
Assessment of MicroRNAs Associated with Tumor Purity by Random Forest Regression


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Dataset Description for Model Construction
2.2. Random Forest Regression and Optimizing Parameters
2.3. Feature Importance of miRNAs with Random Forest Regression
2.4. Target Gene Prediction for miRNA with High Feature Importance
2.5. Enrichment Analysis of Target Genes
2.6. Validation with Independent Datasets
3. Results
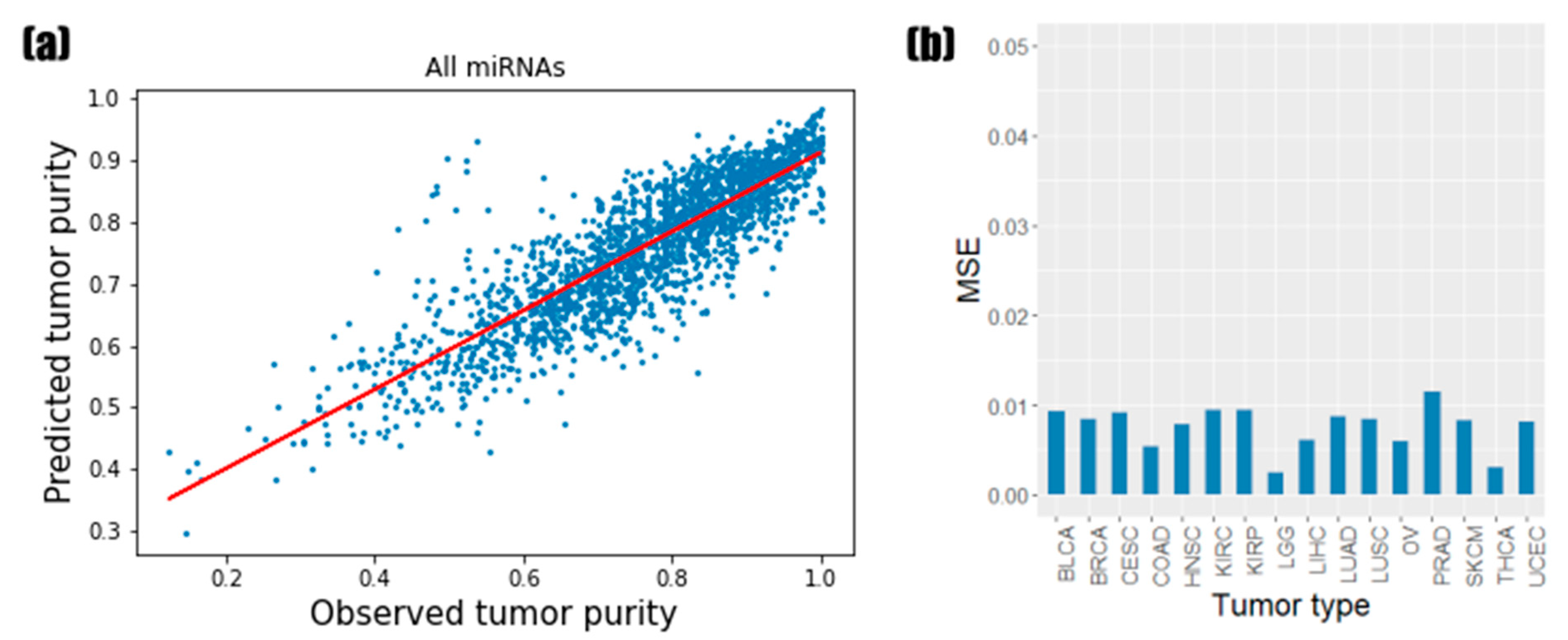
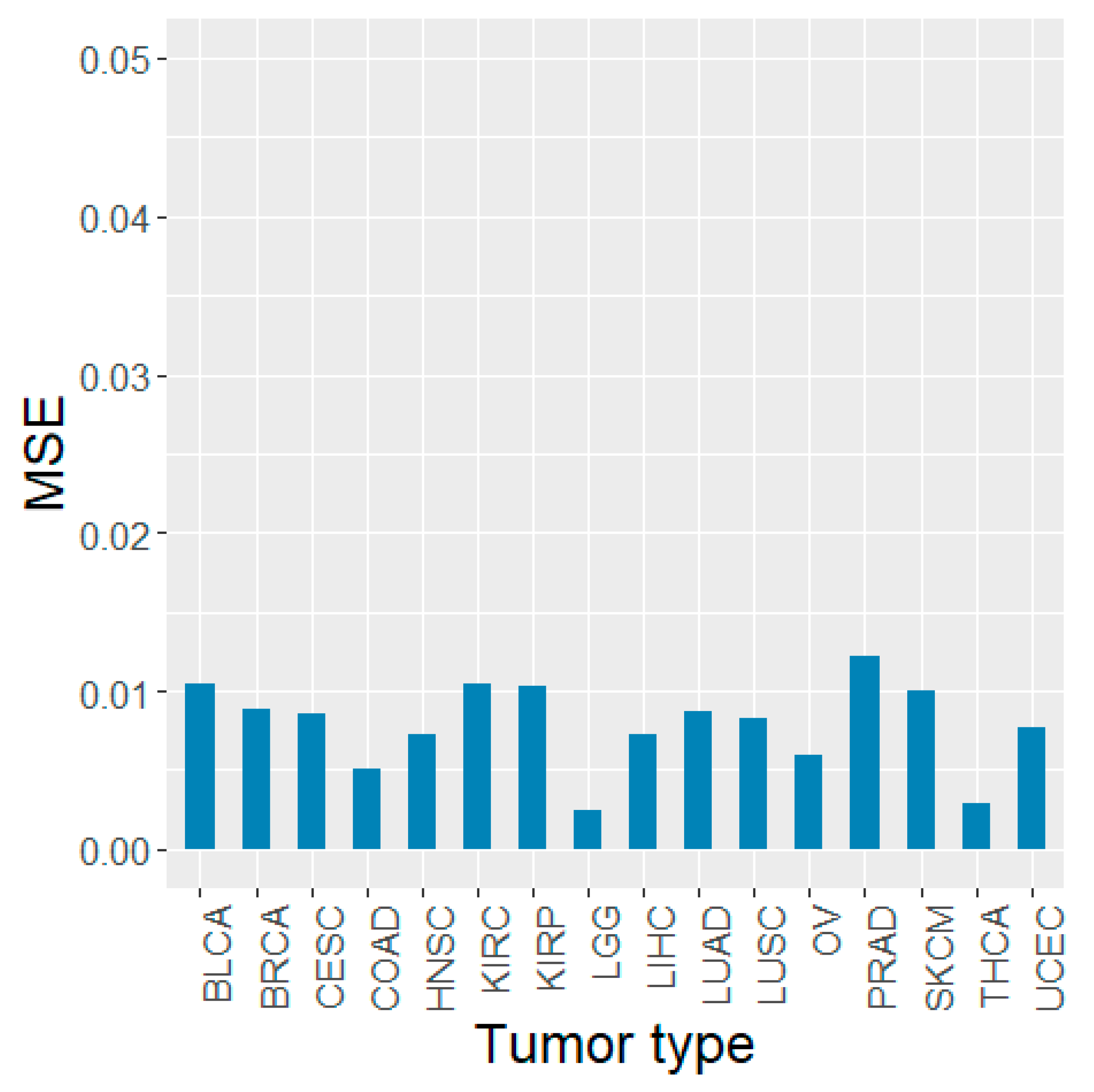
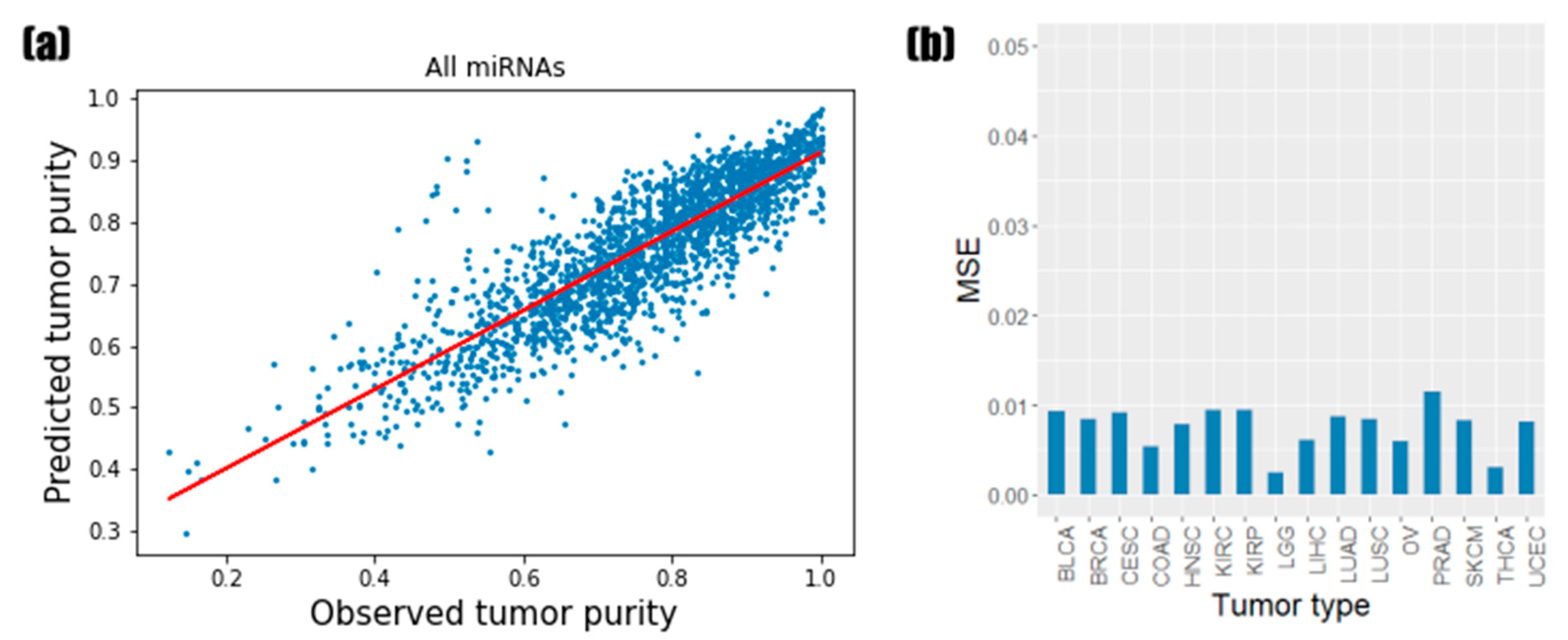
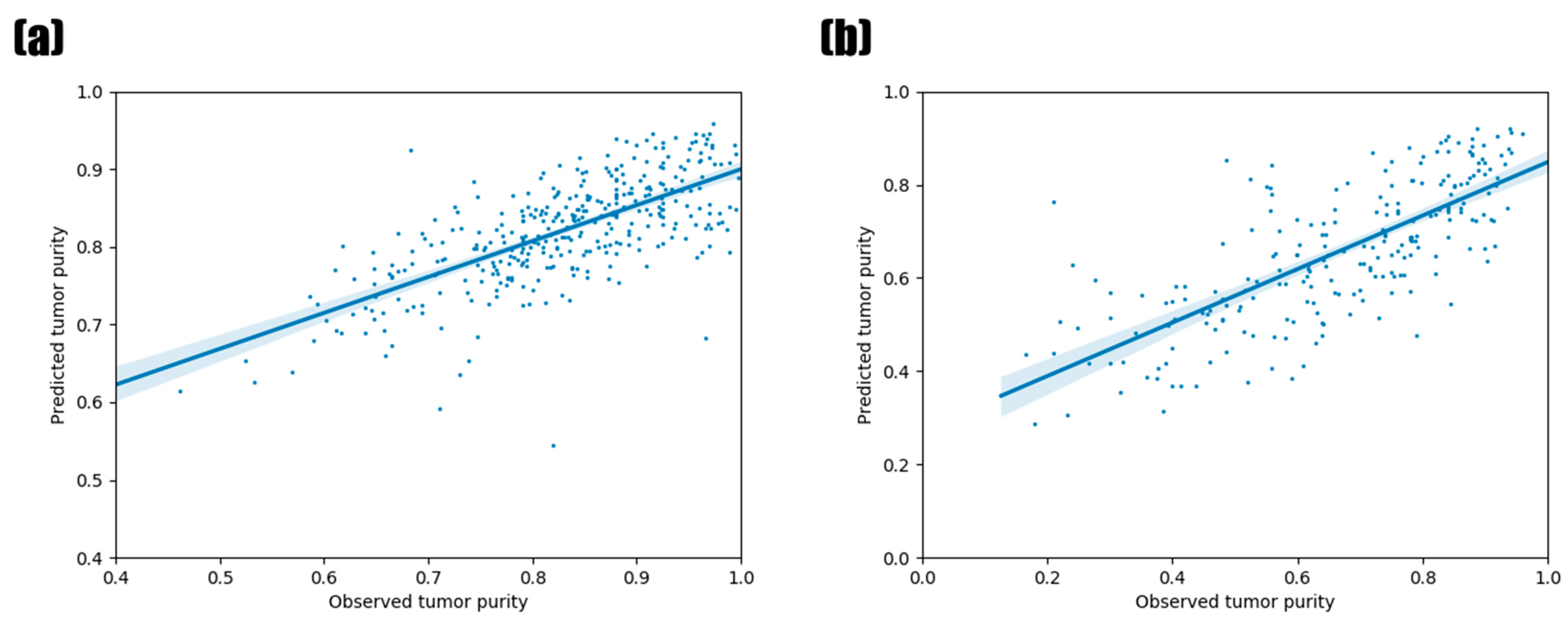
3.1. Predictive Performance of Random Forest Regression Model
3.2. Identification of the Informative miRNAs with Feature Importance
