

Renal Microcirculation Injury as the Main Cause of Ischemic Acute Kidney Injury Development

{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
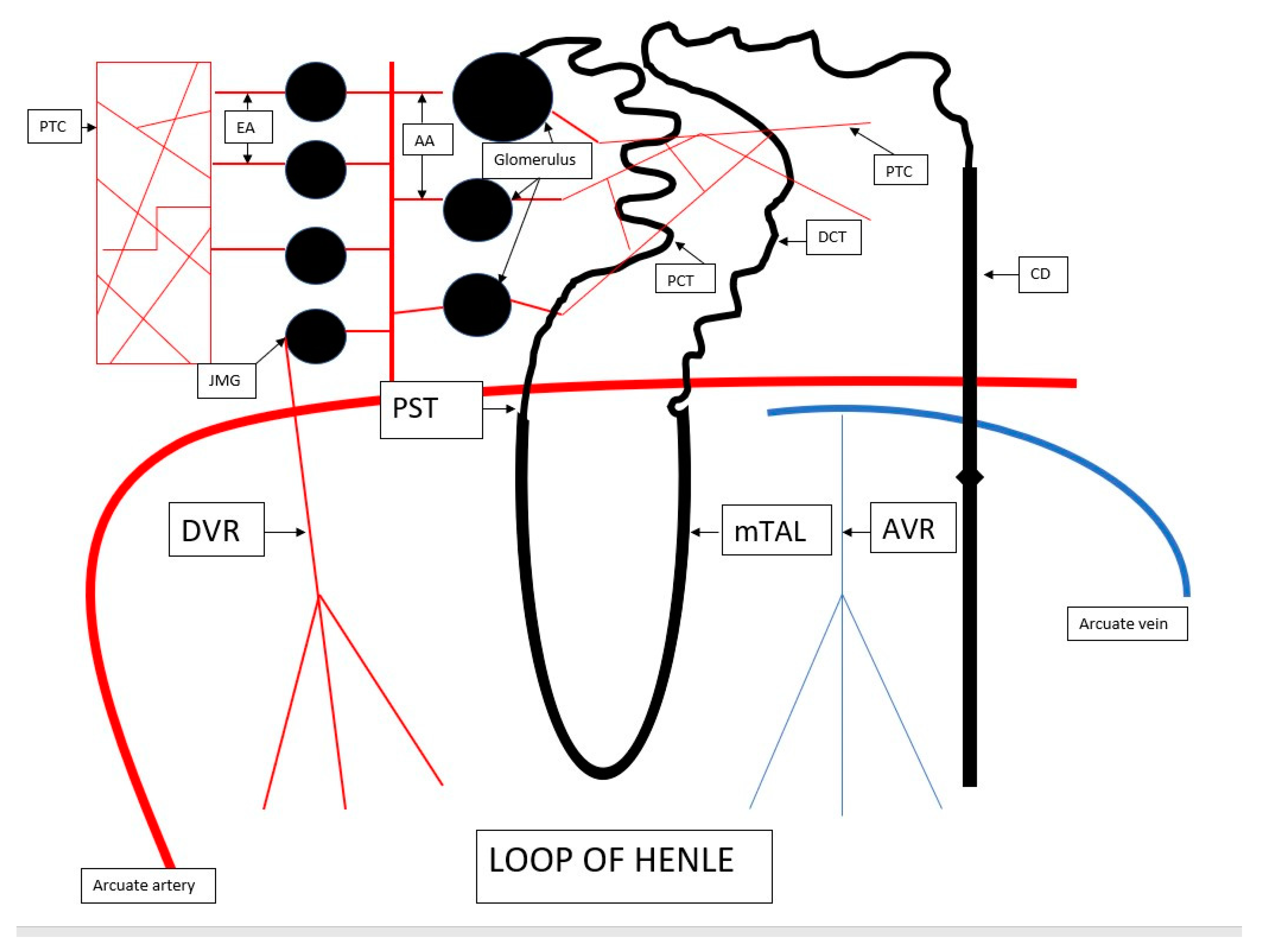
2. The Anatomy of Renal Microcirculation
3. Outer Medulla of the Kidney—A Region Most Vulnerable to Ischemia
4. Regulation of Renal Circulation-Microcirculation
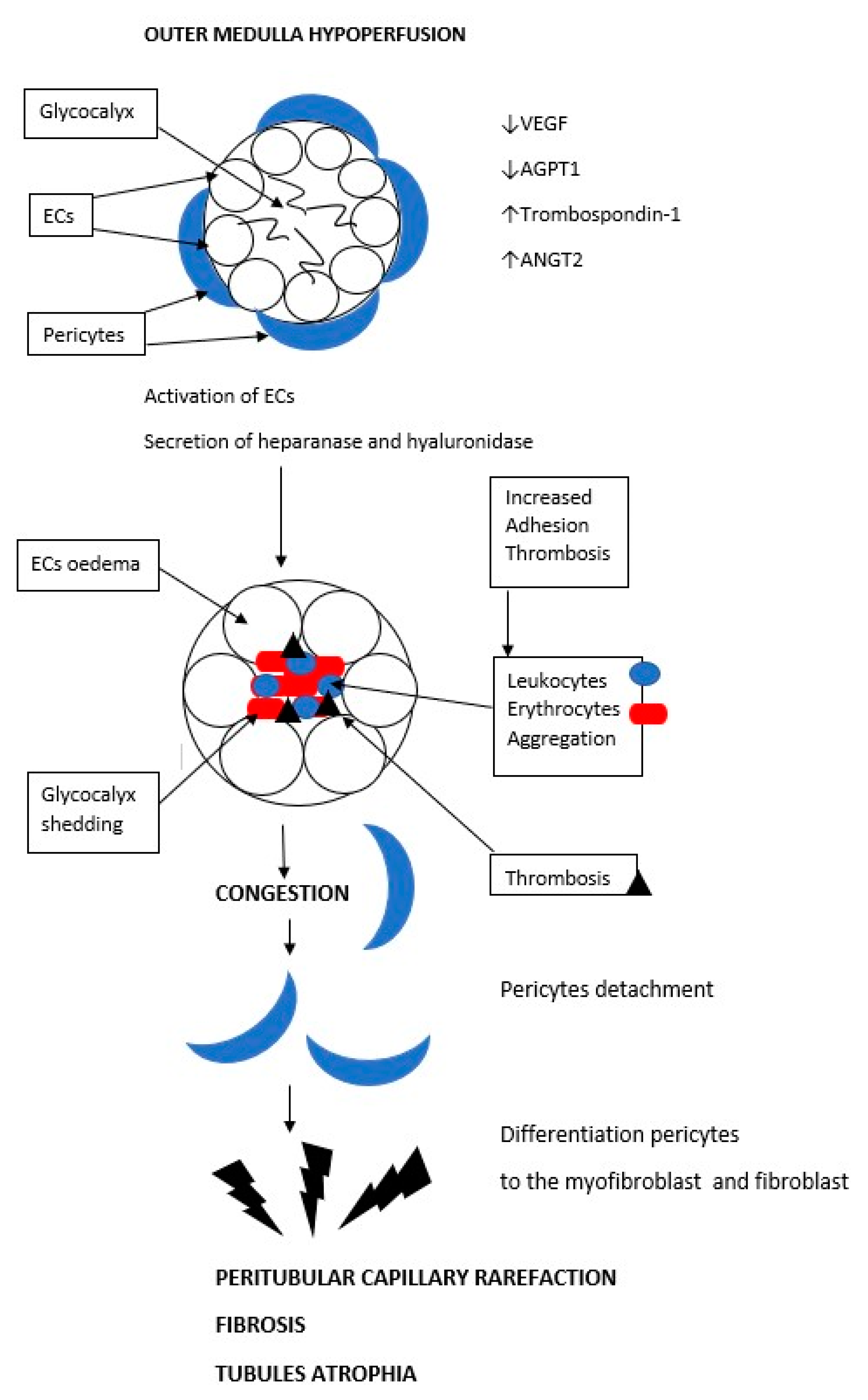
5. Microcirculation in AKI
6. Peritubular Capillary Rarefaction as a Consequence of Acute Ischemia and a Cause of Progression to Chronic Kidney Disease
7. Therapy for Medullary Hypoperfusion
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of acute kidney injury. Compr. Physiol. 2012, 2, 1303–1353. [Google Scholar] [PubMed]
- Chertow, G.M.; Burdick, E.; Honour, M.; Bonventre, J.V.; Bates, D.W. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J. Am. Soc. Nephrol. 2005, 16, 3365–3370. [Google Scholar] [CrossRef] [PubMed]
- Singbartl, K.; Kellum, J.A. AKI in the ICU: Definition, epidemiology, risk stratification, and outcomes. Kidney Int. 2012, 81, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Li, P.K.; Burdmann, E.A.; Mehta, R.L.; World Kidney Day Steering Committee. Acute kidney injury: Global health alert. Transplantation 2013, 95, 653–657. [Google Scholar] [CrossRef]
- Susantitaphong, P.; Cruz, D.N.; Cerda, J.; Abulfaraj, M.; Alqahtani, F.; Koulouridis, I.; Jaber, B.L. World incidence of AKI: A meta-analysis. Clin. J. Am. Soc. Nephrol. 2013, 8, 1482–1493. [Google Scholar] [CrossRef]
- Liu, Z.Z.; Bullen, A.A.; Li, Y.; Singh, P. Renal Oxygenation in the Pathophysiology of Chronic Kidney Disease. Front. Physiol. 2017, 8, 385. [Google Scholar] [CrossRef]
- Jensen, P.K.; Steven, K. Influence of intratubular pressure on proximal tubular compliance and capillary diameter in the rat kidney. Pflug. Arch. Eur. J. Physiol. 1979, 382, 179–187. [Google Scholar] [CrossRef]
- Holliger, C.; Lemley, K.V.; Schmitt, S.L.; Thomas, F.C.; Robertson, C.R.; Jamison, R.L. Direct determination of vasa recta blood flow in the rat renal papilla. Circ. Res. 1983, 53, 401–413. [Google Scholar] [CrossRef]
- Shiva, N.; Sharma, N.; Kulkarni, Y.A.; Mulay, S.R.; Gaikwad, A.B. Renal ischemia/reperfusion injury: An insight on in vitro and in vivo models. Life Sci. 2020, 256, 117860. [Google Scholar] [CrossRef]
- Evans, R.G.; Smith, D.W.; Lee, C.J.; Ngo, J.P.; Gardiner, B.S. What Makes the Kidney Susceptible to Hypoxia? Anat. Rec. 2020, 303, 2544–2552. [Google Scholar] [CrossRef]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Invest. 2011, 121, 4210–4221. [Google Scholar] [CrossRef]
- Shanley, P.F.; Rosen, M.D.; Brezis, M.; Silva, P.; Epstein, F.H.; Rosen, S. Topography of focal proximal tubular necrosis after ischemia with reflow in the rat kidney. Am. J. Pathol. 1986, 122, 462–468. [Google Scholar] [PubMed]
- Torhorst, J.; de Rougemont, D.; Brunner, F.P.; Thiel, G. Morphology of the renal medulla in ischemic acute renal failure in the rat. Nephron 1982, 31, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, M.A.; Bernard, D.B.; Donohoe, J.F.; Levinsky, N.G. Ischemic damage and repair in the rat proximal tubule: Differences among the S1, S2, and S3 segments. Kidney Int. 1978, 14, 31–49. [Google Scholar] [CrossRef] [PubMed]
- Brezis, M.; Rosen, S. Hypoxia of the renal medulla--its implications for disease. N. Engl. J. Med. 1995, 332, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Carlström, M.; Wilcox, C.S.; Arendshorst, W.J. Renal Autoregulation in Health and Disease. Physiol. Rev. 2015, 95, 405–511. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.P.; Skøtt, O.; Schnermann, J. Cellular mechanisms within the juxtaglomerular apparatus. Am. J. Hypertens. 1990, 3, 76–80. [Google Scholar] [CrossRef]
