
Extracellular Vesicles: New Classification and Tumor Immunosuppression


Abstract
Simple Summary
Abstract
1. Introduction
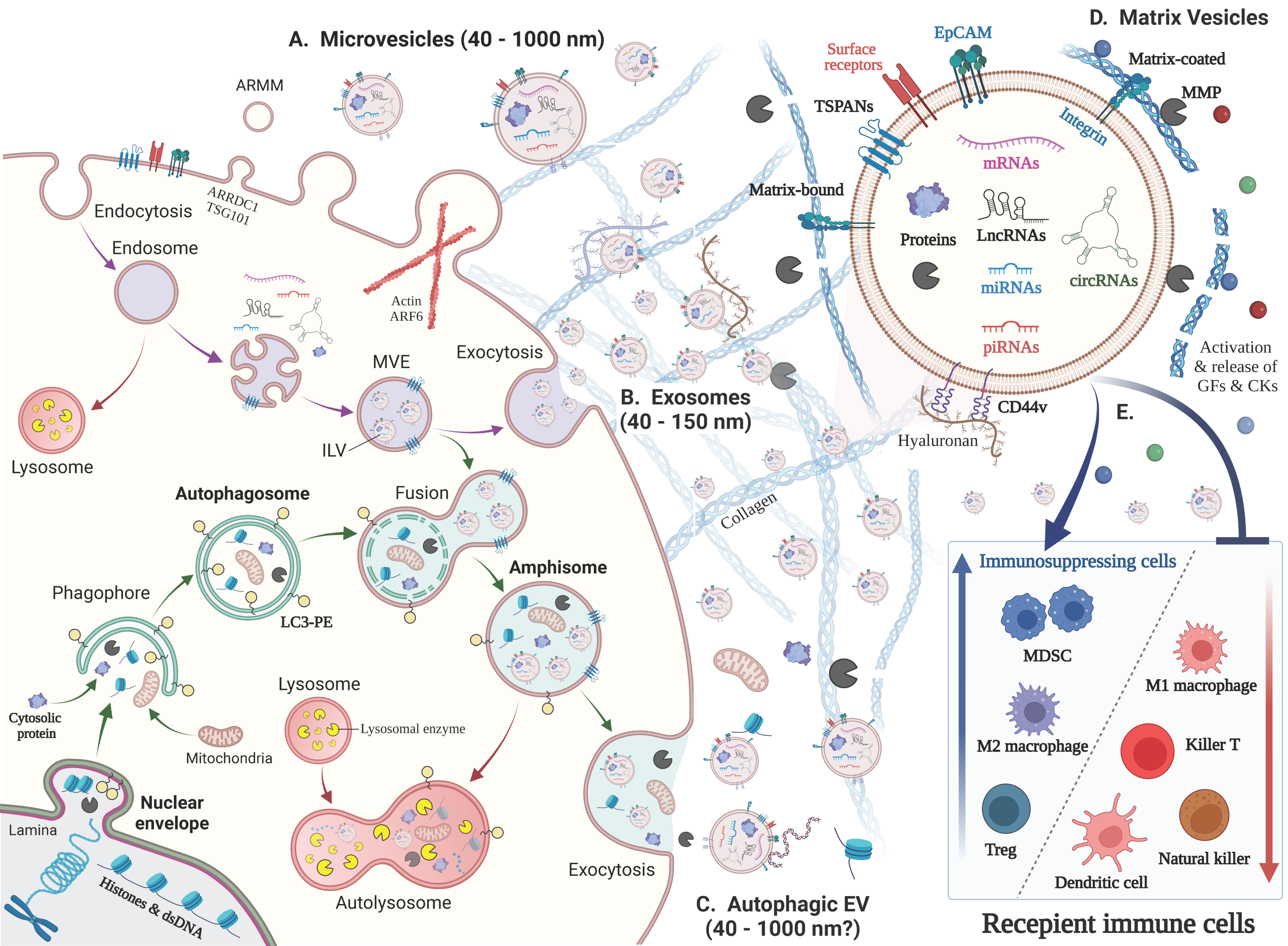
2. New Classification, Biogenesis and Functions of EVs
2.1. New Classification and Terminology of EVs
2.2. Exosome
2.2.1. Multivesicular Endosome (MVE) Biogenesis and Exosome Secretion
2.2.2. Chaperones and Kinases Promote Exosome Biogenesis and Secretion
2.3. Autophagic EVs: Autophagosome–Endosome Fusion to Secrete Amphisomes
2.4. EMT Is Associated with EV Release and Immunosuppression
2.5. Matrix Vesicles
2.5.1. ECM in the Tumor Microenvironment
2.5.2. ECM-Rich Microenvironment Is a Risk of Poor Prognosis in Cancer
2.5.3. ECM Mediate Tumor Malignancy
2.5.4. ECM–EVs Interaction
2.5.5. Matrix Moonlighting Metalloproteinases (MMPs)
2.6. EV-Mediated Molecular Transfer
2.6.1. Protein S-Palmitoylation Regulates EV Protein Sorting and Molecular Transfer
2.6.2. Transfer of Oncogenic Factors
2.7. Markers Defining EVs and Finding Biomarkers from EVs
2.7.1. Protein Markers of EVs
2.7.2. Stem Cell Markers on EV Surface
2.7.3. EV as a Source of Biomarkers
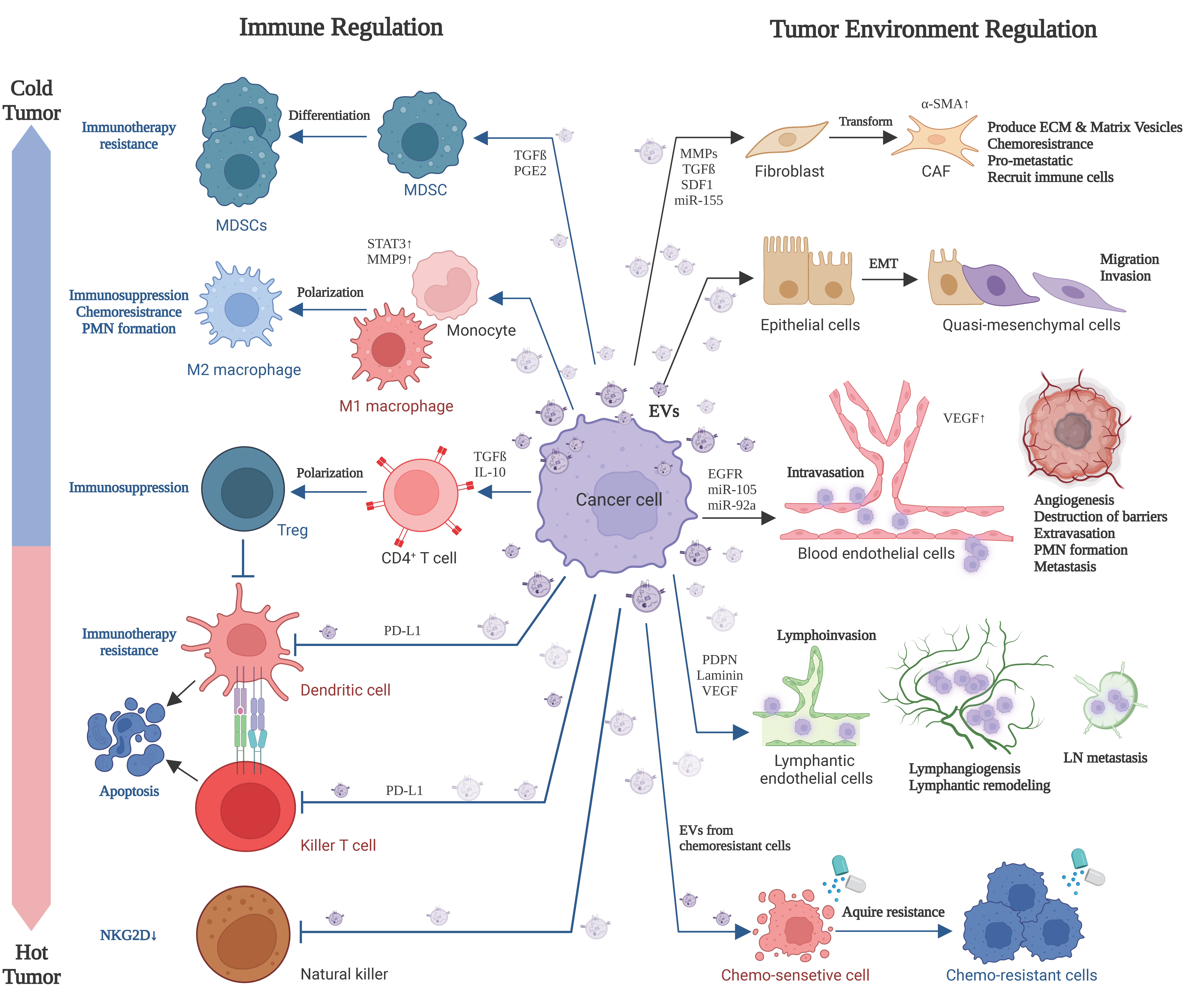
3. Tumor EVs Develop the Immunosuppressive and Resistant Microenvironment
3.1. Tumor–CAF Communication to Develop Chemoresistance
3.1.1. CAF Differentiation
3.1.2. CAF-Derived Exosomal miRNAs Promote Chemoresistance and Metastasis
3.2. Angiogenesis, Extravasation, and Intravasation Induced by EVs
3.2.1. Blood Endothelial Cells Support Tumor Progression and Metastasis
3.2.2. Chemokines and Growth Factors Induce Angiogenesis
3.2.3. Angiogenesis Promoted by Exosomal Noncoding RNAs
3.3. Tumor-Associated Macrophages Affected by Cancer EVs
3.3.1. M2 TAM Mediates Immunosuppression
3.3.2. M2 TAM-Derived EVs Induce Chemoresistance
3.3.3. Potential Exosomal Oncolipid
3.4. T Cells Affected by Cancer EVs
3.4.1. Tumor-Infiltrating Lymphocytes (TIL)
3.4.2. Apoptosis of Killer T Cells Induced by Cancer EVs
3.4.3. Treg Cells Induced by Cancer EVs
3.5. MDSCs Potentiated by Cancer EVs
3.5.1. MDSC—A Master Regulator of Immunosuppression
3.5.2. MDSC Differentiation and Recruitment Promoted by Tumor EVs
3.6. Tumor EVs Downregulate a Killing Factor of NK Cells
3.6.1. NK Cells Express a Killing Factor NKG2D
3.6.2. Tumor EVs Downregulate NKG2D Expression on NK Cells
3.7. Tumor EVs Induce the Immune Checkpoint of DCs
3.8. Tumor EVs Regulate Lymphatic Endothelial Cells (LECs) and Lymph Node Metastasis
3.8.1. Tumor Lymphangiogenesis and Lymphoinvasion
3.8.2. Podoplanin (PDPN) Regulates Tumor Lymphangiogenesis and Lymphoinvasion
3.8.3. Tumor EVs Regulate Lymphangiogenesis
3.8.4. Tumor EVs Promote LN Metastasis
3.8.5. Exosomal Biomarkers of Lymph Node Metastasis (LNM)
4. EVs Contribute to Immunosuppression and Chemoresistance
4.1. Hot Tumors and Cold Tumors
4.2. Exosomal PD-L1 Contributes to Immunosuppression
4.3. Exosomal EGFR Contributes to Immune Evasion
4.4. Tumor Microenvironmental EVs Contribute to Chemoresistance
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445.e18. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, T.; Sogawa, C.; Ono, K.; Matsumoto, M.; Tran, M.T.; Okusha, Y.; Lang, B.J.; Okamoto, K.; Calderwood, S.K. Cell Stress Induced Stressome Release Including Damaged Membrane Vesicles and Extracellular HSP90 by Prostate Cancer Cells. Cells 2020, 9, 755. [Google Scholar] [CrossRef] [PubMed]
- Hikita, T.; Kuwahara, A.; Watanabe, R.; Miyata, M.; Oneyama, C. Src in endosomal membranes promotes exosome secretion and tumor progression. Sci. Rep. 2019, 9, 3265. [Google Scholar] [CrossRef]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef]
- Ono, K.; Sogawa, C.; Kawai, H.; Tran, M.T.; Taha, E.A.; Lu, Y.; Oo, M.W.; Okusha, Y.; Okamura, H.; Ibaragi, S.; et al. Triple knockdown of CDC37, HSP90-alpha and HSP90-beta diminishes extracellular vesicles-driven malignancy events and macrophage M2 polarization in oral cancer. J. Extracell. Vesicles 2020, 9, 1769373. [Google Scholar] [CrossRef]
- Okusha, Y.; Eguchi, T.; Tran, M.T.; Sogawa, C.; Yoshida, K.; Itagaki, M.; Taha, E.A.; Ono, K.; Aoyama, E.; Okamura, H.; et al. Extracellular Vesicles Enriched with Moonlighting Metalloproteinase Are Highly Transmissive, Pro-Tumorigenic, and Trans-Activates Cellular Communication Network Factor (CCN2/CTGF): CRISPR against Cancer. Cancers 2020, 12, 881. [Google Scholar] [CrossRef]
- Taha, E.A.; Sogawa, C.; Okusha, Y.; Kawai, H.; Oo, M.W.; Elseoudi, A.; Lu, Y.; Nagatsuka, H.; Kubota, S.; Satoh, A.; et al. Knockout of MMP3 Weakens Solid Tumor Organoids and Cancer Extracellular Vesicles. Cancers 2020, 12, 1260. [Google Scholar] [CrossRef]
- Eguchi, T.; Sheta, M.; Fujii, M.; Calderwood, S.K. Cancer extracellular vesicles, tumoroid models, and tumor microenvironment. Semin. Cancer Biol. 2022, 86, 112–126. [Google Scholar] [CrossRef]
- Eguchi, T.; Sogawa, C.; Okusha, Y.; Uchibe, K.; Iinuma, R.; Ono, K.; Nakano, K.; Murakami, J.; Itoh, M.; Arai, K.; et al. Organoids with cancer stem cell-like properties secrete exosomes and HSP90 in a 3D nanoenvironment. PLoS ONE 2018, 13, e0191109. [Google Scholar] [CrossRef]
- Minciacchi, V.R.; Spinelli, C.; Reis-Sobreiro, M.; Cavallini, L.; You, S.; Zandian, M.; Li, X.; Mishra, R.; Chiarugi, P.; Adam, R.M.; et al. MYC Mediates Large Oncosome-Induced Fibroblast Reprogramming in Prostate Cancer. Cancer Res. 2017, 77, 2306–2317. [Google Scholar] [CrossRef]
- Huang, Y.; Zucker, B.; Zhang, S.; Elias, S.; Zhu, Y.; Chen, H.; Ding, T.; Li, Y.; Sun, Y.; Lou, J.; et al. Migrasome formation is mediated by assembly of micron-scale tetraspanin macrodomains. Nat. Cell Biol. 2019, 21, 991–1002. [Google Scholar] [CrossRef]
- Minamizaki, T.; Nakao, Y.; Irie, Y.; Ahmed, F.; Itoh, S.; Sarmin, N.; Yoshioka, H.; Nobukiyo, A.; Fujimoto, C.; Niida, S.; et al. The matrix vesicle cargo miR-125b accumulates in the bone matrix, inhibiting bone resorption in mice. Commun. Biol. 2020, 3, 30. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Meldolesi, J. Exosomes and Ectosomes in Intercellular Communication. Curr. Biol. 2018, 28, R435–R444. [Google Scholar] [CrossRef]
- Lu, Y.; Eguchi, T. HSP Stimulation on Macrophages and Dendritic Cells Activates Innate Immune System. In Heat Shock Proteins in Inflammatory Diseases. Heat Shock Proteins; Asea, A.A.A., Kaur, P., Eds.; Springer: Cham, Switzerland, 2021; Volume 22, pp. 53–67. [Google Scholar]
- Lu, Y.; Eguchi, T.; Sogawa, C.; Taha, E.A.; Tran, M.T.; Nara, T.; Wei, P.; Fukuoka, S.; Miyawaki, T.; Okamoto, K. Exosome-based molecular transfer activity of macrophage-like cells involves viability of oral carcinoma cells: Size exclusion chromatography and concentration filter method. Cells 2021, 10, 1328. [Google Scholar] [CrossRef]
- Zhang, Q.; Higginbotham, J.N.; Jeppesen, D.K.; Yang, Y.P.; Li, W.; McKinley, E.T.; Graves-Deal, R.; Ping, J.; Britain, C.M.; Dorsett, K.A.; et al. Transfer of Functional Cargo in Exomeres. Cell Rep. 2019, 27, 940–954.e6. [Google Scholar] [CrossRef]
- Dall’Olio, F.; Chiricolo, M.; Mariani, E.; Facchini, A. Biosynthesis of the cancer-related sialyl-alpha 2,6-lactosaminyl epitope in colon cancer cell lines expressing beta-galactoside alpha 2,6-sialyltransferase under a constitutive promoter. Eur. J. Biochem. 2001, 268, 5876–5884. [Google Scholar] [CrossRef]
- Schultz, M.J.; Holdbrooks, A.T.; Chakraborty, A.; Grizzle, W.E.; Landen, C.N.; Buchsbaum, D.J.; Conner, M.G.; Arend, R.C.; Yoon, K.J.; Klug, C.A.; et al. The Tumor-Associated Glycosyltransferase ST6Gal-I Regulates Stem Cell Transcription Factors and Confers a Cancer Stem Cell Phenotype. Cancer Res. 2016, 76, 3978–3988. [Google Scholar] [CrossRef]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Théry, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef] [PubMed]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brügger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Webber, J.P.; Spary, L.K.; Sanders, A.J.; Chowdhury, R.; Jiang, W.G.; Steadman, R.; Wymant, J.; Jones, A.T.; Kynaston, H.; Mason, M.D.; et al. Differentiation of tumour-promoting stromal myofibroblasts by cancer exosomes. Oncogene 2015, 34, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.; Teis, D. The ESCRT machinery. Curr. Biol. 2012, 22, R116–R120. [Google Scholar] [CrossRef] [PubMed]
