
Gut Bacteriomes and Ecological Niche Divergence: An Example of Two Cryptic Gastropod Species

 , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. 16S rDNA-Metabarcoding
2.1.1. Sampling
2.1.2. Library Preparation and Sequencing
2.1.3. Bioinformatic and Statistical Analysis
2.2. Shotgun-Sequencing and Metabolic Pathway Annotation
2.2.1. Sampling
2.2.2. Sequencing
2.2.3. Assembly and Annotation
3. Results and Discussion
3.1. General Patterns
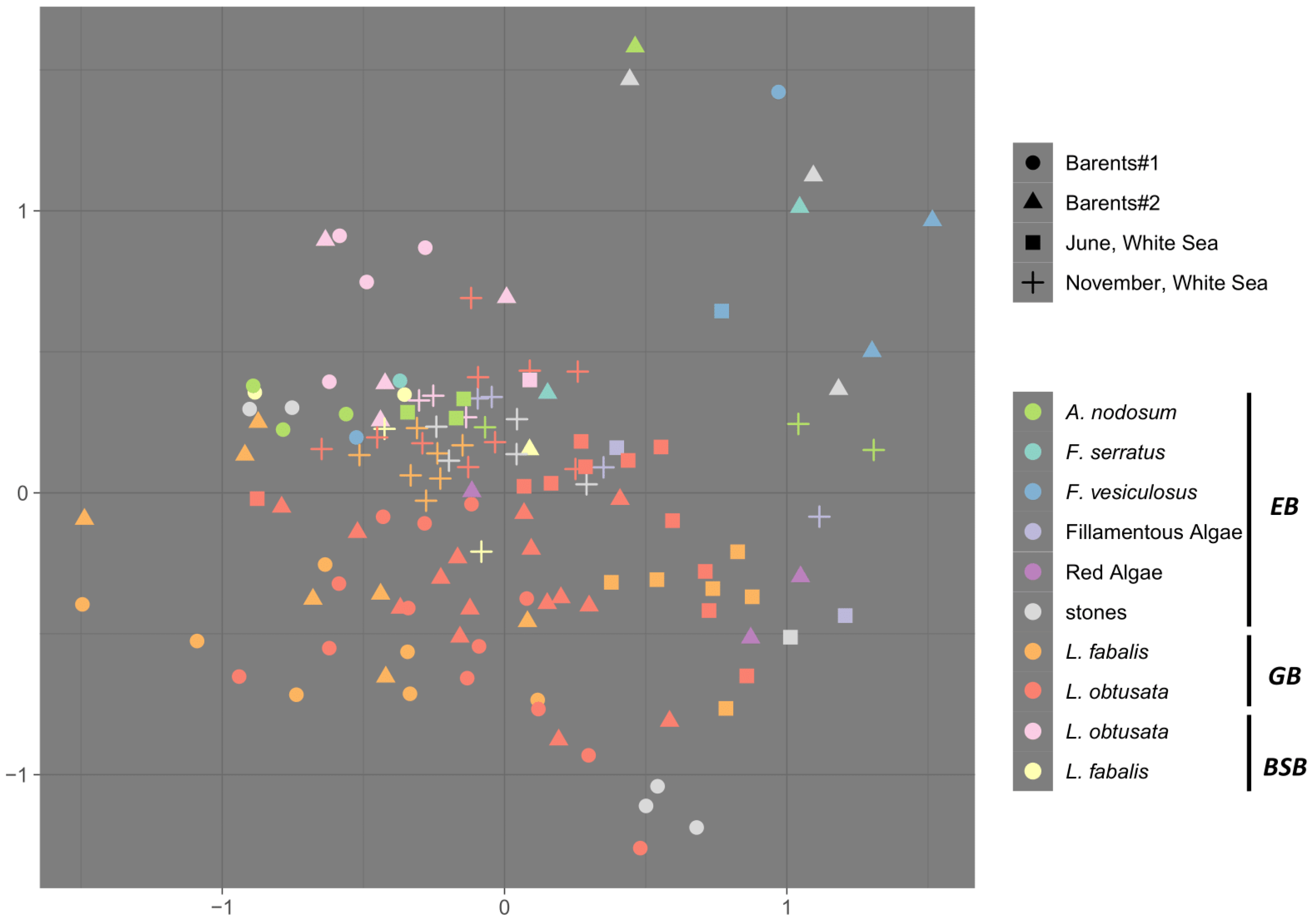