- Mason, J.; Joeris, B.; Welsch, J.; Kriz, W. Vascular congestion in ischemic renal failure: The role of cell swelling. Miner Electrolyte. Metab. 1989, 15, 114–124. [Google Scholar]
- Zhang, B.; Chen, N.; Shi, W.; Wang, W.; Shi, H.; Yu, H. Peritubular Capillary Loss Is Ameliorated by Ramipril or Valsartan Treatment. Microcirculation 2008, 15, 4. [Google Scholar] [CrossRef]
- Yeh, C.-H.; Hsu, S.-P.; Yang, C.-C.; Chien, C.-T.; Wang, N.-P. Hypoxic preconditioning reinforces HIF-alpha-dependent HSP70 signaling to reduce ischemic renal failure-induced renal tubular apoptosis and autophagy. Life Sci. 2010, 86, 115–123. [Google Scholar] [CrossRef]
- Gotshall, R.W.; Miles, D.S.; Sexson, W.R. Renal oxygen delivery and consumption during progressive hypoxemia in the anesthetized dog. Proc. Soc. Exp. Biol. Med. 1983, 174, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Hellberg, P.O.; Bayati, A.; Kallskog, O.; Wolgast, M. Red cell trapping after ischemia and long-term kidney damage. Influence of hematocrit. Kidney Int. 1990, 37, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Hellberg, P.O.; Kallskog, O.; Wolgast, M. Nephron function in the early phase of ischemic renal failure. Significance of erythrocyte trapping. Kidney Int. 1990, 38, 432–439. [Google Scholar] [CrossRef]
- Olof, P.; Hellberg, A.; Kallskog, O.; Wolgast, M. Red cell trapping and postischemic renal blood flow. Differences between the cortex, outer and inner medulla. Kidney Int. 1991, 40, 625–631. [Google Scholar] [PubMed]
- Caraba, A.; Iurciuc, S.; Munteanu, A.; Iurciuc, M. Hyponatremia and Renal Venous Congestion in Heart Failure Patients. Dis. Markers 2021, 2021, 6499346. [Google Scholar] [CrossRef]
- Ray, S.C.; Mason, B.J.; O’Connor, P.M. Ischemic Renal Injury: Can Renal Anatomy and Associated Vascular Congestion Explain Why the Medulla and Not the Cortex Is Where the Trouble Starts. Semin. Nephrol. 2019, 39, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Owji, S.M.; Nikeghbal, E.; Moosavi, S.M. Comparison of ischaemia-reperfusion-induced acute kidney injury by clamping renal arteries, veins or pedicles in anaesthetized rats. Exp Physiol. 2018, 103, 1390–1402. [Google Scholar] [CrossRef]
- Kenig-Kozlovsky, Y.; Scott, R.P.; Onay, T.; Carota, I.A.; Thomson, B.R.; Gil, H.J.; Ramirez, V.; Yamaguchi, S.; Tanna, C.E.; Heinen, S.; et al. Ascending Vasa Recta Are Angiopoietin/Tie2-Dependent Lymphatic-Like Vessels. J. Am. Soc. Nephrol. 2018, 29, 1097–1107. [Google Scholar] [CrossRef]
- Afsar, B.; Afsar, R.E.; Dagel, T.; Kaya, E.; Erus, S.; Ortiz, A.; Covic, A.; Kanbay, M. Capillary rarefaction from the kidney point of view. Clin. Kidney J. 2018, 11, 295–301. [Google Scholar] [CrossRef]
- Ergin, B.; Akin, S.; Ince, C. Kidney Microcirculation as a Target for Innovative Therapies in AKI. J. Clin. Med. 2021, 10, 4041. [Google Scholar] [CrossRef]
- Ren, Y.; Garvin, J.L.; Liu, R.; Carretero, O.A. Crosstalk between the Connecting Tubule and the Afferent Arteriole Regulates Renal Microcirculation. Kidney Int. 2007, 71, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.; Pabbidi, M.R.; Farley, J.; Roman, R.J. Molecular Mechanisms of Renal Blood Flow Autoregulation. Curr. Vasc. Pharmacol. 2014, 12, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Guerci, P.; Ergin, B.; Ince, C. The macro- and microcirculation of the kidney. Best Pr. Res. Clin. Anaesthesiol. 2017, 31, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Cowley, A.W.; Skelton, M.M.; Kurth, T.M. Effects of long-term vasopressin receptor stimulationon medullary blood flow and arterial pressure. Am. J. Physiol. 1998, 275, R1420–R1424. [Google Scholar] [CrossRef] [PubMed]