- Gatta, A.T.; Carlton, J.G. The ESCRT-machinery: Closing holes and expanding roles. Curr. Opin. Cell Biol. 2019, 59, 121–132. [Google Scholar] [CrossRef]
- Marat, A.L.; Haucke, V. Phosphatidylinositol 3-phosphates-at the interface between cell signalling and membrane traffic. EMBO J. 2016, 35, 561–579. [Google Scholar] [CrossRef]
- Fujiwara, T.; Eguchi, T.; Sogawa, C.; Ono, K.; Murakami, J.; Ibaragi, S.; Asaumi, J.I.; Calderwood, S.K.; Okamoto, K.; Kozaki, K.I. Carcinogenic epithelial-mesenchymal transition initiated by oral cancer exosomes is inhibited by anti-EGFR antibody cetuximab. Oral Oncol. 2018, 86, 251–257. [Google Scholar] [CrossRef]
- Ono, K.; Eguchi, T.; Sogawa, C.; Calderwood, S.K.; Futagawa, J.; Kasai, T.; Seno, M.; Okamoto, K.; Sasaki, A.; Kozaki, K.I. HSP-enriched properties of extracellular vesicles involve survival of metastatic oral cancer cells. J. Cell Biochem. 2018, 119, 7350–7362. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Liu, L.; Yan, L.; Liao, N.; Wu, W.Q.; Shi, J.L. A Review of ULK1-Mediated Autophagy in Drug Resistance of Cancer. Cancers 2020, 12, 352. [Google Scholar] [CrossRef]
- Noman, M.Z.; Parpal, S.; van Moer, K.; Xiao, M.; Yu, Y.; Viklund, J.; de Milito, A.; Hasmim, M.; Andersson, M.; Amaravadi, R.K.; et al. Inhibition of Vps34 reprograms cold into hot inflamed tumors and improves anti-PD-1/PD-L1 immunotherapy. Sci. Adv. 2020, 6, eaax7881. [Google Scholar] [CrossRef]
- Joo, J.H.; Dorsey, F.C.; Joshi, A.; Hennessy-Walters, K.M.; Rose, K.L.; McCastlain, K.; Zhang, J.; Iyengar, R.; Jung, C.H.; Suen, D.F.; et al. Hsp90-Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy. Mol. Cell 2011, 43, 572–585. [Google Scholar] [CrossRef]
- Eguchi, T.; Ono, K.; Kawata, K.; Okamoto, K.; Calderwood, S.K. Regulatory Roles of HSP90-Rich Extracellular Vesicles. In Heat Shock Protein 90 in Human Diseases and Disorders, Heat Shock Proteins; Asea, A.A.A., Kaur, P., Eds.; Springer: Cham, Switzerland, 2019; Volume 19, pp. 3–17. [Google Scholar]
- Taha, S.; Volkmer, E.; Haas, E.; Alberton, P.; Straub, T.; David-Rus, D.; Aszodi, A.; Giunta, R.; Saller, M.M. Differences in the Inflammatory Response of White Adipose Tissue and Adipose-Derived Stem Cells. Int. J. Mol. Sci. 2020, 21, 1086. [Google Scholar] [CrossRef]
- Eguchi, T.; Prince, T.L.; Tran, M.T.; Sogawa, C.; Lang, B.J.; Calderwood, S.K. MZF1 and SCAND1 reciprocally regulate CDC37 Gene expression in prostate cancer. Cancers 2019, 11, 792. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Gong, J.; Murshid, A. Extracellular HSPs: The Complicated Roles of Extracellular HSPs in Immunity. Front. Immunol. 2016, 7, 159. [Google Scholar] [CrossRef]
- Taha, E.A.; Ono, K.; Eguchi, T. Roles of Extracellular HSPs as Biomarkers in Immune Surveillance and Immune Evasion. Int. J. Mol. Sci. 2019, 20, 4588. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Fader, C.M.; Colombo, M.I. Autophagy and multivesicular bodies: Two closely related partners. Cell Death Differ. 2009, 16, 70–78. [Google Scholar] [CrossRef]
- Xu, J.; Camfield, R.; Gorski, S.M. The interplay between exosomes and autophagy-partners in crime. J. Cell Sci. 2018, 131, jcs215210. [Google Scholar] [CrossRef]
- Chen, T.; Long, W.; Zhang, C.; Liu, S.; Zhao, L.; Hamaker, B.R. Fiber-utilizing capacity varies in Prevotella- versus Bacteroides-dominated gut microbiota. Sci. Rep. 2017, 7, 2594. [Google Scholar] [CrossRef]
- Fujiwara, T.; Eguchi, T.; Sogawa, C.; Ono, K.; Murakami, J.; Ibaragi, S.; Asaumi, J.I.; Okamoto, K.; Calderwood, S.K.; Kozaki, K.I. Anti-EGFR antibody cetuximab is secreted by oral squamous cell carcinoma and alters EGF-driven mesenchymal transition. Biochem. Biophys. Res. Commun. 2018, 503, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Namba, Y.; Sogawa, C.; Okusha, Y.; Kawai, H.; Itagaki, M.; Ono, K.; Murakami, J.; Aoyama, E.; Ohyama, K.; Asaumi, J.I.; et al. Depletion of lipid efflux pump ABCG1 triggers the intracellular accumulation of extracellular vesicles and reduces aggregation and tumorigenesis of metastatic cancer cells. Front. Oncol. 2018, 8, 376. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, T.; Taha, E.A.; Calderwood, S.K.; Ono, K. A novel model of cancer drug resistance: Oncosomal release of cytotoxic and antibody-based drugs. Biology 2020, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhan, H. Communication between EMT and PD-L1 signaling: New insights into tumor immune evasion. Cancer Lett. 2020, 468, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.Y.J.; Jackson, R.A.A.; Thiery, J.P.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef]
- Eguchi, T.; Csizmadia, E.; Kawai, H.; Sheta, M.; Yoshida, K.; Prince, T.L.; Wegiel, B.; Calderwood, S.K. SCAND1 Reverses Epithelial-to-Mesenchymal Transition (EMT) and Suppresses Prostate Cancer Growth and Migration. Cells 2022, 11, 3993. [Google Scholar] [CrossRef]
- Brabletz, S.; Schuhwerk, H.; Brabletz, T.; Stemmler, M.P. Dynamic EMT: A multi-tool for tumor progression. EMBO J. 2021, 40, e108647. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, L.; Wan, D.; Zhou, L.; Zheng, S.; Lin, S.; Qiao, Y. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct. Target Ther. 2021, 6, 153. [Google Scholar] [CrossRef]
- Karamanos, N.K.; Piperigkou, Z.; Passi, A.; Götte, M.; Rousselle, P.; Vlodavsky, I. Extracellular matrix-based cancer targeting. Trends Mol. Med. 2021, 27, 1000–1013. [Google Scholar] [CrossRef]
- Clayton, A.; Turkes, A.; Dewitt, S.; Steadman, R.; Mason, M.D.; Hallett, M.B. Adhesion and signaling by B cell-derived exosomes: The role of integrins. FASEB J. 2004, 18, 977–979. [Google Scholar] [CrossRef]
- Rieu, S.; Géminard, C.; Rabesandratana, H.; Sainte-Marie, J.; Vidal, M. Exosomes released during reticulocyte maturation bind to fibronectin via integrin alpha4beta1. Eur. J. Biochem. 2000, 267, 583–590. [Google Scholar] [CrossRef]
- Parker, A.L.; Bowman, E.; Zingone, A.; Ryan, B.M.; Cooper, W.A.; Kohonen-Corish, M.; Harris, C.C.; Cox, T.R. Extracellular matrix profiles determine risk and prognosis of the squamous cell carcinoma subtype of non-small cell lung carcinoma. Genome Med. 2022, 14, 126. [Google Scholar] [CrossRef]
- Al-Akkad, W.; Acedo, P.; Vilia, M.G.; Frenguelli, L.; Ney, A.; Rodriguez-Hernandez, I.; Labib, P.L.; Tamburrino, D.; Spoletini, G.; Hall, A.R.; et al. Tissue-Specific Human Extracellular Matrix Scaffolds Promote Pancreatic Tumour Progression and Chemotherapy Resistance. Cells 2022, 11, 3652. [Google Scholar] [CrossRef]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef]
- Deng, B.; Zhao, Z.; Kong, W.; Han, C.; Shen, X.; Zhou, C. Biological role of matrix stiffness in tumor growth and treatment. J. Transl. Med. 2022, 20, 540. [Google Scholar] [CrossRef]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Kim, M.; Lee, C.; Park, J. Extracellular matrix remodeling facilitates obesity-associated cancer progression. Trends Cell Biol. 2022, 32, 825–834. [Google Scholar] [CrossRef]
- Rilla, K.; Mustonen, A.M.; Arasu, U.T.; Härkönen, K.; Matilainen, J.; Nieminen, P. Extracellular vesicles are integral and functional components of the extracellular matrix. Matrix Biol. 2019, 75–76, 201–219. [Google Scholar] [CrossRef]
- Jong, A.Y.; Wu, C.H.; Li, J.; Sun, J.; Fabbri, M.; Wayne, A.S.; Seeger, R.C. Large-scale isolation and cytotoxicity of extracellular vesicles derived from activated human natural killer cells. J. Extracell. Vesicles 2017, 6, 1294368. [Google Scholar] [CrossRef]
- Adnani, L.; Spinelli, C.; Tawil, N.; Rak, J. Role of extracellular vesicles in cancer-specific interactions between tumour cells and the vasculature. Semin. Cancer Biol. 2022, 87, 196–213. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N.; et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015, 523, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, T.; Taha, E.A. Extracellular Vesicle-Associated Moonlighting Proteins: Heat Shock Proteins and Metalloproteinases. In Heat Shock Proteins in Inflammatory Diseases, Heat Shock Proteins; Asea, A.A.A., Kaur, P., Eds.; Springer: Cham, Switzerland, 2021; Volume 22, pp. 1–18. [Google Scholar]