3.2. Sources of Variability in the Bacteriome Composition
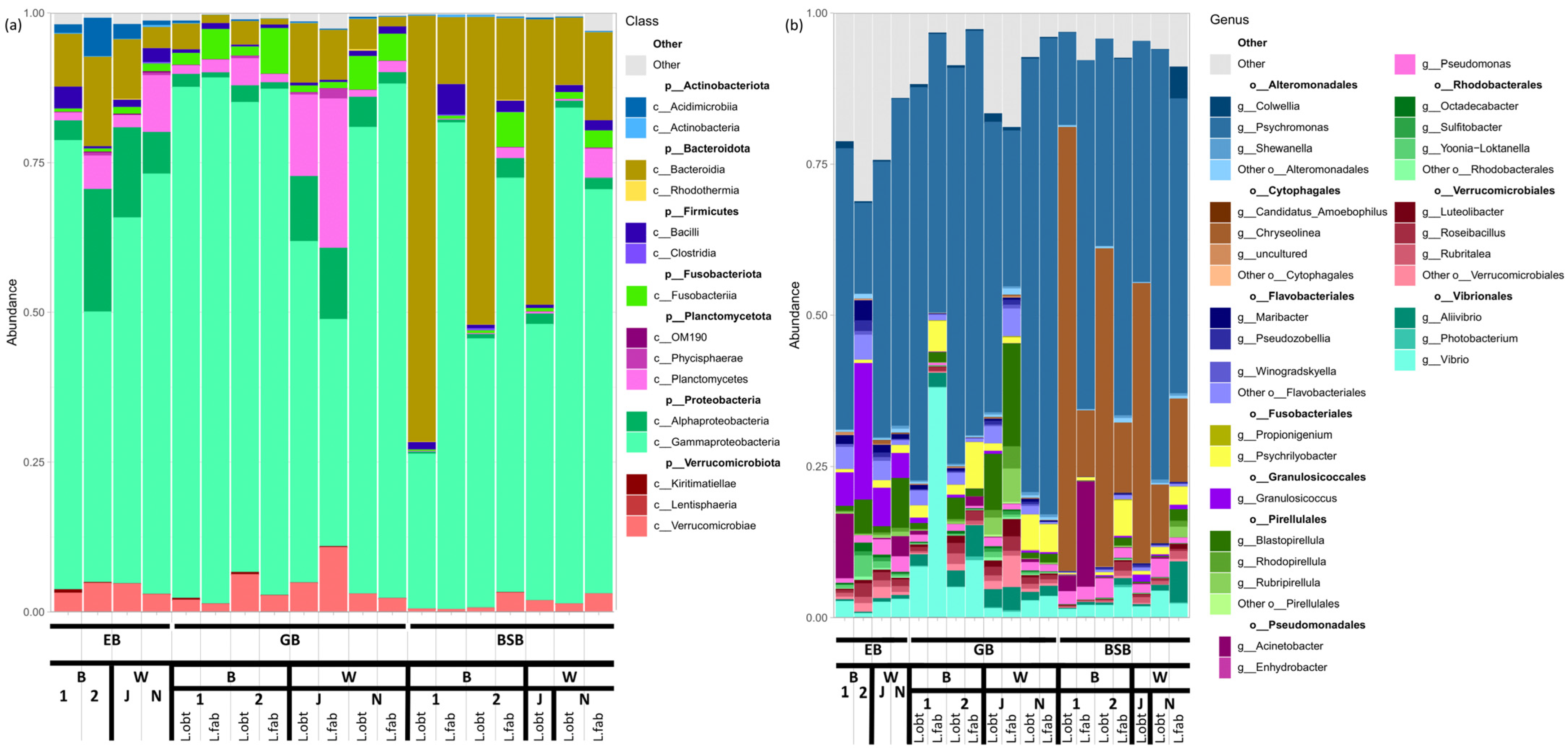
3.3. Associated Bacteriome Composition
- The community is dominated by a limited number of bacterial lineages (3–5);
- The most abundant taxon is Proteobacteria, especially Gammaproteobacteria;
- Those less abundant, but inevitably present in the GB groups, are Fusobacteria (Fusobacteriales), Bacteroides (Flavobacteriales) and Planctomycetes (Pirellulales);
- Cytophagales are predominantly enriched in the BSB (Figure 3a).