- Cowley, A.W. Control of the renal medullary circulation by vasopressin V1 and V2 receptors in the rat. Exp. Physiol. 2000, 85, 223S–231S. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.M.; Trizna, W.; Kinter, L.B. Renal microvascular effects of vasopressin and vasopressin antagonists. Am. J. Physiol. 1989, 256, F274–F278. [Google Scholar] [CrossRef]
- Wang, J.; Shi, M.; Huang, L.; Li, Q.; Meng, S.; Xu, J.; Xue, M.; Xie, J.; Liu, S.; Huang, Y. Addition of terlipressin to norepinephrine in septic shock and effect of renal perfusion: A pilot study. Ren. Fail. 2022, 44, 1207–1215. [Google Scholar] [CrossRef]
- Russell, J.A.; Walley, K.R.; Singer, J.; Gordon, A.C.; Hébert, P.C.; Cooper, D.J.; Holmes, C.L.; Mehta, S.; Granton, J.T.; Storms, M.M.; et al. VASST Investigators: Vasopressin versus norepinephrine infusion in patients with septic shock. N. EngI. J. Med. 2008, 358, 877–887. [Google Scholar] [CrossRef]
- Gordon, A.C.; Russell, J.A.; Walley, K.R.; Singer, J.; Ayers, D.; Storms, M.M.; Holmes, C.L.; Hébert, P.C.; Cooper, D.J.; Mehta, S.; et al. The effects of vasopressin on acute kidney injury in septic shock. Intensive Care Med. 2010, 36, 83–91. [Google Scholar] [CrossRef]
- Murphy, S.; Williams, J.M. Impaired Renal Autoregulation in Susceptible Models of Renal Disease. Curr. Vasc. Pharmacol. 2014, 12, 859–866. [Google Scholar] [CrossRef]
- Barajas, L.; Müller, J. The Innervation of the Juxtaglomerular Apparatus and Surrounding Tubules: A Quantitative Analysis by Serial Section Electron Microscopy. J. Ultrastruct. Res. 1973, 43, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Barajas, L.; Liu, L.; Powers, K. Anatomy of the Renal Innervation: Intrarenal Aspects and Ganglia of Origin. Can. J. Physiol. Pharmacol. 1992, 70, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Garland, C.J.; Hiley, C.R.; Dora, K.A. EDHF: Spreading the Influence of the Endothelium. Br. J. Pharmacol. 2011, 164, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Marti, C.N.; Gheorghiade, M.; Kalogeropoulos, A.P.; Georgiopoulou, V.V.; Quyyumi, A.A.; Butler, J. Endothelial Dysfunction, Arterial Stiffness, and Heart Failure. J. Am. Coll Cardiol. 2012, 60, 1455–1469. [Google Scholar] [CrossRef] [PubMed]
- Lubrano, V.; Balzan, S. Roles of LOX-1 in Microvascular Dysfunction. Microvasc. Res. 2016, 105, 132–140. [Google Scholar] [CrossRef]
- Guan, Z.; VanBeusecum, J.P.; Inscho, E.W. Endothelin and the renal microcirculation. Semin. Nephrol. 2015, 35, 145–155. [Google Scholar] [CrossRef]
- Gellai, M.; DeWolf, R.; Pullen, M.; Nambi, P. Distribution and functional role of renal ET receptor subtypes in normotensive and hypertensive rats. Kidney Int. 1994, 46, 1287–1294. [Google Scholar] [CrossRef]
- Inscho, E.W. P2 receptors in regulation of renal microvascular function. Am. J. Physiol. Ren. Physiol. 2001, 280, F927–F944. [Google Scholar] [CrossRef]
- Eppel, G.A.; Ventura, S.; Evans, R.G. Regional vascular responses to ATP and ATP analogues in the rabbit kidney in vivo: Roles for adenosine receptors and prostanoids. Br. J. Pharmacol. 2006, 149, 523–531. [Google Scholar] [CrossRef]
- Ince, C. Hemodynamic Coherence and the Rationale for Monitoring the Microcirculation. Crit. Care 2015, 19, 8. [Google Scholar] [CrossRef]