- Okusha, Y.; Eguchi, T.; Sogawa, C.; Okui, T.; Nakano, K.; Okamoto, K.; Kozaki, K.I. The intranuclear PEX domain of MMP involves proliferation, migration, and metastasis of aggressive adenocarcinoma cells. J. Cell Biochem. 2018, 119, 7363–7376. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, T.; Calderwood, S.K.; Takigawa, M.; Kubota, S.; Kozaki, K.I. Intracellular MMP3 Promotes HSP Gene Expression in Collaboration with Chromobox Proteins. J. Cell Biochem. 2017, 118, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, T.; Kubota, S.; Kawata, K.; Mukudai, Y.; Uehara, J.; Ohgawara, T.; Ibaragi, S.; Sasaki, A.; Kuboki, T.; Takigawa, M. Novel Transcription Factor-Like Function of Human Matrix Metalloproteinase 3 Regulating the CTGF/CCN2 Gene. Mol. Cell Biol. 2008, 28, 2391. [Google Scholar] [CrossRef]
- Eguchi, T.; Kubota, S.; Kawata, K.; Mukudai, Y.; Uehara, J.; Ohgawara, T.; Ibaragi, S.; Sasaki, A.; Kuboki, T.; Takigawa, M. Novel Transcriptional Regulation of CCN2/CTGF by Nuclear Translocation of MMP3. In CCN Proteins in Health and Disease: An Overview of the Fifth International Workshop on the CCN Family of Genes; Perbal, A., Takigawa, M., Perbal, B., Eds.; Springer: Dordrecht, The Netherlands, 2010; pp. 255–264. [Google Scholar]
- Ono, K.; Okusha, Y.; Tran, M.T.; Umemori, K.; Eguchi, T. Western Blot Protocols for Analysis of CCN Proteins and Fragments in Exosomes, Vesicle-Free Fractions, and Cells. In CCN Proteins in Health and Disease: An Overview of the Fifth International Workshop on the CCN Family of Genes; Takigawa, M., Ed.; Humana: New York, NY, USA, 2023; Volume 2582, pp. 39–57. [Google Scholar]
- Sung, B.H.; Ketova, T.; Hoshino, D.; Zijlstra, A.; Weaver, A.M. Directional cell movement through tissues is controlled by exosome secretion. Nat. Commun. 2015, 6, 7164. [Google Scholar] [CrossRef]
- Sanderson, M.P.; Keller, S.; Alonso, A.; Riedle, S.; Dempsey, P.J.; Altevogt, P. Generation of Novel, Secreted Epidermal Growth Factor Receptor (EGFR/ErbB1) Isoforms Via Metalloprotease-Dependent Ectodomain Shedding and Exosome Secretion. J. Cell Biochem. 2008, 103, 1783. [Google Scholar] [CrossRef]
- Rilla, K.; Siiskonen, H.; Tammi, M.; Tammi, R. Hyaluronan-coated extracellular vesicles—A novel link between hyaluronan and cancer. Adv. Cancer Res. 2014, 123, 121–148. [Google Scholar]
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev. Cell 2019, 49, 347–360. [Google Scholar] [CrossRef]
- Purushothaman, A.; Bandari, S.K.; Liu, J.; Mobley, J.A.; Brown, E.A.; Sanderson, R.D. Fibronectin on the Surface of Myeloma Cell-derived Exosomes Mediates Exosome-Cell Interactions. J. Biol Chem. 2016, 291, 1652–1663. [Google Scholar] [CrossRef]
- Lima, L.G.; Ham, S.; Shin, H.; Chai, E.P.Z.; Lek, E.S.H.; Lobb, R.J.; Müller, A.F.; Mathivanan, S.; Yeo, B.; Choi, Y.; et al. Tumor microenvironmental cytokines bound to cancer exosomes determine uptake by cytokine receptor-expressing cells and biodistribution. Nat. Commun. 2021, 12, 3543. [Google Scholar] [CrossRef]
- Costa Verdera, H.; Gitz-Francois, J.J.; Schiffelers, R.M.; Vader, P. Cellular uptake of extracellular vesicles is mediated by clathrin-independent endocytosis and macropinocytosis. J. Control Release 2017, 266, 100–108. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Mariscal, J.; Vagner, T.; Kim, M.; Zhou, B.; Chin, A.; Zandian, M.; Freeman, M.R.; You, S.; Zijlstra, A.; Yang, W.; et al. Comprehensive palmitoyl-proteomic analysis identifies distinct protein signatures for large and small cancer-derived extracellular vesicles. J. Extracell. Vesicles 2020, 9, 1764192. [Google Scholar] [CrossRef]
- Yu, L.; Wei, Y.; Duan, J.; Schmitz, D.A.; Sakurai, M.; Wang, L.; Wang, K.; Zhao, S.; Hon, G.C.; Wu, J. Blastocyst-like structures generated from human pluripotent stem cells. Nature 2021, 591, 620–626. [Google Scholar] [CrossRef]
- Romancino, D.P.; Buffa, V.; Caruso, S.; Ferrara, I.; Raccosta, S.; Notaro, A.; Campos, Y.; Noto, R.; Martorana, V.; Cupane, A.; et al. Palmitoylation is a post-translational modification of Alix regulating the membrane organization of exosome-like small extracellular vesicles. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2879–2887. [Google Scholar] [CrossRef]
- Sun, C.; Wang, P.; Dong, W.; Liu, H.; Sun, J.; Zhao, L. LncRNA PVT1 promotes exosome secretion through YKT6, RAB7, and VAMP3 in pancreatic cancer. Aging 2020, 12, 10427. [Google Scholar] [CrossRef]
- Zhou, B.; Hao, Q.; Liang, Y.; Kong, E. Protein palmitoylation in cancer: Molecular functions and therapeutic potential. Mol. Oncol. 2022, 17, 3–26. [Google Scholar] [CrossRef]
- Beckler, M.D.; Higginbotham, J.N.; Franklin, J.L.; Ham, A.J.; Halvey, P.J.; Imasuen, I.E.; Whitwell, C.; Li, M.; Liebler, D.C.; Coffey, R.J. Proteomic analysis of exosomes from mutant KRAS colon cancer cells identifies intercellular transfer of mutant KRAS. Mol. Cell Proteom. 2013, 12, 343–355. [Google Scholar] [CrossRef]
- Mu, J.; Zhuang, X.; Wang, Q.; Jiang, H.; Deng, Z.B.; Wang, B.; Zhang, L.; Kakar, S.; Jun, Y.; Miller, D.; et al. Interspecies communication between plant and mouse gut host cells through edible plant derived exosome-like nanoparticles. Mol. Nutr. Food Res. 2014, 58, 1561–1573. [Google Scholar] [CrossRef]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Al-Nedawi, K.; Meehan, B.; Kerbel, R.S.; Allison, A.C.; Rak, J. Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc. Natl. Acad. Sci. USA 2009, 106, 3794. [Google Scholar] [CrossRef] [PubMed]
- Braicu, C.; Tomuleasa, C.; Monroig, P.; Cucuianu, A.; Berindan-Neagoe, I.; Calin, G.A. Exosomes as divine messengers: Are they the Hermes of modern molecular oncology? Cell Death Differ. 2015, 22, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Skog, J.; Würdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Curry, W.T.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Fujita, Y.; Yoshioka, Y.; Ochiya, T. Extracellular vesicle transfer of cancer pathogenic components. Cancer Sci. 2016, 107, 385–390. [Google Scholar] [CrossRef]
- Pan, B.T.; Teng, K.; Wu, C.; Adam, M.; Johnstone, R.M. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J. Cell Biol. 1985, 101, 942–948. [Google Scholar] [CrossRef]
- Chiba, M.; Kimura, M.; Asari, S. Exosomes secreted from human colorectal cancer cell lines contain mRNAs, microRNAs and natural antisense RNAs, that can transfer into the human hepatoma HepG2 and lung cancer A549 cell lines. Oncol. Rep. 2012, 28, 1551–1558. [Google Scholar] [CrossRef]
- Kalluri, R.; Lebleu, V.S. Discovery of Double-Stranded Genomic DNA in Circulating Exosomes. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 275–280. [Google Scholar] [CrossRef]
- Mathivanan, S.; Fahner, C.J.; Reid, G.E.; Simpson, R.J. ExoCarta 2012: Database of exosomal proteins, RNA and lipids. Nucleic Acids Res. 2012, 40, D1241–D1244. [Google Scholar] [CrossRef]
- Mears, R.; Craven, R.A.; Hanrahan, S.; Totty, N.; Upton, C.; Young, S.L.; Patel, P.; Selby, P.J.; Banks, R.E. Proteomic analysis of melanoma-derived exosomes by two-dimensional polyacrylamide gel electrophoresis and mass spectrometry. Proteomics 2004, 4, 4019–4031. [Google Scholar] [CrossRef]
- Gastpar, R.; Gehrmann, M.; Bausero, M.A.; Asea, A.; Gross, C.; Schroeder, J.A.; Multhoff, G. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res. 2005, 65, 5238–5247. [Google Scholar] [CrossRef]
- Mathivanan, S.; Ji, H.; Simpson, R.J. Exosomes: Extracellular organelles important in intercellular communication. J. Proteom. 2010, 73, 1907–1920. [Google Scholar] [CrossRef]
- Nakamura, K.; Sawada, K.; Kinose, Y.; Yoshimura, A.; Toda, A.; Nakatsuka, E.; Hashimoto, K.; Mabuchi, S.; Morishige, K.I.; Kurachi, H.; et al. Exosomes Promote Ovarian Cancer Cell Invasion through Transfer of CD44 to Peritoneal Mesothelial Cells. Mol. Cancer Res. 2017, 15, 78–92. [Google Scholar] [CrossRef]
- Sharghi-Namini, S.; Tan, E.; Ong, L.L.S.; Ge, R.; Asada, H.H. Dll4-containing exosomes induce capillary sprout retraction in a 3D microenvironment. Sci. Rep. 2014, 4, 4031. [Google Scholar] [CrossRef]
- Kang, L.J.; Yu, Z.H.; Cai, J.; He, R.; Lu, J.T.; Hou, C.; Wang, Q.S.; Li, X.Q.; Zhang, R.; Feng, Y.M. Reciprocal transrepression between FOXF2 and FOXQ1 controls basal-like breast cancer aggressiveness. FASEB J. 2019, 33, 6564–6573. [Google Scholar] [CrossRef]