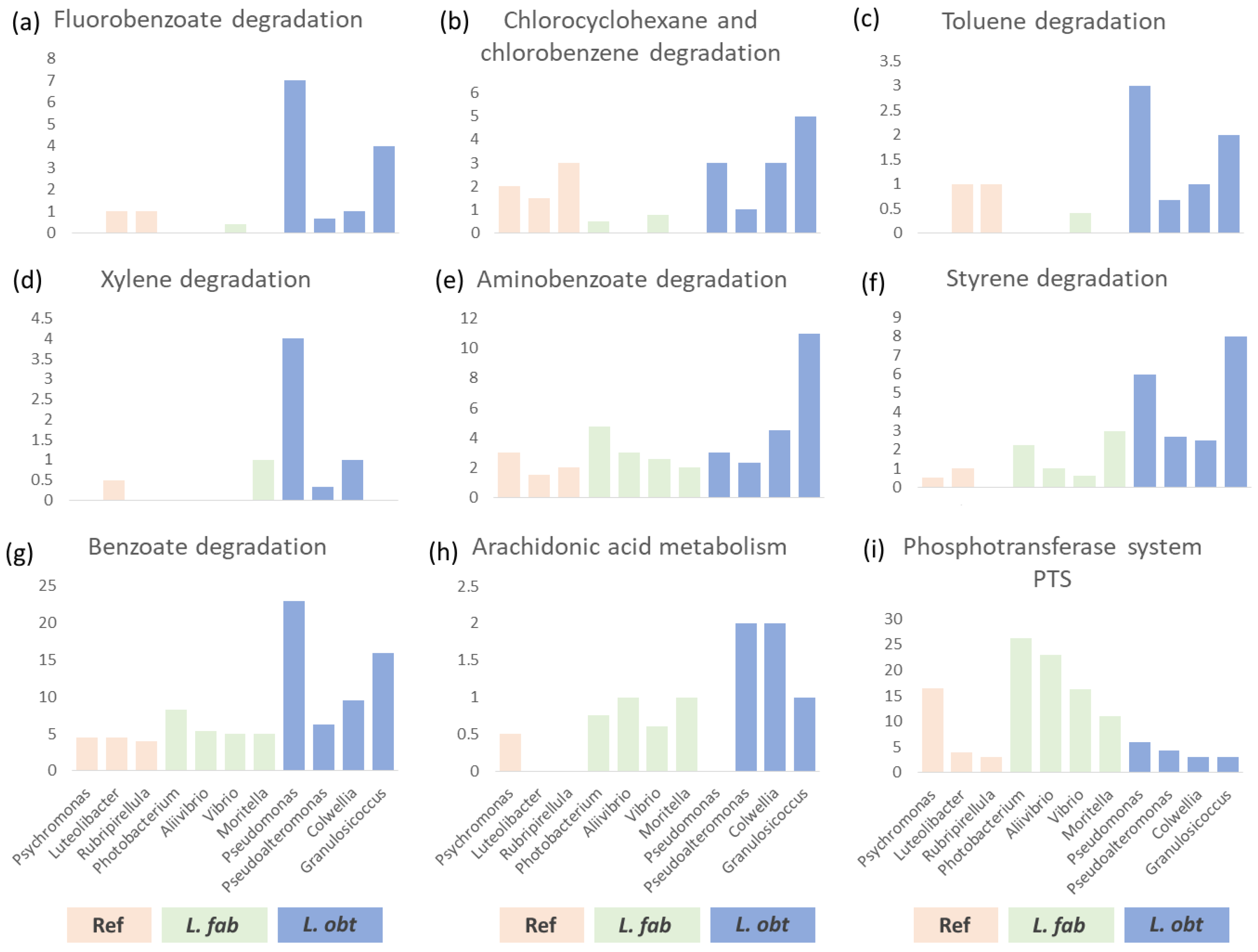
3.4. Interspecies Differences in the Gut Bacteriome Composition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Attardo, G.M.; Lohs, C.; Heddi, A.; Alam, U.H.; Yildirim, S.; Aksoy, S. Analysis of milk gland structure and function in Glossina morsitans: Milk protein production, symbiont populations and fecundity. J. Insect Physiol. 2008, 54, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.K.; Moran, N.A. Aphid genome expression reveals host-symbiont cooperation in the production of amino acids. Proc. Natl. Acad. Sci. USA 2011, 108, 2849–2854. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Ceja-Navarro, J.A.; Vega, F.E.; Karaoz, U.; Hao, Z.; Jenkins, S.; Lim, H.C.; Kosina, P.; Infante, F.; Northen, T.R.; Brodie, E.L. Gut microbiota mediate caffeine detoxification in the primary insect pest of coffee. Nat. Commun. 2015, 6, 7618. [Google Scholar] [CrossRef] [PubMed]
- Shapira, M. Gut Microbiotas and Host Evolution: Scaling up Symbiosis. Trends Ecol. Evol. 2016, 31, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Aronson, H.S.; Zellmer, A.J.; Goffredi, S.K. The specific and exclusive microbiome of the deep-sea bone-eating snail, Rubyspira osteovora. FEMS Microbiol. Ecol. 2017, 93, fiw250. [Google Scholar] [CrossRef]
- O’Hara, A.M.; Shanahan, F. The gut flora as a forgotten organ. EMBO Rep. 2006, 7, 688–693. [Google Scholar] [CrossRef]
- Gerardo, N.M.; Altincicek, B.; Anselme, C.; Atamian, H.; Barribeau, S.M.; de Vos, M.; Duncan, E.J.; Evans, J.D.; Gabaldon, T.; Ghanim, M.; et al. Immunity and other defenses in pea aphids, Acyrthosiphon pisum. Genome Biol. 2010, 11, R21. [Google Scholar] [CrossRef]
- Laughton, A.M.; Garcia, J.R.; Altincicek, B.; Strand, M.R.; Gerardo, N.M. Characterisation of immune responses in the pea aphid, Acyrthosiphon pisum. J. Insect Physiol. 2011, 57, 830–839. [Google Scholar] [CrossRef]
- Sommer, F.; Backhed, F. The gut microbiota--masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef]
- Kamada, N.; Seo, S.U.; Chen, G.Y.; Nunez, G. Role of the gut microbiota in immunity and inflammatory disease. Nat. Rev. Immunol. 2013, 13, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Sharon, G.; Segal, D.; Ringo, J.M.; Hefetz, A.; Zilber-Rosenberg, I.; Rosenberg, E. Commensal Bacteria Play a Role in Mating Preference of Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2010, 107, 20051–20056. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.A.; McVey Neufeld, K.-A. Gut–Brain Axis: How the Microbiome Influences Anxiety and Depression. Trends Neurosci. 2013, 36, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota Modulate Behavioral and Physiological Abnormalities Associated with Neurodevelopmental Disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef] [PubMed]
- Lize, A.; McKay, R.; Lewis, Z. Gut microbiota and kin recognition. Trends Ecol. Evol. 2013, 28, 325–326. [Google Scholar] [CrossRef] [PubMed]
- Desbonnet, L.; Clarke, G.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. Microbiota Is Essential for Social Development in the Mouse. Mol. Psychiatry 2014, 19, 146–148. [Google Scholar] [CrossRef]
- Cryan, J.F.; Dinan, T.G. More than a Gut Feeling: The Microbiota Regulates Neurodevelopment and Behavior. Neuropsychopharmacol 2015, 40, 241–242. [Google Scholar] [CrossRef]
- Valles-Colomer, M.; Falony, G.; Darzi, Y.; Tigchelaar, E.F.; Wang, J.; Tito, R.Y.; Schiweck, C.; Kurilshikov, A.; Joossens, M.; Wijmenga, C.; et al. The neuroactive potential of the human gut microbiota in quality of life and depression. Nat. Microbiol. 2019, 4, 623–632. [Google Scholar] [CrossRef]