- Abuelo, J.G. Normotensive ischemic acute renal failure. N. Engl. J. Med. 2007, 357, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Ergin, B.; Guerci, P.; Zafrani, L.; Nocken, F.; Kandil, A.; Gurel-Gurevin, E.; Demirci-Tansel, C.; Ince, C. Effects of N-Acetylcysteine (NAC) Supplementation in Resuscitation Fluids on Renal Microcirculatory Oxygenation, Inflammation, and Function in a Rat Model of Endotoxemia. Intensive Care Med. Exp. 2016, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Aksu, U.; Bezemer, R.; Demirci, C.; Ince, C. Acute Effects of Balanced versus Unbalanced Colloid Resuscitation on Renal Macrocirculatory and Microcirculatory Perfusion during Endotoxemic Shock. Shock 2012, 37, 205–209. [Google Scholar] [CrossRef]
- Johannes, T.; Mik, E.G.; Nohé, B.; Raat, N.J.H.; Unertl, K.E.; Ince, C. Influence of Fluid Resuscitation on Renal Microvascular PO2 in a Normotensive Rat Model of Endotoxemia. Crit. Care 2006, 10, R88. [Google Scholar] [CrossRef] [PubMed]
- Lima, A.; van Rooij, T.; Ergin, B.; Sorelli, M.; Ince, Y.; Specht, P.A.C.; Mik, E.G.; Bocchi, L.; Kooiman, K.; de Jong, N.; et al. Dynamic Contrast-Enhanced Ultrasound Identifies Microcirculatory Alterations in Sepsis-Induced Acute Kidney Injury. Crit. Care Med. 2018, 46, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Chvojka, J.; Sykora, R.; Krouzecky, A.; Radej, J.; Varnerova, V.; Karvunidis, T.; Hes, O.; Novak, I.; Radermacher, P.; Matejovic, M. Renal haemodynamic, microcirculatory, metabolic and histopathological responses to peritonitis-induced septic shock in pigs. Crit Care. 2008, 12, R164. [Google Scholar] [CrossRef] [PubMed]
- Maiden, M.J.; Otto, S.; Brealey, J.K.; Finnis, M.E.; Chapman, M.J.; Kuchel, T.R.; Nash, C.H.; Edwards, J.; Bellomo, R. Structure 575 and Function of the Kidney in Septic Shock: A Prospective Controlled Experimental Study. Am. J. Respir. Crit. Care Med. 2016, 194, 692–700. [Google Scholar] [CrossRef]
- Chou, Y.-H.; Chu, T.-S.; Lin, S.-L. Role of renin-angiotensin system in acute kidney injury-chronic kidney disease transition. Nephrology 2018, 23, 121–125. [Google Scholar] [CrossRef]
- Coca, S.G.; Singanamala, S.; Parikh, C.R. Chronic kidney disease afteracute kidney injury: A systematic review and meta-analysis. Kidney Int. 2012, 81, 442–448. [Google Scholar] [CrossRef]
- Lindenmeyer, M.T.; Kretzler, M.; Boucherot, A.; Berra, S.; Yasuda, Y.; Henger, A.; Eichinger, F.; Gaiser, S.; Schmid, H.; Rastaldi, M.P.; et al. Cohen, Interstitial vascular rarefaction and reduced VEGF-A expression in human diabetic nephropathy. J. Am. Soc. Nephrol. 2007, 18, 1765–1776. [Google Scholar] [CrossRef]
- Lombardi, D.; Gordon, K.L.; Polinsky, P.; Suga, S.; Schwartz, S.M.; Johnson, R.J. Salt-sensitive hypertension develops after short-term exposure to Angiotensin II. Hypertension 1999, 33, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Iwazu, Y.; Muto, S.; Fujisawa, G.; Nakazawa, E.; Okada, K.; Ishibashi, S.; Kusano, E. Spironolactone suppresses peritubular capillary loss and prevents deoxycorticosterone acetate/salt-induced tubulointerstitial fibrosis. Hypertension 2008, 51, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Namikoshi, T.; Satoh, M.; Horike, H.; Fujimoto, S.; Arakawa, S.; Sasaki, T.; Kashihara, N. Implication of peritubular capillary loss and altered expression of vascular endothelial growth factor in IgA nephropathy. Nephron. Physiol. 2006, 102, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Kaukinen, A.; Lautenschlager, I.; Helin, H.; Karikoski, R.; Jalanko, H. Peritubular capillaries are rarefied in congenital nephrotic syndrome of the Finnish type. Kidney Int. 2009, 75, 1099–1108. [Google Scholar] [CrossRef]