- Hu, Y.B.; Yan, C.; Mu, L.; Mi, Y.-L.; Zhao, H.; Hu, H.; Li, X.L.; Tao, D.D.; Wu, Y.Q.; Gong, J.P.; et al. Correction: Exosomal Wnt-induced dedifferentiation of colorectal cancer cells contributes to chemotherapy resistance. Oncogene 2019, 38, 6319–6321. [Google Scholar] [CrossRef]
- Ramos, T.L.; Sánchez-Abarca, L.I.; Muntión, S.; Preciado, S.; Puig, N.; López-Ruano, G.; Hernández-Hernández, Á.; Redondo, A.; Ortega, R.; Rodríguez, C.; et al. MSC surface markers (CD44, CD73, and CD90) can identify human MSC-derived extracellular vesicles by conventional flow cytometry. Cell Commun. Signal 2016, 14, 2. [Google Scholar] [CrossRef]
- las Heras, K.; Royo, F.; Garcia-Vallicrosa, C.; Igartua, M.; Santos-Vizcaino, E.; Falcon-Perez, J.M.; Hernandez, R.M. Extracellular vesicles from hair follicle-derived mesenchymal stromal cells: Isolation, characterization and therapeutic potential for chronic wound healing. Stem Cell Res. Ther. 2022, 13, 147. [Google Scholar] [CrossRef]
- Elsner, C.; Ergün, S.; Wagner, N. Biogenesis and release of endothelial extracellular vesicles: Morphological aspects. Ann. Anat. 2023, 245, 152006. [Google Scholar] [CrossRef]
- Hiraga, C.; Yamamoto, S.; Hashimoto, S.; Kasahara, M.; Minamisawa, T.; Matsumura, S.; Katakura, A.; Yajima, Y.; Nomura, T.; Shiba, K. Pentapartite fractionation of particles in oral fluids by differential centrifugation. Sci. Rep. 2021, 11, 3326. [Google Scholar] [CrossRef]
- Park, J.E.; Dutta, B.; Tse, S.W.; Gupta, N.; Tan, C.F.; Low, J.K.; Yeoh, K.W.; Kon, O.L.; Tam, J.P.; Sze, S.K. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift. Oncogene 2019, 38, 5158–5173. [Google Scholar] [CrossRef]
- Li, J.; Tian, T.; Zhou, X. The role of exosomal shuttle RNA (esRNA) in lymphoma. Crit. Rev. Oncol. Hematol. 2019, 137, 27–34. [Google Scholar] [CrossRef]
- Li, S.; Li, Y.; Chen, B.; Zhao, J.; Yu, S.; Tang, Y.; Zheng, Q.; Li, Y.; Wang, P.; He, X.; et al. ExoRBase: A database of circRNA, lncRNA and mRNA in human blood exosomes. Nucleic Acids Res. 2018, 46, D106–D112. [Google Scholar] [CrossRef] [PubMed]
- Hessvik, N.P.; Sandvig, K.; Llorente, A. Exosomal miRNAs as biomarkers for prostate cancer. Front. Genet. 2013, 4, 36. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Vickers, K.C. Intercellular transport of MicroRNAs. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Chen, J.; Su, F.; Yu, B.; Su, F.; Lin, L.; Liu, Y.; Huang, J.D.; Song, E. Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs into breast cancer cells. Mol. Cancer 2011, 10, 117. [Google Scholar] [CrossRef] [PubMed]
- Sasaya, T.; Kubo, T.; Murata, K.; Mizue, Y.; Sasaki, K.; Yanagawa, J.; Imagawa, M.; Kato, H.; Tsukahara, T.; Kanaseki, T.; et al. Cisplatin-induced HSF1-HSP90 axis enhances the expression of functional PD-L1 in oral squamous cell carcinoma. Cancer Med. 2022. [Google Scholar] [CrossRef]
- Boedtkjer, E.; Pedersen, S.F. The Acidic Tumor Microenvironment as a Driver of Cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef]
- Anckar, J.; Sistonen, L. Regulation of HSF1 function in the heat stress response: Implications in aging and disease. Annu. Rev. Biochem. 2011, 80, 1089–1115. [Google Scholar] [CrossRef]
- Murshid, A.; Eguchi, T.; Calderwood, S.K. Stress proteins in aging and life span. Int. J. Hyperthermia 2013, 29, 442–447. [Google Scholar] [CrossRef]
- van den Tempel, N.; Zelensky, A.N.; Odijk, H.; Laffeber, C.; Schmidt, C.K.; Brandsma, I.; Demmers, J.; Krawczyk, P.M.; Kanaar, R. On the Mechanism of Hyperthermia-Induced BRCA2 Protein Degradation. Cancers 2019, 11, 97. [Google Scholar] [CrossRef]
- Gong, J.L.; Lang, B.J.; Weng, D.S.; Eguchi, T.; Murshid, A.; Borges, T.J.; Doshi, S.; Song, B.Z.; Stevenson, M.A.; Calderwood, S.K. Genotoxic stress induces Sca-1-expressing metastatic mammary cancer cells. Mol. Oncol. 2018, 12, 1249–1263. [Google Scholar] [CrossRef]
- Hitomi, K.; Okada, R.; Loo, T.M.; Miyata, K.; Nakamura, A.J.; Takahashi, A. DNA Damage Regulates Senescence-Associated Extracellular Vesicle Release via the Ceramide Pathway to Prevent Excessive Inflammatory Responses. Int. J. Mol. Sci. 2020, 21, 3720. [Google Scholar] [CrossRef]
- Gomez-Pastor, R.; Burchfiel, E.T.; Thiele, D.J. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 4–19. [Google Scholar] [CrossRef]
- Guang, M.H.Z.; Kavanagh, E.L.; Dunne, L.P.; Dowling, P.; Zhang, L.; Lindsay, S.; Bazou, D.; Goh, C.Y.; Hanley, C.; Bianchi, G.; et al. Targeting Proteotoxic Stress in Cancer: A Review of the Role that Protein Quality Control Pathways Play in Oncogenesis. Cancers 2019, 11, 66. [Google Scholar] [CrossRef]
- Morimoto, R.I. The heat shock response: Systems biology of proteotoxic stress in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 91–99. [Google Scholar] [CrossRef]
- Kugeratski, F.G.; Kalluri, R. Exosomes as mediators of immune regulation and immunotherapy in cancer. FEBS J. 2021, 288, 10–35. [Google Scholar] [CrossRef]
- Huang, Z.; Feng, Y. Exosomes derived from hypoxic colorectal cancer cells promote angiogenesis through Wnt4-Induced β-catenin signaling in endothelial cells. Oncol. Res. 2017, 25, 651–661. [Google Scholar] [CrossRef]
- Xiang, X.; Poliakov, A.; Liu, C.; Liu, Y.; Deng, Z.B.; Wang, J.; Cheng, Z.; Shah, S.V.; Wang, G.J.; Zhang, L.; et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int. J. Cancer 2009, 124, 2621–2633. [Google Scholar] [CrossRef] [PubMed]
- Oo, M.W.; Kawai, H.; Takabatake, K.; Tomida, S.; Eguchi, T.; Ono, K.; Shan, Q.; Ohara, T.; Yoshida, S.; Omori, H.; et al. Resident stroma-secreted chemokine CCL2 governs myeloid-derived suppressor cells in the tumor microenvironment. JCI Insight 2022, 7, e148960. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.A.; Park, H.; Lim, E.H.; Kim, K.H.; Choi, J.S.; Lee, J.H.; Shin, J.W.; Lee, K.W. Exosomes from ovarian cancer cells induce adipose tissue-derived mesenchymal stem cells to acquire the physical and functional characteristics of tumor-supporting myofibroblasts. Gynecol. Oncol. 2011, 123, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Webber, J.; Steadman, R.; Mason, M.D.; Tabi, Z.; Clayton, A. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. 2010, 70, 9621–9630. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Miyata, H.; Sugimura, K.; Fukuda, S.; Kanemura, T.; Yamashita, K.; Miyazaki, Y.; Takahashi, T.; Kurokawa, Y.; Yamasaki, M.; et al. miR-27 is associated with chemoresistance in esophageal cancer through transformation of normal fibroblasts to cancer-associated fibroblasts. Carcinogenesis 2015, 36, 894–903. [Google Scholar] [CrossRef]
- Kong, J.; Tian, H.; Zhang, F.; Zhang, Z.; Li, J.; Liu, X.; Li, X.; Liu, J.; Li, X.; Jin, D.; et al. Extracellular vesicles of carcinoma-associated fibroblasts creates a pre-metastatic niche in the lung through activating fibroblasts. Mol. Cancer 2019, 18, 175. [Google Scholar] [CrossRef]
- Zeng, Z.; Li, Y.; Pan, Y.; Lan, X.; Song, F.; Sun, J.; Zhou, K.; Liu, X.; Ren, X.; Wang, F.; et al. Cancer-derived exosomal miR-25-3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nat. Commun. 2018, 9, 5395. [Google Scholar] [CrossRef]
- Beider, K.; Bitner, H.; Leiba, M.; Gutwein, O.; Koren-Michowitz, M.; Ostrovsky, O.; Abraham, M.; Wald, H.; Galun, E.; Peled, A.; et al. Multiple myeloma cells recruit tumor-supportive macrophages through the CXCR4/CXCL12 axis and promote their polarization toward the M2 phenotype. Oncotarget 2014, 5, 11283. [Google Scholar] [CrossRef]
- Cheng, H.Y.; Hsieh, C.H.; Lin, P.H.; Chen, Y.T.; Hsu, D.S.S.; Tai, S.K.; Chu, P.Y.; Yang, M.H. Snail-regulated exosomal microRNA-21 suppresses NLRP3 inflammasome activity to enhance cisplatin resistance. J. Immunother. Cancer 2022, 10, e004832. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, H.; Yuan, X.; Jiang, P.; Qian, H.; Xu, W. Tumor-derived exosomes induce N2 polarization of neutrophils to promote gastric cancer cell migration. Mol. Cancer 2018, 17, 146. [Google Scholar] [CrossRef]
- Ning, Y.; Shen, K.; Wu, Q.; Sun, X.; Bai, Y.; Xie, Y.; Pan, J.; Qi, C. Tumor exosomes block dendritic cells maturation to decrease the T cell immune response. Immunol. Lett. 2018, 199, 36–43. [Google Scholar] [CrossRef]