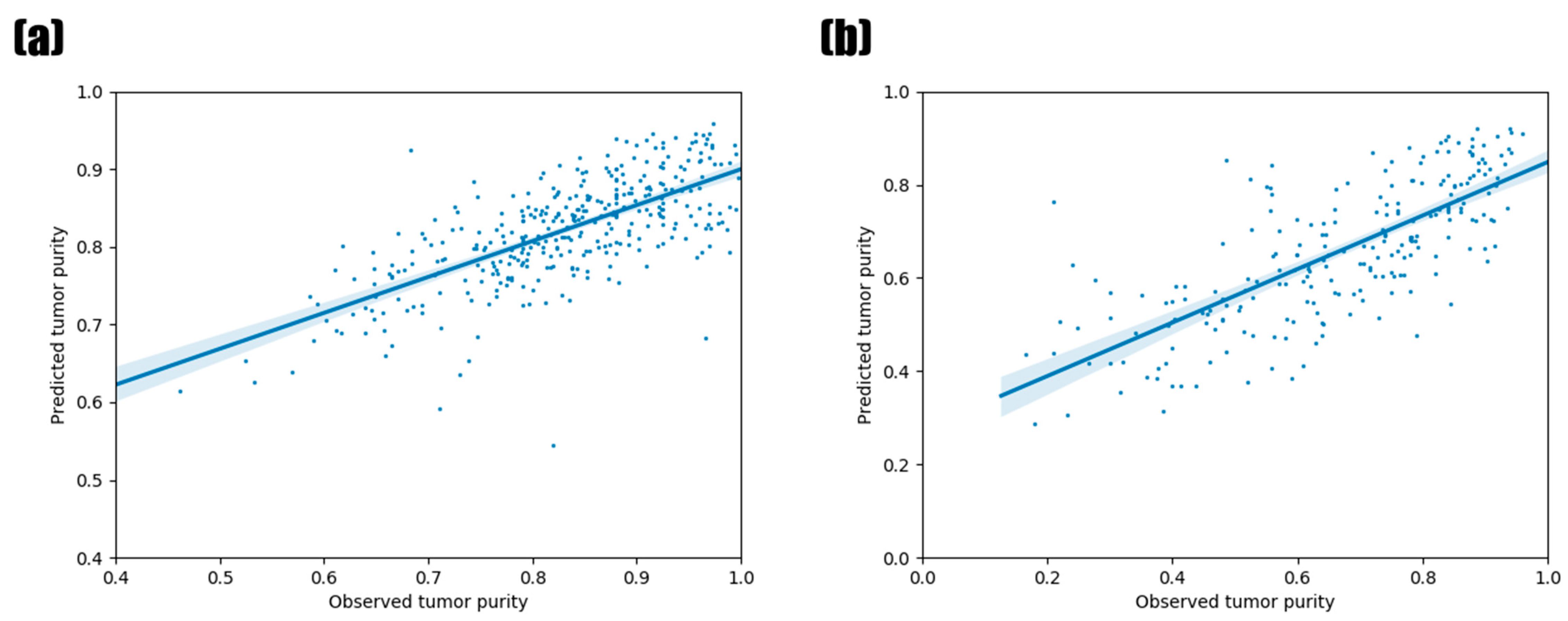
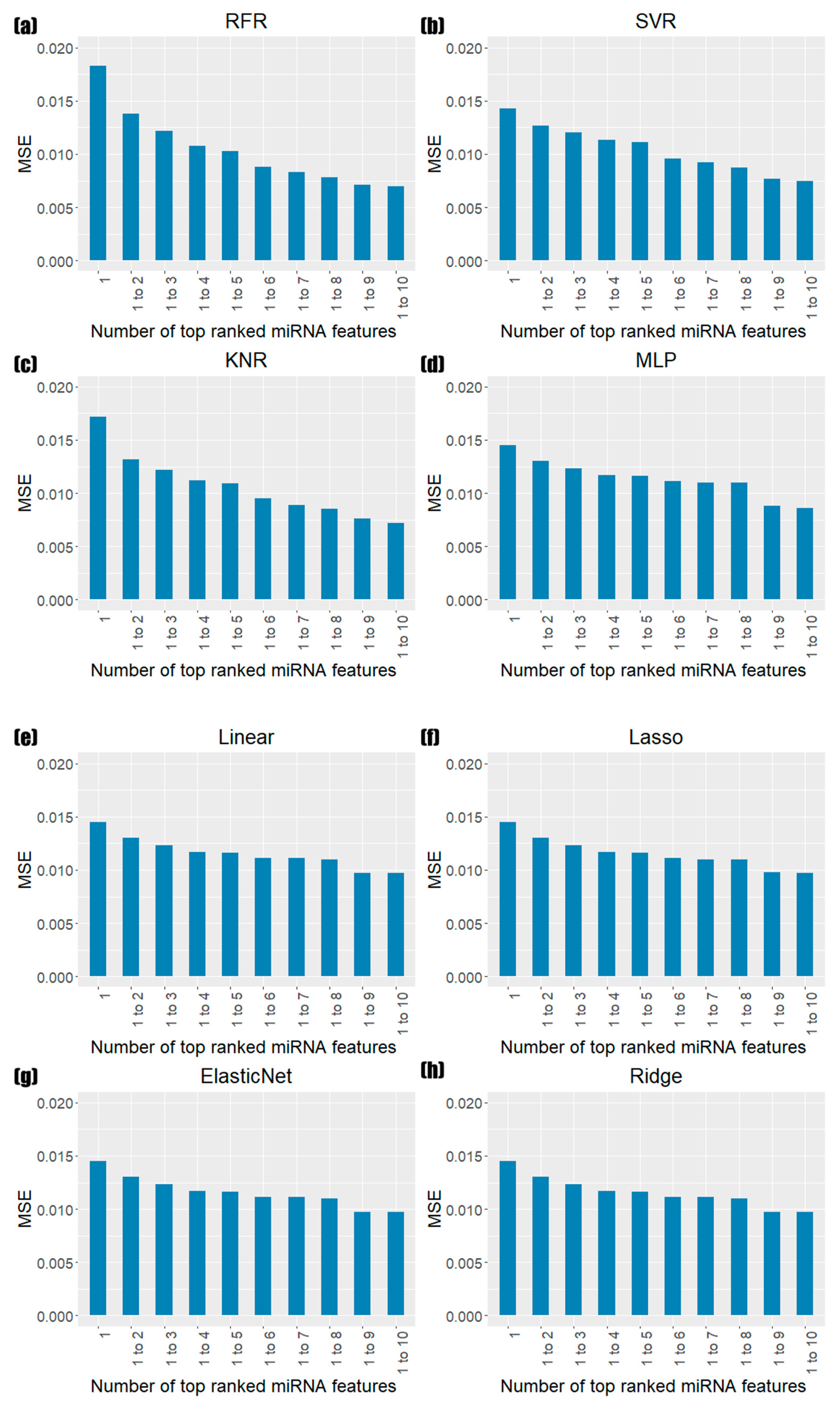
3.3. Prediction Results of Models Based on miRNA Feature Importance
3.4. Enrichment Analysis of Target Genes Predicted by Top Ranked miRNAs
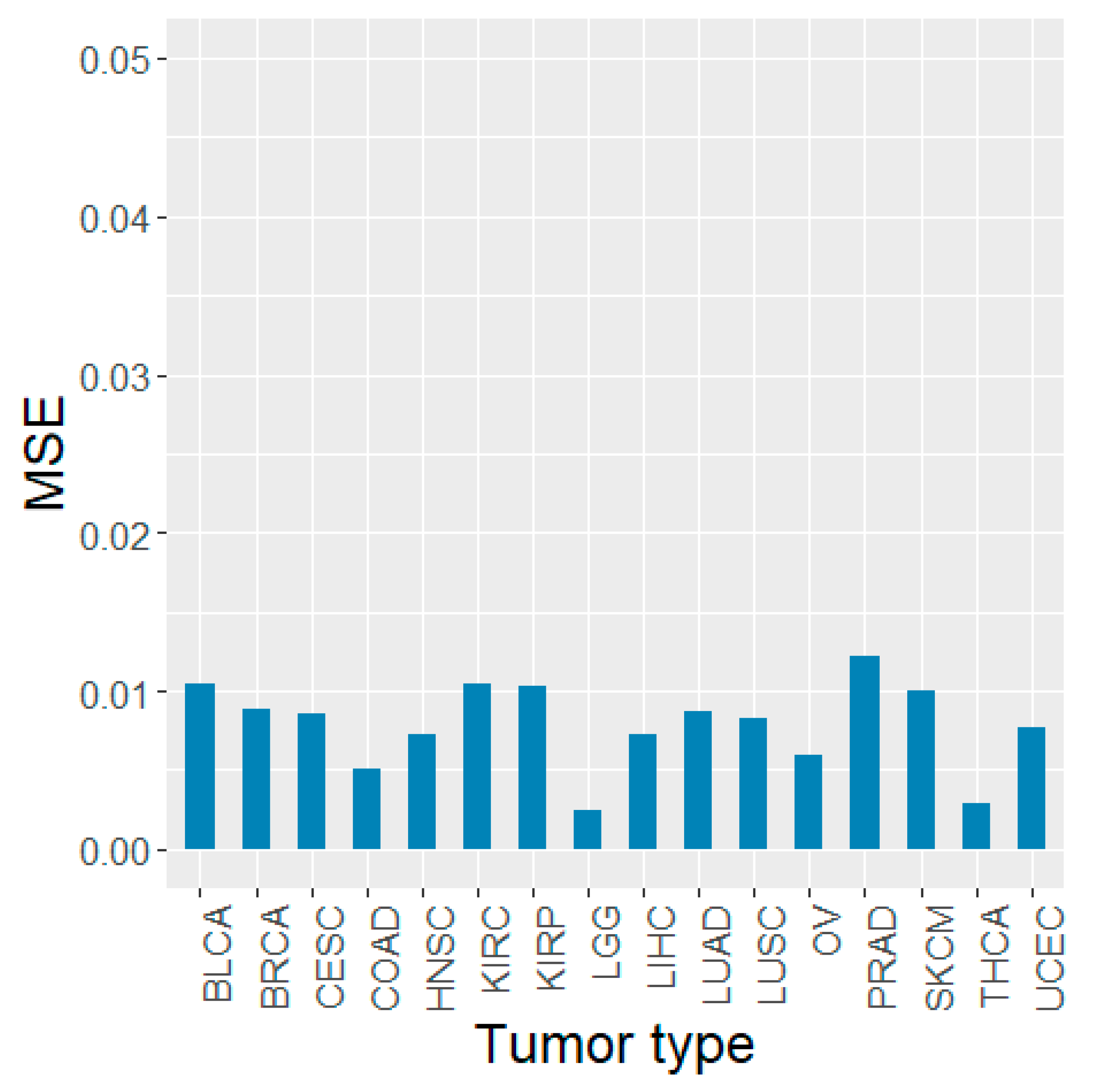
3.5. Validation Using Other TCGA Tumor Types and PCAWG Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Kowdley, K.V. MicroRNAs in common human diseases. Genom. Proteom. Bioinform. 2012, 10, 246–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Xie, D.; Zhao, Q.; You, Z. MicroRNAs and complex diseases: From experimental results to computational models. Brief. Bioinform. 2019, 20, 515–539. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Hsu, S.; Wang, X.; Kutay, H.; Bid, H.K.; Yu, J.; Ganju, R.K.; Jacob, S.T.; Yuneva, M.; Ghoshal, K. Reciprocal regulation of microRNA-122 and c-myc in hepatocellular cancer: Role of E2F1 and transcription factor dimerization partner 2. Hepatology 2014, 59, 555–566. [Google Scholar] [CrossRef] [Green Version]
- Hayashita, Y.; Osada, H.; Tatematsu, Y.; Yamada, H.; Yanagisawa, K.; Tomida, S.; Yatabe, Y.; Kawahara, K.; Sekido, Y.; Takahashi, T. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 2005, 65, 9628–9632. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.; Wang, X.; Sun, B.; Zhang, X.D.; Zhu, X.H.; Yu, Z.J.; Luo, H. Expression, regulation and mechanism of action of the miR-17-92 cluster in tumor cells (review). Int. J. Mol. Med. 2017, 40, 1624–1630. [Google Scholar] [CrossRef] [Green Version]
- Chou, J.; Shahi, P.; Werb, Z. microRNA-mediated regulation of the tumor microenvironment. Cell Cycle 2013, 12, 3262–3271. [Google Scholar] [CrossRef] [Green Version]
- Kohlhapp, F.J.; Mitra, A.K.; Lengyel, E.; Peter, M.E. MicroRNAs as mediators and communicators between cancer cells and the tumor microenvironment. Oncogene 2015, 34, 5857–5868. [Google Scholar] [CrossRef] [Green Version]
- Mitra, A.K.; Zillhardt, M.; Hua, Y.; Tiwari, P.; Murmann, A.E.; Peter, M.E.; Lengyel, E. MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov. 2012, 2, 1100–1108. [Google Scholar] [CrossRef] [Green Version]
- Aprelikova, O.; Yu, X.; Palla, J.; Wei, B.R.; John, S.; Yi, M.; Stephens, R.; Simpson, R.M.; Risinger, J.I.; Jazaeri, A.; et al. The