- Anutrakulchai, S.; Titipungul, T.; Pattay, T.; Mesung, P.; Puapairoj, A.; Sirivongs, D.; Pongsakul, C.; Futrakul, P.; Thinkhamrop, B.; Johnson, R.J. Relation of peritubular capillary features to class of lupus nephritis. BMC Nephrol. 2016, 17, 169. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Popov, V.; Walocha, J.A.; Wen, J.; Bello-Reuss, E. Evidence of angiogenesis and microvascular regression in autosomal-dominant polycystic kidney disease kidneys: A corrosion cast study. Kidney Int. 2006, 70, 1261–1268. [Google Scholar] [CrossRef]
- O’Brien, K.; Saravanabavan, S.; Zhang, J.Q.J.; Wong, A.T.Y.; Munt, A.; Burgess, J.S.; Rangan, G.K. Regression of Peritubular Capillaries Coincides with Angiogenesis and Renal Cyst Growth in Experimental Polycystic Kidney Disease. Int. J. Nephrol. Renovasc. Dis. 2020, 13, 53–64. [Google Scholar] [CrossRef]
- Ishii, Y.; Sawada, T.; Kubota, K.; Fuchinoue, S.; Teraoka, S.; Shimizu, A. Injury and progressive loss of peritubular capillaries in the development of chronic allograft nephropathy. Kidney Int. 2005, 67, 321–332. [Google Scholar] [CrossRef]
- Steegh, F.M.; Gelens, M.A.; Nieman, F.H.; van Hooff, J.P.; Cleutjens, J.P.; van Suylen, R.J.; Daemen, M.J.; van Heurn, E.L.; Christiaans, M.H.; Peutz-Kootstra, C.J. Early loss of peritubular capillaries after kidney transplantation. J. Am. Soc. Nephrol. 2011, 22, 1024–1029. [Google Scholar] [CrossRef]
- Stan, R.V.; Tse, D.; Deharvengt, S.J.; Smits, N.C.; Xu, Y.; Luciano, M.R.; McGarry, C.L.; Buitendijk, M.; Nemani, K.V.; Elgueta, R.; et al. The diaphragms of fenestrated endothelia: Gatekeepers of vascular permeability and blood composition. Dev. Cell 2012, 23, 1203–1218. [Google Scholar] [CrossRef]
- Stolz, D.B.; Sims-Lucas, S. Unwrapping the origins and roles of the renal endothelium. Pediatr. Nephrol. 2015, 30, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Bearer, E.L.; Orci, L. Endothelial fenestral diaphragms: A quick-freeze, deep-etch study. J. Cell Biol. 1985, 100, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Shimada, S.; Hirose, T.; Takahashi, C.; Sato, E.; Kinugasa, S.; Ohsaki, Y.; Kisu, K.; Sato, H.; Ito, S.; Mori, T. Pathophysiological and molecular mechanisms involved in renal congestion in a novel rat model. Sci. Rep. 2018, 8, 16808. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, R.; Shimizu, A.; Masuda, Y.; Kitamura, H.; Ishizaki, M.; Sugisaki, Y.; Yamanaka, N. Peritubular capillary regression during the progression of experimental obstructive nephropathy. J. Am. Soc. Nephrol. 2002, 13, 1795–1805. [Google Scholar] [CrossRef]
- Loganathan, K.; Said, E.S.; Winterrowd, E.; Orebrand, M.; He, L.; Vanlandewijck, M.; Betsholtz, C.; Quaggin, S.E.; Jeansson, M. Angiopoietin-1 deficiency increases renal capillary rarefaction and tubulointerstitial fibrosis in mice. PLoS ONE 2018, 13, e0189433. [Google Scholar] [CrossRef]
- Kang, D.H.; Joly, A.H.; Oh, S.W.; Hugo, C.; Kerjaschki, D.; Gordon, K.L.; Mazzali, M.; Jefferson, J.A.; Hughes, J.; Madsen, K.M.; et al. Impaired angiogenesis in the remnant kidney model: I. Potential role of vascular endothelial growth factor and thrombospondin-1. J. Am. Soc. Nephrol. 2001, 12, 1434–1447. [Google Scholar] [CrossRef]
- Futrakul, N.; Butthep, P.; Futrakul, P. Altered vascular homeostasis in chronic kidney disease. Clin. Hemorheol. Microcirc. 2008, 38, 201–207. [Google Scholar]