- Leary, N.; Walser, S.; He, Y.; Cousin, N.; Pereira, P.; Gallo, A.; Collado-Diaz, V.; Halin, C.; Garcia-Silva, S.; Peinado, H.; et al. Melanoma-derived extracellular vesicles mediate lymphatic remodelling and impair tumour immunity in draining lymph nodes. J. Extracell. Vesicles 2022, 11, e12197. [Google Scholar] [CrossRef]
- Wang, L.; Li, L.; Zhu, G. Role of Extracellular Vesicles on Cancer Lymphangiogenesis and Lymph Node Metastasis. Front. Oncol. 2021, 11, 721785. [Google Scholar] [CrossRef]
- Andreola, G.; Rivoltini, L.; Castelli, C.; Huber, V.; Perego, P.; Deho, P.; Squarcina, P.; Accornero, P.; Lozupone, F.; Lugini, L.; et al. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J. Exp. Med. 2002, 195, 1303–1316. [Google Scholar] [CrossRef]
- Szajnik, M.; Czystowska, M.; Szczepanski, M.J.; Mandapathil, M.; Whiteside, T.L. Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg). PLoS ONE 2010, 5, e11469. [Google Scholar] [CrossRef]
- Lundholm, M.; Schröder, M.; Nagaeva, O.; Baranov, V.; Widmark, A.; Mincheva-Nilsson, L.; Wikström, P. Prostate tumor-derived exosomes down-regulate NKG2D expression on natural killer cells and CD8+ T cells: Mechanism of immune evasion. PLoS ONE 2014, 9, e108925. [Google Scholar] [CrossRef]
- Jamieson, A.M.; Diefenbach, A.; McMahon, C.W.; Xiong, N.; Carlyle, J.R.; Raulet, D.H. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity 2002, 17, 19–29. [Google Scholar] [CrossRef]
- Zhang, H.; Deng, T.; Liu, R.; Ning, T.; Yang, H.; Liu, D.; Zhang, Q.; Lin, D.; Ge, S.; Bai, M.; et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol. Cancer 2020, 19, 43. [Google Scholar] [CrossRef]
- Qin, X.; Guo, H.; Wang, X.; Zhu, X.; Yan, M.; Wang, X.; Xu, Q.; Shi, J.; Lu, E.; Chen, W.; et al. Exosomal miR-196a derived from cancer-associated fibroblasts confers cisplatin resistance in head and neck cancer through targeting CDKN1B and ING. Genome Biol. 2019, 20, 12. [Google Scholar] [CrossRef]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778. [Google Scholar] [CrossRef]
- Chen, B.; Sang, Y.; Song, X.; Zhang, D.; Wang, L.; Zhao, W.; Liang, Y.; Zhang, N.; Yang, Q. Exosomal miR-500a-5p derived from cancer-associated fibroblasts promotes breast cancer cell proliferation and metastasis through targeting USP28. Theranostics 2021, 11, 3932–3947. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Li, M.; Cao, L.M.; Gu, Q.H.; Deng, P.B.; Tan, Y.; Hu, C.P. Snail1-dependent cancer-associated fibroblasts induce epithelial-mesenchymal transition in lung cancer cells via exosomes. QJM 2019, 112, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Shani, O.; Vorobyov, T.; Monteran, L.; Lavie, D.; Cohen, N.; Raz, Y.; Tsarfaty, G.; Avivi, C.; Barshack, I.; Erez, N. Fibroblast-Derived IL33 Facilitates Breast Cancer Metastasis by Modifying the Immune Microenvironment and Driving Type 2 Immunity. Cancer Res. 2020, 80, 5317–5329. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Wu, Y.; Xu, Y.; Li, G.; Li, Z.; Liu, T. Mesenchymal stem cell-derived exosomes in cancer therapy resistance: Recent advances and therapeutic potential. Mol. Cancer 2022, 21, 179. [Google Scholar] [CrossRef] [PubMed]
- Sohrabi, B.; Dayeri, B.; Zahedi, E.; Khoshbakht, S.; Nezamabadi Pour, N.; Ranjbar, H.; Davari Nejad, A.; Noureddini, M.; Alani, B. Mesenchymal stem cell (MSC)-derived exosomes as novel vehicles for delivery of miRNAs in cancer therapy. Cancer Gene Ther. 2022, 29, 1105–1116. [Google Scholar] [CrossRef]
- Zheng, P.; Chen, L.; Yuan, X.; Luo, Q.; Liu, Y.; Xie, G.; Ma, Y.; Shen, L. Exosomal transfer of tumor-associated macrophage-derived miR-21 confers cisplatin resistance in gastric cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 53. [Google Scholar] [CrossRef]
- Binenbaum, Y.; Fridman, E.; Yaari, Z.; Milman, N.; Schroeder, A.; David, G.B.; Shlomi, T.; Gil, Z. Transfer of miRNA in Macrophage-Derived Exosomes Induces Drug Resistance in Pancreatic Adenocarcinoma. Cancer Res. 2018, 78, 5287–5299. [Google Scholar] [CrossRef]
- Zhang, J.; Ji, C.; Zhang, H.; Shi, H.; Mao, F.; Qian, H.; Xu, W.; Wang, D.; Pan, J.; Fang, X.; et al. Engineered neutrophil-derived exosome-like vesicles for targeted cancer therapy. Sci. Adv. 2022, 8, eabj8207. [Google Scholar] [CrossRef]
- Basak, M.; Chaudhary, D.K.; Takahashi, R.; Yamamoto, Y.; Tiwari, S.; Tahara, H.; Mittal, A. Immunocyte Derived Exosomes: Insight into the Potential Chemo-immunotherapeutic Nanocarrier Targeting the Tumor Microenvironment. ACS Biomater. Sci. Eng. 2022, 9, 20–39. [Google Scholar] [CrossRef]
- Merjaneh, M.; Langlois, A.; Larochelle, S.; Cloutier, C.B.; Ricard-Blum, S.; Moulin, V.J. Pro-angiogenic capacities of microvesicles produced by skin wound myofibroblasts. Angiogenesis 2017, 20, 385–398. [Google Scholar] [CrossRef]
- Han, K.Y.; Tran, J.A.; Chang, J.H.; Azar, D.T.; Zieske, J.D. Potential role of corneal epithelial cell-derived exosomes in corneal wound healing and neovascularization. Sci. Rep. 2017, 7, 40548. [Google Scholar] [CrossRef]
- Gu, J.; Qian, H.; Shen, L.; Zhang, X.; Zhu, W.; Huang, L.; Yan, Y.; Mao, F.; Zhao, C.; Shi, Y.; et al. Gastric Cancer Exosomes Trigger Differentiation of Umbilical Cord Derived Mesenchymal Stem Cells to Carcinoma-Associated Fibroblasts through TGF-β/Smad Pathway. PLoS ONE 2012, 7, e52465. [Google Scholar] [CrossRef]
- Pang, W.; Su, J.; Wang, Y.; Feng, H.; Dai, X.; Yuan, Y.; Chen, X.; Yao, W. Pancreatic cancer-secreted miR-155 implicates in the conversion from normal fibroblasts to cancer-associated fibroblasts. Cancer Sci. 2015, 106, 1362. [Google Scholar] [CrossRef]
- Chowdhury, R.; Webber, J.P.; Gurney, M.; Mason, M.D.; Tabi, Z.; Clayton, A. Cancer exosomes trigger mesenchymal stem cell differentiation into pro-angiogenic and pro-invasive myofibroblasts. Oncotarget 2015, 6, 715. [Google Scholar] [CrossRef]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef]
- Guo, Q.R.; Wang, H.; Yan, Y.D.; Liu, Y.; Su, C.Y.; Chen, H.B.; Yan, Y.Y.; Adhikari, R.; Wu, Q.; Zhang, J.Y. The Role of Exosomal microRNA in Cancer Drug Resistance. Front. Oncol. 2020, 10, 472. [Google Scholar] [CrossRef]
- Zhong, Y.; Li, H.; Li, P.; Chen, Y.; Zhang, M.; Yuan, Z.; Zhang, Y.; Xu, Z.; Luo, G.; Fang, Y.; et al. Exosomes: A New Pathway for Cancer Drug Resistance. Front. Oncol. 2021, 11, 3846. [Google Scholar] [CrossRef]
- Li, J.; Gao, N.; Gao, Z.; Liu, W.; Pang, B.; Dong, X.; Li, Y.; Fan, T. The Emerging Role of Exosomes in Cancer Chemoresistance. Front. Cell Dev. Biol. 2021, 9, 2985. [Google Scholar] [CrossRef]
- Fang, Y.; Zhou, W.; Rong, Y.; Kuang, T.; Xu, X.; Wu, W.; Wang, D.; Lou, W. Exosomal miRNA-106b from cancer-associated fibroblast promotes gemcitabine resistance in pancreatic cancer. Exp. Cell Res. 2019, 383, 111543. [Google Scholar] [CrossRef]
- Zhang, L.; Yao, J.; Li, W.; Zhang, C. Micro-RNA-21 Regulates Cancer-Associated Fibroblast-Mediated Drug Resistance in Pancreatic Cancer. Oncol. Res. 2018, 26, 827–836. [Google Scholar] [CrossRef]
- Au Yeung, C.L.; Co, N.N.; Tsuruga, T.; Yeung, T.L.; Kwan, S.Y.; Leung, C.S.; Li, Y.; Lu, E.S.; Kwan, K.; Wong, K.K.; et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat. Commun. 2016, 7, 11150. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Ruan, H.; Zhang, X.; Xu, X.; Zhu, Y.; Peng, H.; Zhang, X.; Kong, F.; Guan, M. Long noncoding RNA CCAL transferred from fibroblasts by exosomes promotes chemoresistance of colorectal cancer cells. Int. J. Cancer 2020, 146, 1700–1716. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Ding, L.; Zhang, D.; Shi, G.; Xu, Q.; Shen, S.; Wang, Y.; Wang, T.; Hou, Y. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics 2018, 8, 3932–3948. [Google Scholar] [CrossRef] [PubMed]
- Dudley, A.C. Tumor endothelial cells. Cold Spring Harb. Perspect. Med. 2012, 2, a006536. [Google Scholar] [CrossRef] [PubMed]
- Baluk, P.; Hashizume, H.; McDonald, D.M. Cellular abnormalities of blood vessels as targets in cancer. Curr. Opin. Genet. Dev. 2005, 15, 102–111. [Google Scholar] [CrossRef]
- Yadav, A.; Kumar, B.; Yu, J.G.; Old, M.; Teknos, T.N.; Kumar, P. Tumor-associated endothelial cells promote tumor metastasis by chaperoning circulating tumor cells and protecting them from anoikis. PLoS ONE 2015, 10, e0141602. [Google Scholar] [CrossRef]