- Greene, L.K.; Williams, C.V.; Junge, R.E.; Mahefarisoa, K.L.; Rajaonarivelo, T.; Rakotondrainibe, H.; O’Connell, T.M.; Drea, C.M. A role for gut microbiota in host niche differentiation. ISME J. 2020, 14, 1675–1687. [Google Scholar] [CrossRef]
- Duperron, S.; Pottier, M.A.; Leger, N.; Gaudron, S.M.; Puillandre, N.; Le Prieur, S.; Sigwart, J.D.; Ravaux, J.; Zbinden, M. A tale of two chitons: Is habitat specialisation linked to distinct associated bacterial communities? FEMS Microbiol. Ecol. 2013, 83, 552–567. [Google Scholar] [CrossRef]
- Derycke, S.; De Meester, N.; Rigaux, A.; Creer, S.; Bik, H.; Thomas, W.K.; Moens, T. Coexisting Cryptic Species of the Litoditis marina Complex (Nematoda) Show Differential Resource Use and Have Distinct Microbiomes with High Intraspecific Variability. Mol. Ecol. 2016, 25, 2093–2110. [Google Scholar] [CrossRef] [PubMed]
- Gause, G.F. The Struggle for Existence; Williams and Wilkins: Baltimore, MD, USA, 1934; p. 163. [Google Scholar]
- Schluter, D. The Ecology of Adaptive Radiation; Oxford University Press: New York, NY, USA, 2000; p. 296. [Google Scholar]
- Ackerly, D.D. Community Assembly, Niche Conservatism, and Adaptive Evolution in Changing Environments. Int. J. Plant Sci. 2003, 164, S165–S184. [Google Scholar] [CrossRef]
- Haygood, M.G.; Schmidt, E.W.; Davidson, S.K.; Faulkner, D.J. Microbial symbionts of marine invertebrates: Opportunities for microbial biotechnology. J. Mol. Microbiol. Biotechnol. 1999, 1, 33–43. [Google Scholar] [PubMed]
- Reveillaud, J.; Maignien, L.; Eren, A.M.; Huber, J.A.; Apprill, A.; Sogin, M.L.; Vanreusel, A. Host-Specificity among Abundant and Rare Taxa in the Sponge Microbiome. ISME J. 2014, 8, 1198–1209. [Google Scholar] [CrossRef] [PubMed]
- Weiland-Brauer, N.; Neulinger, S.C.; Pinnow, N.; Kunzel, S.; Baines, J.F.; Schmitz, R.A. Composition of Bacterial Communities Associated with Aurelia aurita Changes with Compartment, Life Stage, and Population. Appl. Environ. Microbiol. 2015, 81, 6038–6052. [Google Scholar] [CrossRef]
- Bost, A.; Martinson, V.G.; Franzenburg, S.; Adair, K.L.; Albasi, A.; Wells, M.T.; Douglas, A.E. Functional variation in the gut microbiome of wild Drosophila populations. Mol. Ecol. 2018, 27, 2834–2845. [Google Scholar] [CrossRef]
- Jehrke, L.; Stewart, F.A.; Droste, A.; Beller, M. The impact of genome variation and diet on the metabolic phenotype and microbiome composition of Drosophila melanogaster. Sci. Rep. 2018, 8, 6215. [Google Scholar] [CrossRef]
- Johannesson, K.; Panova, M.; Kemppainen, P.; André, C.; Rolan-Alvarez, E.; Butlin, R.K. Repeated evolution of reproductive isolation in a marine snail: Unveiling mechanisms of speciation. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 1735–1747. [Google Scholar] [CrossRef]
- Butlin, R.K.; Saura, M.; Charrier, G.; Jackson, B.; André, C.; Caballero, A.; Coyne, J.A.; Galindo, J.; Grahame, J.W.; Hollander, J. Parallel evolution of local adaptation and reproductive isolation in the face of gene flow. Evolution 2014, 68, 935–949. [Google Scholar] [CrossRef]
- Panova, M.; Johansson, T.; Canbäck, B.; Bentzer, J.; Rosenblad, M.A.; Johannesson, K.; Tunlid, A.; André, C. Species and gene divergence in Littorina snails detected by array comparative genomic hybridization. BMC Genom. 2014, 15, 687. [Google Scholar] [CrossRef]
- Mittermayer, F.; Helmerson, C.; Duvetorp, M.; Johannesson, K.; Panova, M. The molecular background of the aspartate aminotransferase polymorphism in Littorina snails maintained by strong selection on small spatial scales. Gene 2023, 876, 147517. [Google Scholar] [CrossRef] [PubMed]
- Johannesson, K.; Butlin, R.K.; Panova, M.; Westram, A.M. Mechanisms of adaptive divergence and speciation in Littorina saxatilis: Integrating knowledge from ecology and genetics with new data emerging from genomic studies. In Population Genomics: Marine Organisms; Springer: Cham, Switzerland, 2020; pp. 277–301. [Google Scholar] [CrossRef]