- Koller, G.M.; Schafer, C.; Kemp, S.S.; Aguera, K.N.; Lin, P.K.; Forgy, J.C.; Griffin, C.T.; Davis, G.E. Proinflammatory Mediators, IL (Interleukin)-1β, TNF (Tumor Necrosis Factor) α, and Thrombin Directly Induce Capillary Tube Regression. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 365–377. [Google Scholar] [CrossRef]
- Babickova, J.; Klinkhammer, B.M.; Buhl, E.M.; Djudjaj, S.; Hoss, M.; Heymann, F.; Tacke, F.; Floege, J.; Becker, J.U.; Boor, P. Regardless of etiology, progressive renal disease causes ultrastructural and functional alterations of peritubular capillaries. Kidney Int. 2017, 91, 70–85. [Google Scholar] [CrossRef]
- Ehling, J.; Babickova, J.; Gremse, F.; Klinkhammer, B.M.; Baetke, S.; Knuechel, R.; Kiessling, F.; Floege, J.; Lammers, T.; Boor, P. Quantitative Micro-Computed Tomography Imaging of Vascular Dysfunction in Progressive Kidney Diseases. J. Am. Soc. Nephrol. 2016, 27, 520–532. [Google Scholar] [CrossRef]
- Rouschop, K.M.; Claessen, N.; Pals, S.T.; Weening, J.J.; Florquin, S. CD44 disruption prevents degeneration of the capillary network in obstructive nephropathy via reduction of TGF-beta1-induced apoptosis. J. Am. Soc. Nephrol. 2006, 17, 746–753. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.T.H.; Aburatani, T.; Marsh, G.A.; Johnson, B.G.; Alimperti, S.; Yoon, C.J.; Huang, A.; Szak, S.; Nakagawa, N.; Gomez, I.; et al. Hyperactive FOXO1 results in lack of tip stalk identity and deficient microvascular regeneration during kidney injury. Biomaterials 2017, 141, 314–329. [Google Scholar] [CrossRef] [PubMed]
- Kida, Y. Peritubular Capillary Rarefaction: An Underappreciated Regulator of CKD Progression. Int. J. Mol. Sci. 2020, 21, 8255. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.-T.; Li, X.-Z.; Pitera, J.E.; Long, D.A.; Woolf, A.S. Peritubular Capillary Loss after Mouse Acute Nephrotoxicity Correlates with Down-Regulation of Vascular Endothelial Growth Factor-A and HypoxiaInducible Factor-1. Am. J. Pathol. 2003, 163, 2289–2301. [Google Scholar] [CrossRef]
- Kairaitis, L.K.; Wang, Y.; Gassmann, M.; Tay, Y.-C.; Harris, D.C.H. HIF-1alpha expression follows microvascular loss in advanced murine adriamycin nephrosis. Am. J. Physiol. Ren. Physiol. 2005, 288, F198–F206. [Google Scholar] [CrossRef]
- Matsumoto, M.; Tanaka, T.; Yamamoto, T.; Noiri, E.; Miyata, T.; Inagi, R.; Fujita, T.; Nangaku, M. Hypoperfusion of peritubular capillaries induces chronic hypoxia before progression of tubulointerstitial injury in a progressive model of rat glomerulonephritis. J. Am. Soc. Nephrol. 2004, 15, 1574–1581. [Google Scholar] [CrossRef]
- Ohashi, R.; Kitamura, H.; Yamanaka, N. Peritubular capillary injury during the progression of experimental glomerulonephritis in rats. J. Am. Soc. Nephrol. 2000, 11, 47–56. [Google Scholar] [CrossRef]
- Kobori, H.; Nangaku, M.; Navar, L.G.; Nishiyama, A. The intrarenalrenin-angiotensin system: From physiology to the pathobiology ofhypertension and kidney disease. Pharmacol. Rev. 2007, 59, 251–287. [Google Scholar] [CrossRef]
- Ruster, C.; Wolf, G. Renin-angiotensin-aldosterone system andprogression of renal disease. J. Am. Soc. Nephrol. 2006, 17, 2985–2991. [Google Scholar] [CrossRef]
- Cao, W.; Jin, L.; Zhou, Z.; Yang, M.; Wu, C.; Wu, L.; Cui, S. Overexpression of intrarenal renin-angiotensin system in human acute tubular necrosis. Kidney Blood Press. Res. 2016, 41, 746–756. [Google Scholar] [CrossRef]