- Butler, J.M.; Kobayashi, H.; Rafii, S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat. Rev. Cancer 2010, 10, 138–146. [Google Scholar] [CrossRef]
- Maishi, N.; Hida, K. Tumor endothelial cells accelerate tumor metastasis. Cancer Sci. 2017, 108, 1921–1926. [Google Scholar] [CrossRef]
- Taylor, S.M.; Nevis, K.R.; Park, H.L.; Rogers, G.C.; Rogers, S.L.; Cook, J.G.; Bautch, V.L. Angiogenic factor signaling regulates centrosome duplication in endothelial cells of developing blood vessels. Blood 2010, 116, 3108–3117. [Google Scholar] [CrossRef]
- van Zijl, F.; Krupitza, G.; Mikulits, W. Initial steps of metastasis: Cell invasion and endothelial transmigration. Mutat. Res. 2011, 728, 23–34. [Google Scholar] [CrossRef]
- Bern, M.M. Extracellular vesicles: How they interact with endothelium, potentially contributing to metastatic cancer cell implants. Clin. Transl. Med. 2017, 6, 33. [Google Scholar] [CrossRef]
- Matsuda, K.; Ohga, N.; Hida, Y.; Muraki, C.; Tsuchiya, K.; Kurosu, T.; Akino, T.; Shih, S.C.; Totsuka, Y.; Klagsbrun, M.; et al. Isolated tumor endothelial cells maintain specific character during long-term culture. Biochem. Biophys. Res. Commun. 2010, 394, 947–954. [Google Scholar] [CrossRef]
- Adya, R.; Tan, B.K.; Punn, A.; Chen, J.; Randeva, H.S. Visfatin induces human endothelial VEGF and MMP-2/9 production via MAPK and PI3K/Akt signalling pathways: Novel insights into visfatin-induced angiogenesis. Cardiovasc. Res. 2008, 78, 356–365. [Google Scholar] [CrossRef]
- Yoshida, S.; Kawai, H.; Eguchi, T.; Sukegawa, S.; Oo, M.W.; Anqi, C.; Takabatake, K.; Nakano, K.; Okamoto, K.; Nagatsuka, H. Tumor angiogenic inhibition triggered necrosis (TAITN) in oral cancer. Cells 2019, 8, 761. [Google Scholar] [CrossRef]
- Ludwig, N.; Yerneni, S.S.; Razzo, B.M.; Whiteside, T.L. Exosomes from HNSCC Promote Angiogenesis through Reprogramming of Endothelial Cells. Mol. Cancer Res. 2018, 16, 1798–1808. [Google Scholar] [CrossRef]
- Cao, Z.; Ding, B.S.; Guo, P.; Lee, S.B.; Butler, J.M.; Casey, S.C.; Simons, M.; Tam, W.; Felsher, D.W.; Shido, K.; et al. Angiocrine factors deployed by tumor vascular niche induce B cell lymphoma invasiveness and chemoresistance. Cancer Cell 2014, 25, 350–365. [Google Scholar] [CrossRef]
- Amin, D.N.; Hida, K.; Bielenberg, D.R.; Klagsbrun, M. Tumor endothelial cells express epidermal growth factor receptor (EGFR) but not ErbB3 and are responsive to EGF and to EGFR kinase inhibitors. Cancer Res. 2006, 66, 2173–2180. [Google Scholar] [CrossRef]
- Tsuchiya, K.; Hida, K.; Hida, Y.; Muraki, C.; Ohga, N.; Akino, T.; Kondo, T.; Miseki, T.; Nakagawa, K.; Shindoh, M.; et al. Adrenomedullin antagonist suppresses tumor formation in renal cell carcinoma through inhibitory effects on tumor endothelial cells and endothelial progenitor mobilization. Int. J. Oncol. 2010, 36, 1379–1386. [Google Scholar]
- Bussolati, B.; Assenzio, B.; Deregibus, M.C.; Camussi, G. The proangiogenic phenotype of human tumor-derived endothelial cells depends on thrombospondin-1 downregulation via phosphatidylinositol 3-kinase/Akt pathway. J. Mol. Med. 2006, 84, 852–863. [Google Scholar] [CrossRef]
- Lopatina, T.; Grange, C.; Fonsato, V.; Tapparo, M.; Brossa, A.; Fallo, S.; Pitino, A.; Herrera-Sanchez, M.B.; Kholia, S.; Camussi, G.; et al. Extracellular vesicles from human liver stem cells inhibit tumor angiogenesis. Int. J. Cancer 2019, 144, 322–333. [Google Scholar] [CrossRef]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.F.; Chin, A.R.; et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Umezu, T.; Ohyashiki, K.; Kuroda, M.; Ohyashiki, J.H. Leukemia cell to endothelial cell communication via exosomal miRNAs. Oncogene 2013, 32, 2747–2755. [Google Scholar] [CrossRef] [PubMed]
- Conigliaro, A.; Costa, V.; lo Dico, A.; Saieva, L.; Buccheri, S.; Dieli, F.; Manno, M.; Raccosta, S.; Mancone, C.; Tripodi, M.; et al. CD90+ liver cancer cells modulate endothelial cell phenotype through the release of exosomes containing H19 lncRNA. Mol. Cancer 2015, 14, 155. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Yan, Y.; Hu, M.; Chen, X.; Wang, Y.; Dai, Y.; Wu, D.; Wang, Y.; Zhuang, Z.; Xia, H. Increased level of H19 long noncoding RNA promotes invasion, angiogenesis, and stemness of glioblastoma cells. J. Neurosurg. 2016, 124, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Dai, X.; Yang, T.; Zhang, N.; Liu, Z.; Jiang, Y. Low Long Noncoding RNA Growth Arrest-Specific Transcript 5 Expression in the Exosomes of Lung Cancer Cells Promotes Tumor Angiogenesis. J. Oncol. 2019, 2019, 2476175. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.; Lima, L.G.; Chai, E.P.Z.; Muller, A.; Lobb, R.J.; Krumeich, S.; Wen, S.W.; Wiegmans, A.P.; Möller, A. Breast cancer-derived exosomes alter macrophage polarization via gp130/STAT3 signaling. Front. Immunol. 2018, 9, 871. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Bromberg, J.; Wang, T.C. Inflammation and Cancer: IL-6 and STAT3 Complete the Link. Cancer Cell 2009, 15, 79–80. [Google Scholar] [CrossRef]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.T.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 Blocks CD8+ T Cell-Dependent Responses to Chemotherapy by Suppressing IL-12 Expression in Intratumoral Dendritic Cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef]
- Roca, H.; Varcos, Z.S.; Sud, S.; Craig, M.J.; Pienta, K.J. CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. J. Biol. Chem. 2009, 284, 34342–34354. [Google Scholar] [CrossRef]
- Lu, T.; Zhang, Z.; Zhang, J.; Pan, X.; Zhu, X.; Wang, X.; Li, Z.; Ruan, M.; Li, H.; Chen, W.; et al. CD73 in small extracellular vesicles derived from HNSCC defines tumour-associated immunosuppression mediated by macrophages in the microenvironment. J. Extracell. Vesicles 2022, 11, e12218. [Google Scholar] [CrossRef]
- Borst, J.; Ahrends, T.; Bąbała, N.; Melief, C.J.M.; Kastenmüller, W. CD4+ T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef]
- Bedoui, S.; Heath, W.R.; Mueller, S.N. CD4+ T-cell help amplifies innate signals for primary CD8+ T-cell immunity. Immunol. Rev. 2016, 272, 52–64. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, H.; Zhao, J. The role of CD4 T cell help for CD8 CTL activation. Biochem. Biophys. Res. Commun. 2009, 384, 405–408. [Google Scholar] [CrossRef]
- Yang, C.; Kim, S.H.; Bianco, N.R.; Robbins, P.D. Tumor-derived exosomes confer antigen-specific immunosuppression in a murine delayed-type hypersensitivity model. PLoS ONE 2011, 6, e22517. [Google Scholar] [CrossRef]
- Klibi, J.; Niki, T.; Riedel, A.; Pioche-Durieu, C.; Souquere, S.; Rubinstein, E.; Moulec, S.L.E.; Guigay, J.; Hirashima, M.; Guemira, F.; et al. Blood diffusion and Th1-suppressive effects of galectin-9-containing exosomes released by Epstein-Barr virus-infected nasopharyngeal carcinoma cells. Blood 2009, 113, 1957–1966. [Google Scholar] [CrossRef]
- Zhang, H.-G.; Liu, C.; Su, K.; Yu, S.; Zhang, L.; Zhang, S.; Wang, J.; Cao, X.; Grizzle, W.; Kimberly, R.P. A membrane form of TNF-alpha presented by exosomes delays T cell activation-induced cell death. J. Immunol. 2006, 176, 7385–7393. [Google Scholar] [CrossRef]
- Papa, A.; Chen, M.; Pandolfi, P.P. Pills of PTEN? in and out for tumor suppression. Cell Res. 2013, 23, 1155–1156. [Google Scholar] [CrossRef]
- Putz, U.; Howitt, J.; Doan, A.; Goh, C.P.; Low, L.H.; Silke, J.; Tan, S.S. The tumor suppressor PTEN is exported in exosomes and has phosphatase activity in recipient cells. Sci. Signal 2012, 5, ra70. [Google Scholar] [CrossRef]
- Czystowska, M.; Han, J.; Szczepanski, M.J.; Szajnik, M.; Quadrini, K.; Brandwein, H.; Hadden, J.W.; Signorelli, K.; Whiteside, T.L. IRX-2, a novel immunotherapeutic, protects human T cells from tumor-induced cell death. Cell Death Differ. 2009, 16, 708–718. [Google Scholar] [CrossRef]
- Liu, Z.M.; Wang, Y.B.; Yuan, X.H. Exosomes from murine-derived GL26 cells promote glioblastoma tumor growth by reducing number and function of CD8+T cells. Asian Pac. J. Cancer Prev. 2013, 14, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Mitchell, J.P.; Court, J.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes selectively impair lymphocyte responses to interleukin-2. Cancer Res. 2007, 67, 7458–7466. [Google Scholar] [CrossRef] [PubMed]