role of miR-31 and its target gene SATB2 in cancer-associated fibroblasts. Cell Cycle 2010, 9, 4387–4398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Liu, J.; Liu, Y.; Wu, W.; Li, X.; Wu, Y.; Chen, H.; Zhang, K.; Gu, L. miR-101 represses lung cancer by inhibiting interaction of fibroblasts and cancer cells by down-regulating CXCL12. Biomed. Pharmacother. 2015, 74, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef]
- Carter, S.L.; Cibulskis, K.; Helman, E.; McKenna, A.; Shen, H.; Zack, T.; Laird, P.W.; Onofrio, R.C.; Winckler, W.; Weir, B.A.; et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012, 30, 413–421. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Koo, B.; Rhee, J. Prediction of tumor purity from gene expression data using machine learning. Brief. Bioinform. 2021, 22, bbab163. [Google Scholar] [CrossRef]
- Li, Y.; Umbach, D.M.; Bingham, A.; Li, Q.; Zhuang, Y.; Li, L. Putative biomarkers for predicting tumor sample purity based on gene expression data. BMC Genom. 2019, 20, 1021. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Aran, D.; Sirota, M.; Butte, A.J. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 2015, 6, 8971. [Google Scholar] [CrossRef] [Green Version]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A. Scikit-learn: Machine learning in python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Dudda, J.C.; Salaun, B.; Ji, Y.; Palmer, D.C.; Monnot, G.C.; Merck, E.; Boudousquie, C.; Utzschneider, D.T.; Escobar, T.M.; Perret, R.; et