- Chen, C.; Yang, X.; Lei, Y.; Zha, Y.; Liu, H.; Ma, C.; Tian, J.; Chen, P.; Yang, T.; Hou, F.F. Urinary biomarkers at the time of AKIdiagnosis as predictors of progression of AKI among patients withacute cardiorenal syndrome. Clin. J. Am. Soc. Nephrol. 2016, 11, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Tang, B.; Zhang, C. Signaling pathways of chronic kidney diseases, implications for therapeutics. Signal Transduct. Target Ther. 2022, 7, 182. [Google Scholar] [CrossRef]
- Su, S.-A.; Yang, D.; Wu, Y.; Xie, Y.; Zhu, W.; Cai, Z.; Shen, J.; Fu, Z.; Wang, Y.; Jia, L.; et al. EphrinB2 Regulates Cardiac Fibrosis Through Modulating the Interaction of Stat3 and TGF-β/Smad3 Signaling. Circ. Res. 2017, 121, 617–627. [Google Scholar] [CrossRef] [PubMed]
- van Beusekom, C.D.; Zimmering, T.M. Profibrotic effects of angiotensin II and transforming growth factor beta on feline kidney epithelial cells. J. Feline Med. Surg. 2019, 21, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Yamakoshi, S.; Nakamura, T.; Mori, N.; Suda, C.; Kohzuki, M.; Ito, O. Effects of exercise training on renal interstitial fibrosis and renin-angiotensin system in rats with chronic renal failure. J. Hypertens. 2021, 39, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.-Y.; Li, H.-M.; Yan, Z.-X.; Li, M.-C.; Wei, J.-P.; Zheng, W.-X.; Liu, S.-Q.; Deng, Y.-T.; Xie, H.-F.; Li, C.-G. Renin-angiotensin system activation and imbalance of matrix metalloproteinase-9/tissue inhibitor of matrix metalloproteinase-1 in cold-induced stroke. Life Sci. 2019, 231, 116563. [Google Scholar] [CrossRef] [PubMed]
- Stoian, M.; Stoica, V. Current Trends on Glomerulosclerosis Regression. J. Med. Life. 2020, 13, 116–118. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Ma, Z.; Ding, Y.; Bedarida, T.; Chen, L.; Xie, Z.; Song, P.; Zou, M.-H. Circulating miR-103a-3p contributes to angiotensin II-induced renal inflammation and fibrosis via a SNRK/NF-κB/p65 regulatory axis. Nat. Commun. 2019, 10, 2145. [Google Scholar] [CrossRef]
- Lang, P.-P.; Bai, J.; Zhang, Y.-L.; Yang, X.-L.; Xia, Y.-L.; Lin, Q.-Y.; Li, H.-H. Blockade of intercellular adhesion molecule-1 prevents angiotensin II-induced hypertension and vascular dysfunction. Lab. Invest. 2020, 100, 378–386. [Google Scholar] [CrossRef]
- Schnee, J.M.; Hsueh, W.A. Angiotensin II, adhesion, and cardiac fibrosis. Cardiovasc. Res. 2000, 46, 264–268. [Google Scholar] [CrossRef]
- Wu, J.; Chu, Y.; Jiang, Z.; Yu, Q. Losartan protects against intermittent hypoxia-induced peritubular capillary loss by modulating the renal renin–angiotensin system and angiogenesis factors. Acta Biochim. Et Biophys. Sin. 2020, 52, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Kontogiannis, J.; Burns, K.D. Role of AT1 angiotensin II receptors in renal ischemic injury. Am. J. Physiol. 1998, 274, F79–F90. [Google Scholar] [CrossRef] [PubMed]
- Patschan, D.; Patschan, S.; Buschmann, I.; Ritter, O. Loop Diuretics in Acute Kidney Injury Prevention, Therapy, and Risk Stratification. Kidney Blood Press. Res. 2019, 44, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Benedetto, U.; Melina, G.; Capuano, F.; Comito, C.; Bianchini, R.; Simon, C.; Refice, S.; Angeloni, E.; Sinatra, R. Preoperative angiotensin-converting enzyme inhibitors protect myocardium from ischemia during coronary artery bypass graft surgery. J. Cardiovasc. Med. 2008, 9, 1098–1103. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.H.; Huang, T.M.; Pan, S.Y.; Chang, C.H.; Lai, C.F.; Wu, V.C.; Wu, M.S.; Wu, K.D.; Chu, T.S.; Lin, S.L. Renin-angiotensin system inhibitor is associated with lower risk of ensuing chronic kidneydisease after functional recovery from acute kidney injury. Sci. Rep. 2017, 7, 46518. [Google Scholar] [CrossRef]