- Fleming, V.; Hu, X.; Weber, R.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front. Immunol. 2018, 9, 398. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Mitchell, J.P.; Court, J.; Linnane, S.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes down-modulate NKG2D expression. J. Immunol. 2008, 180, 7249–7258. [Google Scholar] [CrossRef] [PubMed]
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Whiteside, T.L.; Boyiadzis, M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-β1. Haematologica 2011, 96, 1302–1309. [Google Scholar] [CrossRef]
- Pyfferoen, L.; Brabants, E.; Everaert, C.; de Cabooter, N.; Heyns, K.; Deswarte, K.; Vanheerswynghels, M.; de Prijck, S.; Waegemans, G.; Dullaers, M.; et al. The transcriptome of lung tumor-infiltrating dendritic cells reveals a tumor-supporting phenotype and a microRNA signature with negative impact on clinical outcome. Oncoimmunology 2017, 6, e1253655. [Google Scholar] [CrossRef]
- Keirsse, J.; van Damme, H.; van Ginderachter, J.A.; Laoui, D. Exploiting tumor-associated dendritic cell heterogeneity for novel cancer therapies. J. Leukoc. Biol. 2017, 102, 317–324. [Google Scholar] [CrossRef]
- Motta, J.M.; Rumjanek, V.M. Sensitivity of dendritic cells to microenvironment signals. J. Immunol. Res. 2016, 2016, 4753607. [Google Scholar] [CrossRef]
- Skobe, M.; Hawighorst, T.; Jackson, D.G.; Prevo, R.; Janes, L.; Velasco, P.; Riccardi, L.; Alitalo, K.; Claffey, K.; Detmar, M. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat. Med. 2001, 7, 192–198. [Google Scholar] [CrossRef]
- Stacker, S.A.; Williams, S.P.; Karnezis, T.; Shayan, R.; Fox, S.B.; Achen, M.G. Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat. Rev. Cancer 2014, 14, 159–172. [Google Scholar] [CrossRef]
- Zheng, W.; Aspelund, A.; Alitalo, K. Lymphangiogenic factors, mechanisms, and applications. J. Clin. Investig. 2014, 124, 878–887. [Google Scholar] [CrossRef]
- Steinman, R.M.; Pack, M.; Inaba, K. Dendritic cells in the T-cell areas of lymphoid organs. Immunol. Rev. 1997, 156, 25–37. [Google Scholar] [CrossRef]
- Kawada, K.; Hosogi, H.; Sonoshita, M.; Sakashita, H.; Manabe, T.; Shimahara, Y.; Sakai, Y.; Takabayashi, A.; Oshima, M.; Taketo, M.M. Chemokine receptor CXCR3 promotes colon cancer metastasis to lymph nodes. Oncogene 2007, 26, 4679–4688. [Google Scholar] [CrossRef]
- Kawada, K.; Sonoshita, M.; Sakashita, H.; Takabayashi, A.; Yamaoka, Y.; Manabe, T.; Inaba, K.; Minato, N.; Oshima, M.; Taketo, M.M. Pivotal role of CXCR3 in melanoma cell metastasis to lymph nodes. Cancer Res. 2004, 64, 4010–4017. [Google Scholar] [CrossRef]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef]
- Uhrin, P.; Zaujec, J.; Breuss, J.M.; Olcaydu, D.; Chrenek, P.; Stockinger, H.; Fuertbauer, E.; Moser, M.; Haiko, P.; Fässler, R.; et al. Novel function for blood platelets and podoplanin in developmental separation of blood and lymphatic circulation. Blood 2010, 115, 3997–4005. [Google Scholar] [CrossRef]
- Ugorski, M.; Dziegiel, P.; Suchanski, J. Podoplanin—A small glycoprotein with many faces. Am. J. Cancer Res. 2016, 6, 370. [Google Scholar]
- Cueni, L.N.; Hegyi, I.; Shin, J.W.; Albinger-Hegyi, A.; Gruber, S.; Kunstfeld, R.; Moch, H.; Detmar, M. Tumor lymphangiogenesis and metastasis to lymph nodes induced by cancer cell expression of podoplanin. Am. J. Pathol. 2010, 177, 1004–1016. [Google Scholar] [CrossRef]
- Suzuki, H.; Onimaru, M.; Koga, T.; Takeshita, M.; Yano, T.; Maehara, Y.; Nakamura, S.; Sueishi, K. High podoplanin expression in cancer cells predicts lower incidence of nodal metastasis in patients with lung squamous cell carcinoma. Pathol. Res. Pract. 2011, 207, 111–115. [Google Scholar] [CrossRef]
- Carrasco-Ramírez, P.; Greening, D.W.; Andrés, G.; Gopal, S.K.; Martín-Villar, E.; Renart, J.; Simpson, R.J.; Quintanilla, M. Podoplanin is a component of extracellular vesicles that reprograms cell-derived exosomal proteins and modulates lymphatic vessel formation. Oncotarget 2016, 7, 16070–16089. [Google Scholar] [CrossRef]
- Bieniasz-Krzywiec, P.; Martín-Pérez, R.; Ehling, M.; García-Caballero, M.; Pinioti, S.; Pretto, S.; Kroes, R.; Aldeni, C.; di Matteo, M.; Prenen, H.; et al. Podoplanin-Expressing Macrophages Promote Lymphangiogenesis and Lymphoinvasion in Breast Cancer. Cell Metab. 2019, 30, 917–936.e10. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Liou, G.G.; Liu, S.H.; Chang, J.S.; Hsiao, J.R.; Yen, Y.C.; Chen, Y.L.; Wu, W.L.; Chang, J.Y.; Chen, Y.W. Laminin γ2-enriched extracellular vesicles of oral squamous cell carcinoma cells enhance in vitro lymphangiogenesis via integrin α3-dependent uptake by lymphatic endothelial cells. Int. J. Cancer 2019, 144, 2795–2810. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Lu, Y.; Xu, Y.; Wang, J.; Zhang, C.; Du, Y.; Wang, L.; Li, L.; Wang, B.; Shen, J.; et al. Horizontal transfer of exosomal CXCR4 promotes murine hepatocarcinoma cell migration, invasion and lymphangiogenesis. Gene 2018, 676, 101–109. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Zhong, G.; Jiang, N.; Wang, B.; Fan, X.; Chen, C.; Chen, X.; Huang, J.; Lin, T. Long noncoding RNA BLACAT2 promotes bladder cancer–associated lymphangiogenesis and lymphatic metastasis. J. Clin. Investig. 2018, 128, 861. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Ou, C.; Ren, W.; Xie, X.; Li, X.; Li, G. Downregulation of long non-coding RNA ANRIL suppresses lymphangiogenesis and lymphatic metastasis in colorectal cancer. Oncotarget 2016, 7, 47536–47555. [Google Scholar] [CrossRef]
- Chen, C.; He, W.; Huang, J.; Wang, B.; Li, H.; Cai, Q.; Su, F.; Bi, J.; Liu, H.; Zhang, B.; et al. LNMAT1 promotes lymphatic metastasis of bladder cancer via CCL2 dependent macrophage recruitment. Nat. Commun. 2018, 9, 3826. [Google Scholar] [CrossRef]
- Jung, T.; Castellana, D.; Klingbeil, P.; Hernández, I.C.; Vitacolonna, M.; Orlicky, D.J.; Roffler, S.R.; Brodt, P.; Zöller, M. CD44v6 dependence of premetastatic niche preparation by exosomes. Neoplasia 2009, 11, 1093–1105. [Google Scholar] [CrossRef]
- Wang, J.; Man, Q.-W.; Fu, Q.-Y.; Zhong, N.-N.; Wang, H.-Q.; Li, S.-R.; Gao, X.; Lin, H.; Su, F.-C.; Bu, L.-L.; et al. Preliminary Extracellular Vesicle Profiling in Drainage Fluid After Neck Dissection in OSCC. J. Dent. Res. 2022, 002203452211300. [Google Scholar] [CrossRef]
- He, F.; Zhong, X.; Lin, Z.; Lin, J.; Qiu, M.; Li, X.; Hu, Z. Plasma exo-hsa_circRNA_0056616: A potential biomarker for lymph node metastasis in lung adenocarcinoma. J. Cancer 2020, 11, 4037–4046. [Google Scholar] [CrossRef]
- Jiang, K.; Li, G.; Chen, W.; Song, L.; Wei, T.; Li, Z.; Gong, R.; Lei, J.; Shi, H.; Zhu, J. Plasma Exosomal miR-146b-5p and miR-222-3p are Potential Biomarkers for Lymph Node Metastasis in Papillary Thyroid Carcinomas. Onco Targets Ther. 2020, 13, 1311–1319. [Google Scholar] [CrossRef]
- Li, L.; Li, C.; Wang, S.; Wang, Z.; Jiang, J.; Wang, W.; Li, X.; Chen, J.; Liu, K.; Li, C.; et al. Exosomes Derived from Hypoxic Oral Squamous Cell Carcinoma Cells Deliver miR-21 to Normoxic Cells to Elicit a Prometastatic Phenotype. Cancer Res. 2016, 76, 1770–1780. [Google Scholar] [CrossRef]
- Liu, Y.T.; Sun, Z.J. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics 2021, 11, 5365–5386. [Google Scholar] [CrossRef]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Yin, Z.; Yu, M.; Ma, T.; Zhang, C.; Huang, S.; Karimzadeh, M.R.; Momtazi-Borojeni, A.A.; Chen, S. Mechanisms underlying low-clinical responses to PD-1/PD-L1 blocking antibodies in immunotherapy of cancer: A key role of exosomal PD-L1. J. Immunother. Cancer 2021, 9, e001698. [Google Scholar] [CrossRef]
- Cordonnier, M.; Nardin, C.; Chanteloup, G.; Derangere, V.; Algros, M.P.; Arnould, L.; Garrido, C.; Aubin, F.; Gobbo, J. Tracking the evolution of circulating exosomal-PD-L1 to monitor melanoma patients. J. Extracell. Vesicles 2020, 9, 1710899. [Google Scholar] [CrossRef]
- Xin, L.; Zhou, L.-Q.; Liu, C.; Zeng, F.; Yuan, Y.-W.; Zhou, Q.; Li, S.-H.; Wu, Y.; Wang, J.-L.; Wu, D.-Z.; et al. Transfer of LncRNA CRNDE in TAM-derived exosomes is linked with cisplatin resistance in gastric cancer. EMBO Rep. 2021, 22, e52124. [Google Scholar] [CrossRef]