- Maltseva, A.L.; Varfolomeeva, M.A.; Gafarova, E.R.; Panova, M.A.Z.; Mikhailova, N.A.; Granovitch, A.I. Divergence together with microbes: A comparative study of the associated microbiomes in the closely related Littorina species. PLoS ONE 2021, 16, e0260792. [Google Scholar] [CrossRef] [PubMed]
- Panova, M.A.Z.; Varfolomeeva, M.A.; Gafarova, E.R.; Maltseva, A.L.; Mikhailova, N.A.; Granovitch, A.I. First Insights into the Gut Microbiomes and the Diet of the Littorina Snail Ecotypes, a Recently Emerged Marine Evolutionary Model. Evol. Appl. 2023, 16, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Maltseva, A.; Lobov, A.; Pavlova, P.; Panova, M.; Gafarova, E.; Marques, J.; Danilov, L.; Granovitch, A. Orphan gene in Littorina: An unexpected role of symbionts in the host evolution. Gene 2022, 824, 146389. [Google Scholar] [CrossRef] [PubMed]
- Reid, D.G. Systematics and Evolution of Littorina; Ray Society: London, UK, 1996; p. 463. [Google Scholar]
- Williams, G.A. The comparative ecology of the flat periwinkles littorina obtusata l. and littorina mariae sacchi et rastelli. Field Study 1990, 7, 469–482. [Google Scholar]
- Watson, D.C.; Norton, T.A. The habitat and feeding preferences of Littorina obtusata (L.) and L. mariae Sacchi et Rastelli. J. Exp. Mar. Biol. Ecol. 1987, 112, 61–72. [Google Scholar] [CrossRef]
- Norton, T.A.; Hawkins, S.J.; Manley, N.L.; Williams, G.A.; Watson, D.C. Scraping a Living: A Review of Iittorinid Grazing. In Progress in Littorinid and Muricid Biology; Developments in Hydrobiology; Springer: Dordrecht, The Netherlands, 1990; Volume 56, pp. 117–138. [Google Scholar] [CrossRef]
- Maltseva, A.L.; Varfolomeeva, M.A.; Ayanka, R.V.; Gafarova, E.R.; Repkin, E.A.; Pavlova, P.A.; Shavarda, A.L.; Mikhailova, N.A.; Granovitch, A.I. Linking Ecology, Morphology, and Metabolism: Niche Differentiation in Sympatric Populations of Closely Related Species of the Genus Littorina (Neritrema). Ecol. Evol. 2021, 11, 11134–11154. [Google Scholar] [CrossRef]
- Carvalho, J.; Pereira, C.; Sotelo, G.; Costa, D.; Galindo, J.; Faria, R. De Novo Isolation of 17 Microsatellite Loci for Flat Periwinkles (Littorina fabalis and L. Obtusata) and Their Application for Species Discrimination and Hybridization Studies. J. Mollus. Stud. 2015, 81, 421–425. [Google Scholar] [CrossRef]
- Weingarten, E.A.; Atkinson, C.L.; Jackson, C.R. The gut microbiome of freshwater Unionidae mussels is determined by host species and is selectively retained from filtered seston. PLoS ONE 2019, 14, e0224796. [Google Scholar] [CrossRef]
- Schols, R.; Vanoverberghe, I.; Huyse, T.; Decaestecker, E. Host-bacteriome transplants of the schistosome snail host Biomphalaria glabrata reflect species-specific associations. FEMS Microbiol. Ecol. 2023, 99, fiad101. [Google Scholar] [CrossRef]
- Hu, Z.; Chen, X.; Chang, J.; Yu, J.; Tong, Q.; Li, S.; Niu, H. Compositional and predicted functional analysis of the gut microbiota of Radix auricularia (Linnaeus) via high-throughput Illumina sequencing. PeerJ 2018, 6, e5537. [Google Scholar] [CrossRef] [PubMed]
- Zonn, I.S.; Kostianoy, A.G.; Semenov, A.V. The Western Arctic Seas Encyclopedia; Springer International Publishing: Cham, Switzerland, 2017; p. 547. [Google Scholar]
- Golikova, E.; Varfolomeeva, M.; Yakovis, E.; Korsun, S. Saltmarsh foraminifera in the subarctic White Sea: Thrive in summer, endure in winter. Estuar. Coast. Shelf Sci. 2020, 238, 106685. [Google Scholar] [CrossRef]
- White Sea Hydrology and Zooplankton Time-Series: Kartesh D1. 2020. Available online: https://www.st.nmfs.noaa.gov/copepod/time-series/ru-10101/ (accessed on 24 November 2023).
