjugated ternary nanoparticles for acute lung injury therapy. Nanomedicine 2019, 15, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Y.; Gao, J.; Wang, Z. Bioresponsive nanoparticles targeted to infectious microenvironments for sepsis management. Adv. Mater. 2018, 30, e1803618. [Google Scholar] [CrossRef] [PubMed]
- Roki, N.; Tsinas, Z.; Solomon, M.; Bowers, J.; Getts, R.C.; Muro, S. Unprecedently high targeting specificity toward lung ICAM-1 using 3DNA nanocarriers. J. Control. Release 2019, 305, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ashander, L.M.; Appukuttan, B.; Ryan, F.J.; Tan, A.C.R.; Matthews, J.M.; Michael, M.Z.; Lynn, D.J.; Smith, J.R. Selective transcription factor blockade reduces human retinal endothelial cell expression of intercellular adhesion molecule-1 and leukocyte binding. Int. J. Mol. Sci. 2023, 24, 3304. [Google Scholar] [CrossRef]
- Huang, Y.W.; Richardson, J.A.; Vitetta, E.S. Anti-CD54 (ICAM-1) has antitumor activity in SCID mice with human myeloma cells. Cancer Res. 1995, 55, 610–616. [Google Scholar]
- Hansson, M.; Gimsing, P.; Badros, A.; Niskanen, T.M.; Nahi, H.; Offner, F.; Salomo, M.; Sonesson, E.; Mau-Sorensen, M.; Stenberg, Y.; et al. A phase I dose-escalation study of antibody BI-505 in relapsed/refractory multiple myeloma. Clin. Cancer Res. 2015, 21, 2730–2736. [Google Scholar] [CrossRef]
- Wichert, S.; Juliusson, G.; Johansson, Å.; Sonesson, E.; Teige, I.; Wickenberg, A.T.; Frendeus, B.; Korsgren, M.; Hansson, M. A single-arm, open-label, phase 2 clinical trial evaluating disease response following treatment with BI-505, a human anti-intercellular adhesion molecule-1 monoclonal antibody, in patients with smoldering multiple myeloma. PLoS ONE 2017, 12, e0171205. [Google Scholar] [CrossRef]
- Sherbenou, D.W.; Su, Y.; Behrens, C.R.; Aftab, B.T.; de Acha, O.P.; Murnane, M.; Bearrows, S.C.; Hann, B.C.; Wolf, J.L.; Martin, T.G.; et al. Potent activity of an anti-ICAM1 antibody-drug conjugate against multiple myeloma. Clin. Cancer Res. 2020, 26, 6028–6038. [Google Scholar] [CrossRef]
- Fukushima, H.; Kato, T.; Furusawa, A.; Okada, R.; Wakiyama, H.; Furumoto, H.; Okuyama, S.; Kondo, E.; Choyke, P.L.; Kobayashi, H. Intercellular adhesion molecule-1-targeted near-infrared photoimmunotherapy of triple-negative breast cancer. Cancer Sci. 2022, 113, 3180–3192. [Google Scholar] [CrossRef]
- Zhu, L.; Mu, Q.; Yu, J.; Griffin, J.I.; Xu, X.; Ho, R.J.Y. ICAM-1 targeted drug combination nanoparticles enhanced gemcitabine-paclitaxel exposure and breast cancer suppression in mouse models. Pharmaceutics 2021, 14, 89. [Google Scholar] [CrossRef]
- Park, S.; Shevlin, E.; Vedvyas, Y.; Zaman, M.; Park, S.; Hsu, Y.S.; Min, I.M.; Jin, M.M. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci. Rep. 2017, 7, 14366. [Google Scholar] [CrossRef]
- Gao, D.; Cho, C.W.; Kim, J.H.; Bao, H.; Kim, H.M.; Li, X.; Kang, J.S. Phenolic profile and fingerprint analysis of Akebia quinata leaves extract with endothelial protective activity. Molecules 2022, 27, 4636. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Immune Process | ICAM-1 Role |
|---|---|
| Leukocyte transendothelial migration |
|
| |
| |
| |
| |
| Immune synapse formation |
|
| |
| |
| Wound healing |
|
| |
| |
|
| Disease | ICAM-1 Involvement | Reference |
|---|---|---|
| Sepsis |
| [143,144,145] |
| ||
| Malaria |
| [17,146] |
| Rhinovirus infection |
| [147,148] |
| Multiple sclerosis |
| [65,149] |
| ||
| Inflammatory bowel disease |
| [150,151] |
| Non-infectious uveitis |
| [126,127] |
| Cancer |
| [152,153,154,155,156,157] |
| ||
| Cardiovascular disease |
| [158,159] |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haydinger, C.D.; Ashander, L.M.; Tan, A.C.R.; Smith, J.R. Intercellular Adhesion Molecule 1: More than a Leukocyte Adhesion Molecule. Biology 2023, 12, 743. https://doi.org/10.3390/biology12050743
Haydinger CD, Ashander LM, Tan ACR, Smith JR. Intercellular Adhesion Molecule 1: More than a Leukocyte Adhesion Molecule. Biology. 2023; 12(5):743. https://doi.org/10.3390/biology12050743
Chicago/Turabian StyleHaydinger, Cameron D., Liam M. Ashander, Alwin Chun Rong Tan, and Justine R. Smith. 2023. "Intercellular Adhesion Molecule 1: More than a Leukocyte Adhesion Molecule" Biology 12, no. 5: 743. https://doi.org/10.3390/biology12050743
APA StyleHaydinger, C. D., Ashander, L. M., Tan, A. C. R., & Smith, J. R. (2023). Intercellular Adhesion Molecule 1: More than a Leukocyte Adhesion Molecule. Biology, 12(5), 743. https://doi.org/10.3390/biology12050743