- Ou, B.; Liu, Y.; Gao, Z.; Xu, J.; Yan, Y.; Li, Y.; Zhang, J. Senescent neutrophils-derived exosomal piRNA-17560 promotes chemoresistance and EMT of breast cancer via FTO-mediated m6A demethylation. Cell Death Dis. 2022, 13, 905. [Google Scholar] [CrossRef]
- Shi, L.; Zhu, W.; Huang, Y.; Zhuo, L.; Wang, S.; Chen, S.; Zhang, B.; Ke, B. Cancer-associated fibroblast-derived exosomal microRNA-20a suppresses the PTEN/PI3K-AKT pathway to promote the progression and chemoresistance of non-small cell lung cancer. Clin. Transl. Med. 2022, 12, e989. [Google Scholar] [CrossRef]
- Asare-Werehene, M.; Nakka, K.; Reunov, A.; Chiu, C.T.; Lee, W.T.; Abedini, M.R.; Wang, P.W.; Shieh, D.B.; Dilworth, F.J.; Carmona, E.; et al. The exosome-mediated autocrine and paracrine actions of plasma gelsolin in ovarian cancer chemoresistance. Oncogene 2020, 39, 1600–1616. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Qin, Z.; Cai, S.; Yu, L.; Hu, H.; Zeng, S. The role of non-coding RNAs in ABC transporters regulation and their clinical implications of multidrug resistance in cancer. Expert Opin. Drug Metab. Toxicol. 2021, 17, 291–306. [Google Scholar] [CrossRef]
- Codrich, M.; Degrassi, M.; Malfatti, M.C.; Antoniali, G.; Gorassini, A.; Ayyildiz, D.; de Marco, R.; Verardo, G.; Tell, G. APE1 interacts with the nuclear exosome complex protein MTR4 and is involved in cisplatin- and 5-fluorouracil-induced RNA damage response. FEBS J. 2022. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Category | Name | EV Class | Size | Markers | Biogenesis |
|---|---|---|---|---|---|
| Exosome | Classical exosome | Small EV a | 40–150 nm | CD63, CD9, CD81 | Multivesicular endosome |
| Non-classical exosome | Small EV | 40–150 nm | CD63/CD9/CD81-negative | Multivesicular endosome | |
| Microvesicle | Classical microvesicle | Large EV b | ~150–1000 nm | Annexin A1, ARF6 | Plasma membrane shedding |
| Large oncosome | Large EV | 1–10 µm | Annexin A1, ARF6 | Plasma membrane shedding | |
| ARMM | Small EV | ~40–100 nm | ARRDC1, TSG101 | Plasma membrane shedding | |
| Apoptotic EV | Apoptotic body | Large EV | 1–5 µm | Annexin V, PS | Apoptosis |
| Apoptotic vesicle | Small to Large EV | ~100–1000 nm | Annexin V, PS | Apoptosis | |
| Autophagic EV | Autophagic EV | Small to Large EV | 40–1000 nm? | LC3B-PE, p62 dsDNA/Histones | Autophagosome-endosome fusion (Amphisome) |
| Stressed EV (Stressome) | Stressed EV Damaged EV | Small to Large EV | 40–1000 nm? | HSP90, HSPs | Plasma membrane shedding, autophagy |
| Matrix vesicles | Matrix vesicles | Small to Large EV | 40–1000 nm? | Fibronectin, Proteoglycans | Matrix binding and release |
| Exomere | Nano-particle | Non-EV | ~35–50 nm | HSP90, HSPs | Stress? |
| Non-vesicular particles | Nano-particle | Non-EV | ? (vaults: ~70 nm) | Fibronectin, dsDNA/Histones, MVP, HSPs | Unknown, Cell death |
| Scene | Events | Ref. |
|---|---|---|
| 1. Around producer cells | a. CAFs and cancer cells are major producers of matrix proteins. | [51,52] |
| b. EVs are embedded within ECM and accumulated around producer cells. | [70] | |
| c. EVs and ECM mutually promote their accumulation around cells. | [70] | |
| d. sEVs act similar to car wheels to help cells migrate on rails of fibronectin. | [71] | |
| e. MMPs cleave matrix proteins to release matrix vesicles, growth factors, and chemokines. | [72] | |
| f. MMPs destroy ECM to increase the accessibility of proteins, EVs, and drugs to target cells. | [70] | |
| 2. In bodily fluids (or tissue culture supernatant) | a. EVs are often coated with matrix (fibronectin, proteoglycan, agrin, tenascin, hyaluronan). | [64,73] |
| b. EV surface MMPs promote the dissemination of EVs. | [7] | |
| 3. At niches (at local and distant tissues) | a. EV surface integrins bind to ECM, leading to niche formation. | [74] |
| b. EV surface matrices bind to ECM on the surface of recipient cells. | [75] | |
| c. EV surface growth factors, cytokines and chemokines determine uptake and bio distribution. | [76] | |
| d. MMPs loaded in EVs are transferred into recipient cell nuclei and transactivate the CCN2 gene, encoding a matricellular protein. | [7] | |
| e. EV surface MMPs promote the penetration of EVs into target tissues. | [8] |
| Recipient Cells | Influences | Model | Refs. |
|---|---|---|---|
| MSCs | ↑ Differentiation to proangiogenic myofibroblasts ↑ Differentiation to pro-invasive myofibroblasts | In vitro | [125,126] |
| Fibroblasts (CAF) | ↑ Fibroblast differentiation into CAFs ↑ Create premetastatic niche | In vitro In vivo | [127,128] |
| Epithelial cells | ↑ Initiate carcinogenic EMT | In vitro | [6,28] |
| Blood endothelial cells (BEC) | ↑ Reprogram normal endothelial cells to TECs ↑ Promote tumor angiogenesis ↑ Destruct endothelial barrier ↑ Extravasation of tumor cells and EVs ↑ Intravasation and metastasis of tumor cells and EVs ↑ Promote premetastatic niche formation | In vivo, In vitro | [129,130] |
| Monocytes Macrophages (TAM) | ↑ Induce immunosuppressive M2 polarization ↑ Expression of IL-10, CXCR4, and CCL2 ↑ Induce chemoresistance ↑ Initiate premetastatic niche formation ↓ Suppress NLRP3 inflammasome activity | In vitro, Ex vivo | [131,132] |
| Neutrophils (TAN) | ↑ Induce N2 polarization ↑ Promote cancer cell migration | In vitro, In vivo | [133] |
| Dendritic cells (DC) | ↓ Block myeloid precursor cells differentiation to DCs ↓ Induce DC apoptosis ↓ Decrease CD4+ IFN-γ+ Th1 differentiation ↑ Increase the rate of Treg | In vitro | [134] |
| Lymphantic endothelial cells (LEC) | ↑ Lymphatic remodeling ↑ Lymphangiogenesis ↑ Immunosuppression ↑ Premetastatic niche formation ↑ Lymph node metastasis | In vitro In vivo | [135,136] |
| Killer T cells | ↓ Inhibit proliferation and differentiation ↓ Induce apoptosis | Patient samples In vitro | [137] |
| Treg cells (Immunosuppressive) | ↑ Promote the differentiation and proliferation | In vitro | [138] |
| MDSCs (Immunosuppressive) | ↑ Promote MDSC differentiation ↑ Expression of Cox2, IL-6, VEGF, and arginase-1 ↓ Decrease antitumor immunotherapy efficacy | In vivo | [139,140] |
| Natural Killer (NK) cells | ↓ Downregulate NKG2D expression | In vitro | [141,142] |
| Donor EVs | Recipient Cells | Functions | Model | Ref. |
|---|---|---|---|---|
| CAF-EVs | Cancer cells | ↑ Induce chemoresistance ↑ Increase survival and proliferation ↑ Activate EMT ↑ Promote metastasis ↓ Suppress cell death (ferroptosis) | In vitro | [143,144,145,146,147] |
| Immune cells | Additionally called metastasis-associated fibroblasts (MAF) ↑ Upregulation of IL-33 instigating type 2 immunity ↑ Recruitment of eosinophils, neutrophils, and inflammatory monocytes to lung metastasis | In vivo | [148] | |
| MSC-EVs | Cancer cells | ↑ Activate EMT ↑ Evade apoptosis ↑ Increase cancer stemness and dormancy | In vitro | [149] |
| Immune cells | ↑ Increase immunotherapy resistance | In vitro | [149] | |
| Cancer cells | ↓ Vehicles for delivery in cancer therapy | In vitro | [150] | |
| TAM-EVs | Cancer cells | ↑ M2 TAM-EVs induce chemoresistance ↓ M2 TAM-EVs Inhibit immune surveillance ↓ Reduce cancer cells viability | In vitro | [151,152] |
| TAN-EVs | Cancer cells | ↑ Induce chemoresistance ↑ Activate EMT | In vitro, In vivo | [153] |
| (Engineered) Immunocyte-EVs | Cancer cells | Chemoimmunotherapeutic nanocarrier ↓ Reduce cancer cells viability | In vitro In vivo | [153,154] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheta, M.; Taha, E.A.; Lu, Y.; Eguchi, T. Extracellular Vesicles: New Classification and Tumor Immunosuppression. Biology 2023, 12, 110. https://doi.org/10.3390/biology12010110
Sheta M, Taha EA, Lu Y, Eguchi T. Extracellular Vesicles: New Classification and Tumor Immunosuppression. Biology. 2023; 12(1):110. https://doi.org/10.3390/biology12010110
Chicago/Turabian StyleSheta, Mona, Eman A. Taha, Yanyin Lu, and Takanori Eguchi. 2023. "Extracellular Vesicles: New Classification and Tumor Immunosuppression" Biology 12, no. 1: 110. https://doi.org/10.3390/biology12010110
APA StyleSheta, M., Taha, E. A., Lu, Y., & Eguchi, T. (2023). Extracellular Vesicles: New Classification and Tumor Immunosuppression. Biology, 12(1), 110. https://doi.org/10.3390/biology12010110