- Costa, D.; Sotelo, G.; Kaliontzopoulou, A.; Carvalho, J.; Butlin, R.; Hollander, J.; Faria, R. Hybridization patterns between two marine snails, Littorina fabalis and L. obtusata. Ecol. Evol. 2020, 10, 1158–1179. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, J.; Sotelo, G.; Galindo, J.; Faria, R. Genetic characterization of flat periwinkles (Littorinidae) from the Iberian Peninsula reveals interspecific hybridization and different degrees of differentiation. Biol. J. Linn. Soc. 2016, 118, 503–519. [Google Scholar] [CrossRef]
- Neu, A.T.; Allen, E.E.; Roy, K. Diversity and composition of intertidal gastropod microbiomes across a major marine biogeographic boundary. Environ. Microbiol. Rep. 2019, 11, 434–447. [Google Scholar] [CrossRef]
- Hugerth, L. MiSeq Amplicon Sequencing Sample Prep with Dual Barcoding. Available online: https://github.com/EnvGen/LabProtocols/blob/master/Amplicon_dual_index_prep_EnvGen.rst (accessed on 23 October 2023).
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global Patterns of 16S rRNA Diversity at a Depth of Millions of Sequences per Sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 23 October 2023).
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- MacArthur, R. Fluctuations of Animal Populations and a Measure of Community Stability. Ecology 1955, 36, 533. [Google Scholar] [CrossRef]
- Pielou, E.C. Shannon’s Formula as a Measure of Specific Diversity: Its Use and Misuse. Am. Nat. 1966, 100, 463–465. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Kruskal, J.B. Multidimensional Scaling by Optimizing Goodness of Fit to a Nonmetric Hypothesis. Psychometrika 1964, 29, 1–27. [Google Scholar] [CrossRef]
- Wickham, H. A Layered Grammar of Graphics. J. Comput. Graph. Stat. 2010, 19, 3–28. [Google Scholar] [CrossRef]
- Collyer, M.L.; Adams, D.C. RRPP: An r package for fitting linear models to high-dimensional data using residual randomization. Methods Ecol. Evol. 2018, 9, 1772–1779. [Google Scholar] [CrossRef]
- Teunisse, G.M. Fantaxtic: Fantaxtic Plots for Phyloseq Data. 2018. Available online: https://github.com/gmteunisse/fantaxtic (accessed on 23 October 2023).
- Breiman, L.; Breiman, L.; Cutler, R.A. Random Forests Machine Learning. J. Clin. Microbiol. 2001, 2, 199–228. [Google Scholar]
- Dang, T.; Kishino, H. Forward variable selection improves the power of random forest for high-dimensional microbiome data. bioRxiv 2020. [Google Scholar] [CrossRef]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef] [PubMed]
- Andersen, V.D.; Jensen, M.S.; Munk, P.; Vigre, H. Robustness in quantifying the abundance of antimicrobial resistance genes in pooled faeces samples from batches of slaughter pigs using metagenomics analysis. J. Glob. Antimicrob. Resist. 2021, 24, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.J.; Cotter, S.Y.; Arzika, A.M.; Kim, J.; Boubacar, N.; Zhou, Z.; Zhong, L.; Porco, T.C.; Keenan, J.D.; Lietman, T.M. High-throughput sequencing of pooled samples to determine community-level microbiome diversity. Ann. Epidemiol. 2019, 39, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Ruano, S.M.; Juhaňáková, E.; Vávra, J.; Nováková, E. Methodological insight into mosquito microbiome studies. Front. Cell. Infect. Microbiol. 2020, 10, 86. [Google Scholar] [CrossRef]
- Chen, S. Ultrafast One-pass FASTQ Data Preprocessing, Quality Control, and Deduplication Using Fastp. iMeta 2023, 2, e107. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef]
- Kursheva, A.V.; Morgunova, I.P.; Petrova, V.I.; Litvinenko, I.V. Hydrocarbons in the Littoral Sediments and March Soils of the Southwestern Coast of the Barents Sea. Geochem. Int. 2023, 61, 972–988. [Google Scholar] [CrossRef]