- Roberts, D.J.; Smith, S.A.; Tan, Z.; Dixon, E.; Datta, I.; Devrome, A.; Hemmelgarn, B.R.; Tonelli, M.; Pannu, N.; James, M.T. Angiotensin-Converting Enzyme Inhibitor/Receptor Blocker, Diuretic, or Nonsteroidal Anti-inflammatory Drug Use After Major Surgery and Acute Kidney Injury: A Case-Control Study. J. Surg. Res. 2021, 263, 34–43. [Google Scholar] [CrossRef]
- Siew, E.D.; Parr, S.K.; Abdel-Kader, K.; Perkins, A.M.; Greevy, R.A.; Vincz, A.J.; Denton, J.; Wilson, O.D.; Hung, A.M.; Ikizler, T.A.; et al. Renin-angiotensin aldosterone inhibitor use at hospital discharge among patients with moderate to severe acute kidney injury and its association with recurrent acute kidney injury and mortality. Kidney Int. 2021, 99, 1202–1212. [Google Scholar] [CrossRef]
- Bidulka, P.; Fu, E.; Leyrat, C.; Kalogirou, F.; McAllister, K.S.L.; Kingdon, E.J.; Mansfield, K.E.; Iwagami, M.; Smeeth, L.; Clase, C.M.; et al. Stopping renin-angiotensin system blockers after acute kidney injury and risk of adverse outcomes: Parallel population-based cohort studies in English and Swedish routine care. BMC Med. 2020, 18, 195. [Google Scholar] [CrossRef]
- Brar, S.; Liu, K.D.; Go, A.S.; Hsu, R.K.; Chinchilli, V.M.; Coca, S.G.; Garg, A.X.; Himmelfarb, J.; Ikizler, T.A.; Kaufman, J.; et al. Prospective Cohort Study of Renin-Angiotensin System Blocker Usage after Hospitalized Acute Kidney Injury. Clin. J. Am. Soc. Nephrol. 2021, 16, 26–36. [Google Scholar] [CrossRef]
- Whiting, P.; Morden, A.; Tomlinson, L.; Caskey, F.; Blakeman, T.; Tomson, C.; Stone, T.; Richards, A.; Savović, J.; Horwood, J. What are the risks and benefits of temporarily discontinuing medications to prevent acute kidney injury? A systematic review and meta-analysis. BMJ Open 2017, 7, e012674. [Google Scholar] [CrossRef]
- Chen, J.-Y.; Tsai, I.-J.; Pan, H.-C.; Liao, H.-W.; Neyra, J.A.; Wu, V.-C.; Chueh, J.S. The Impact of Angiotensin-Converting Enzyme Inhibitors or Angiotensin II Receptor Blockers on Clinical Outcomes of Acute Kidney Disease Patients: A Systematic Review and Meta-Analysis. Front. Pharmacol. 2021, 12, 665250. [Google Scholar] [CrossRef] [PubMed]
- Legrand, M.; De Backer, D.; Dépret, F.; Ait-Oufella, H. Recruiting the microcirculation in septic shock. Ann. Intensive Care 2019, 9, 102. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwiatkowska, E.; Kwiatkowski, S.; Dziedziejko, V.; Tomasiewicz, I.; Domański, L. Renal Microcirculation Injury as the Main Cause of Ischemic Acute Kidney Injury Development. Biology 2023, 12, 327. https://doi.org/10.3390/biology12020327
Kwiatkowska E, Kwiatkowski S, Dziedziejko V, Tomasiewicz I, Domański L. Renal Microcirculation Injury as the Main Cause of Ischemic Acute Kidney Injury Development. Biology. 2023; 12(2):327. https://doi.org/10.3390/biology12020327
Chicago/Turabian StyleKwiatkowska, Ewa, Sebastian Kwiatkowski, Violetta Dziedziejko, Izabela Tomasiewicz, and Leszek Domański. 2023. "Renal Microcirculation Injury as the Main Cause of Ischemic Acute Kidney Injury Development" Biology 12, no. 2: 327. https://doi.org/10.3390/biology12020327
APA StyleKwiatkowska, E., Kwiatkowski, S., Dziedziejko, V., Tomasiewicz, I., & Domański, L. (2023). Renal Microcirculation Injury as the Main Cause of Ischemic Acute Kidney Injury Development. Biology, 12(2), 327. https://doi.org/10.3390/biology12020327