- Jung, J.; Park, W. Acinetobacter Species as Model Microorganisms in Environmental Microbiology: Current State and Perspectives. Appl. Microbiol. Biotechnol. 2015, 99, 2533–2548. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Price, L.S.; Weinstein, R.A. Acinetobacter infection. N. Engl. J. Med. 2008, 358, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Manchanda, V.; Sanchaita, S.; Singh, N. Multidrug resistant acinetobacter. J. Glob. Infect. Dis. 2010, 2, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Pierce, M.L.; Ward, J.E.; Holohan, B.A.; Zhao, X.; Hicks, R.E. The Influence of Site and Season on the Gut and Pallial Fluid Microbial Communities of the Eastern Oyster, Crassostrea Virginica (Bivalvia, Ostreidae): Community-Level Physiological Profiling and Genetic Structure. Hydrobiologia 2016, 765, 97–113. [Google Scholar] [CrossRef]
- Pierce, M.L.; Ward, J.E. Gut Microbiomes of the Eastern Oyster (Crassostrea virginica) and the Blue Mussel (Mytilus edulis): Temporal Variation and the Influence of Marine Aggregate-Associated Microbial Communities. mSphere 2019, 4, e00730-19. [Google Scholar] [CrossRef]
- Wei, J.; Gao, H.; Yang, Y.; Liu, H.; Yu, H.; Chen, Z.; Dong, B. Seasonal dynamics and starvation impact on the gut microbiome of urochordate ascidian Halocynthia roretzi. Anim. Microbiome 2020, 2, 30. [Google Scholar] [CrossRef]
- Lokmer, A.; Mathias Wegner, K. Hemolymph microbiome of Pacific oysters in response to temperature, temperature stress and infection. ISME J. 2015, 9, 670–682. [Google Scholar] [CrossRef]
- Lokmer, A.; Goedknegt, M.A.; Thieltges, D.W.; Fiorentino, D.; Kuenzel, S.; Baines, J.F.; Wegner, K.M. Spatial and Temporal Dynamics of Pacific oyster Hemolymph Microbiota across Multiple Scales. Front. Microbiol. 2016, 7, 1367. [Google Scholar] [CrossRef]
- Everroad, R.C.; Otaki, H.; Matsuura, K.; Haruta, S. Diversification of Bacterial Community Composition along a Temperature Gradient at a Thermal Spring. Microb. Environ. 2012, 27, 374–381. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, H.; Powell, J.; Delgado-Baquerizo, M.; Wang, J.; Singh, B. Warmer and Drier Ecosystems Select for Smaller Bacterial Genomes in Global Soils. iMeta 2023, 2, e70. [Google Scholar] [CrossRef]
- Jenkins, S.; Arenas, F.; Arrontes, J.; Bussell, J.; Castro, J.; Coleman, R.; Hawkins, S.; Kay, S.; Martínez, B.; Oliveros, J.; et al. European-Scale Analysis of Seasonal Variability in Limpet Grazing Activity and Microalgal Abundance. Mar. Ecol. Prog. Ser. 2001, 211, 193–203. [Google Scholar] [CrossRef]
- Malavenda, S.V. The tolerance of the Barents Sea fucoids for varying salinity. Bot. Zhurnal 2011, 96, 342–349. [Google Scholar]
- Wahl, M.; Jormalainen, V.; Eriksson, B.K.; Coyer, J.A.; Molis, M.; Schubert, H.; Dethier, M.; Karez, R.; Kruse, I.; Lenz, M.; et al. Stress ecology in fucus: Abiotic, biotic and genetic interactions. Adv. Mar. Biol. 2011, 59, 37–105. [Google Scholar] [CrossRef] [PubMed]
- Lachnit, T.; Meske, D.; Wahl, M.; Harder, T.; Schmitz, R. Epibacterial community patterns on marine macroalgae are host-specific but temporally variable. Environ. Microbiol. 2011, 13, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Jung, Y.T.; Won, S.M.; Park, J.M.; Yoon, J.H. Granulosicoccus undariae sp. nov., a member of the family Granulosicoccaceae isolated from a brown algae reservoir and emended description of the genus Granulosicoccus. Antonie Van Leeuwenhoek 2014, 106, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.; Lim, Y.; Cho, J.C. Complete genome sequence of Granulosicoccus antarcticus type strain IMCC3135(T), a marine gammaproteobacterium with a putative dimethylsulfoniopropionate demethylase gene. Mar. Genom. 2018, 37, 176–181. [Google Scholar] [CrossRef]
- Weigel, B.L.; Miranda, K.K.; Fogarty, E.C.; Watson, A.R.; Pfister, C.A. Functional Insights into the Kelp Microbiome from Metagenome-Assembled Genomes. mSystems 2022, 7, e01422-21. [Google Scholar] [CrossRef]
- Chiellini, C.; Lombardo, K.; Mocali, S.; Miceli, E.; Fani, R. Pseudomonas Strains Isolated from Different Environmental Niches Exhibit Different Antagonistic Ability. Ethol. Ecol. Evol. 2019, 31, 399–420. [Google Scholar] [CrossRef]
- Zhang, M.; Sun, Y.; Liu, Y.; Qiao, F.; Chen, L.; Liu, W.-T.; Du, Z.; Li, E. Response of Gut Microbiota to Salinity Change in Two Euryhaline Aquatic Animals with Reverse Salinity Preference. Aquaculture 2016, 454, 72–80. [Google Scholar] [CrossRef]
- Cicala, F.; Lago-Lestón, A.; Gomez-Gil, B.; Gollas-Galván, T.; Chong-Robles, J.; Cortés-Jacinto, E.; Martínez-Porchas, M. Gut Microbiota Shifts in the Giant Tiger Shrimp, Penaeus Monodon, during the Postlarvae, Juvenile, and Adult Stages. Aquac. Int. 2020, 28, 1421–1433. [Google Scholar] [CrossRef]
- Medić, A.B.; Karadžić, I.M. Pseudomonas in environmental bioremediation of hydrocarbons and phenolic compounds-key catabolic degradation enzymes and new analytical platforms for comprehensive investigation. World J. Microbiol. Biotechnol. 2022, 38, 165. [Google Scholar] [CrossRef] [PubMed]
- Yesankar, P.J.; Patil, A.; Kapley, A.; Qureshi, A. Catalytic resilience of multicomponent aromatic ring-hydroxylating dioxygenases in Pseudomonas for degradation of polycyclic aromatic hydrocarbons. World J. Microbiol. Biotechnol. 2023, 39, 166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; An, Z.; Su, X.; Hou, A.; Liu, L.; Zhang, L.; Lai, J. Phenol degradation at high salinity by a resuscitated strain Pseudomonas sp. SAS26: Kinetics and pathway. J. Environ. Chem. Eng. 2023, 11, 110182. [Google Scholar] [CrossRef]
- Ragan, M.A.; Craigie, J.S. Physodes and the phenolic compounds of brown algae. Isolation and characterization of phloroglucinol polymers from Fucus vesiculosus (L.). Can. J. Biochem. 1976, 54, 66–73. [Google Scholar] [CrossRef]
- Imbs, T.I.; Zvyagintseva, T.N. Phlorotannins Are Polyphenolic Metabolites of Brown Algae. Russ. J. Mar. Biol. 2018, 44, 263–273. [Google Scholar] [CrossRef]
- Lomartire, S.; Cotas, J.; Pacheco, D.; Marques, J.C.; Pereira, L.; Goncalves, A.M.M. Environmental Impact on Seaweed Phenolic Production and Activity: An Important Step for Compound Exploitation. Mar. Drugs 2021, 19, 245. [Google Scholar] [CrossRef]
- Bate-Smith, E.C. Haemanalysis of Tannins: The Concept of Relative Astringency. Phytochemistry 1973, 12, 907–912. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Barents#1 | Barents#2 |
|---|---|---|
| Environment | F. vesiculosus scraping (×2) | F. vesiculosus scraping (×2) |
| A. nodosum scraping (×3) | A. nodosum scraping (×1) | |
| F. serratus scraping (×1) | F. serratus scraping (×2) | |
| Stones scraping (×5) | Stones scraping (×3) | |
| Red Algae scraping (×3) | ||
| Snails | L. fabalis (gut ×7, tent. ×2) | L. fabalis (gut ×8, tent. ×2) |
| L. obtusata (gut ×13, tent. ×4) | L. obtusata (gut ×15, tent. ×4) |
| Sample | White, June | White, November |
|---|---|---|
| Environment | F. vesiculosus scraping (×1) | A. nodosum scraping (×3) |
| A. nodosum scraping (×3) | Filamentous Algae scraping (×4) | |
| Filamentous Algae scraping (×2) | Stones scraping (×5) | |
| Stones scraping (×1) | ||
| Snails | L. fabalis (gut ×6) | L. fabalis (gut ×7, tent. ×2) |
| L. obtusata (gut ×11, tent. ×1) | L. obtusata (gut ×12, tent. ×3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gafarova, E.; Kuracji, D.; Sogomonyan, K.; Gorokhov, I.; Polev, D.; Zubova, E.; Golikova, E.; Granovitch, A.; Maltseva, A. Gut Bacteriomes and Ecological Niche Divergence: An Example of Two Cryptic Gastropod Species. Biology 2023, 12, 1521. https://doi.org/10.3390/biology12121521
Gafarova E, Kuracji D, Sogomonyan K, Gorokhov I, Polev D, Zubova E, Golikova E, Granovitch A, Maltseva A. Gut Bacteriomes and Ecological Niche Divergence: An Example of Two Cryptic Gastropod Species. Biology. 2023; 12(12):1521. https://doi.org/10.3390/biology12121521
Chicago/Turabian StyleGafarova, Elizaveta, Dmitrii Kuracji, Karina Sogomonyan, Ivan Gorokhov, Dmitrii Polev, Ekaterina Zubova, Elena Golikova, Andrey Granovitch, and Arina Maltseva. 2023. "Gut Bacteriomes and Ecological Niche Divergence: An Example of Two Cryptic Gastropod Species" Biology 12, no. 12: 1521. https://doi.org/10.3390/biology12121521
APA StyleGafarova, E., Kuracji, D., Sogomonyan, K., Gorokhov, I., Polev, D., Zubova, E., Golikova, E., Granovitch, A., & Maltseva, A. (2023). Gut Bacteriomes and Ecological Niche Divergence: An Example of Two Cryptic Gastropod Species. Biology, 12(12), 1521. https://doi.org/10.3390/biology12121521