al. MicroRNA-155 is required for effector CD8+ T cell responses to virus infection and cancer. Immunity 2013, 38, 742–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Eng, C.; Shen, J.; Lu, Y.; Takata, Y.; Mehdizadeh, A.; Chang, G.J.; Rodriguez-Bigas, M.A.; Li, Y.; Chang, P.; et al. Serum exosomal miR-4772-3p is a predictor of tumor recurrence in stage II and III colon cancer. Oncotarget 2016, 7, 76250–76260. [Google Scholar] [CrossRef] [Green Version]
- Tsang, F.H.; Au, S.L.; Wei, L.; Fan, D.N.Y.; Lee, J.M.F.; Wong, C.C.L.; Ng, I.O.L.; Wong, C.M. MicroRNA-142-3p and microRNA-142-5p are downregulated in hepatocellular carcinoma and exhibit synergistic effects on cell motility. Front. Med. 2015, 9, 331–343. [Google Scholar] [CrossRef]
- Yu, F.; Lu, Z.; Chen, B.; Dong, P.; Zheng, J. microRNA-150: A promising novel biomarker for hepatitis B virus-related hepatocellular carcinoma. Diagn. Pathol. 2015, 10, 129. [Google Scholar] [CrossRef] [Green Version]
- Ito, M.; Teshima, K.; Ikeda, S.; Kitadate, A.; Watanabe, A.; Nara, M.; Yamashita, J.; Ohshima, K.; Sawada, K.; Tagawa, H. MicroRNA-150 inhibits tumor invasion and metastasis by targeting the chemokine receptor CCR6, in advanced cutaneous T-cell lymphoma. Blood 2014, 123, 1499–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffries, J.; Zhou, W.; Hsu, A.Y.; Deng, Q. miRNA-223 at the crossroads of inflammation and cancer. Cancer Lett. 2019, 451, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Qiu, X.; Huang, Y.; Zhang, H.; Chen, K. miR-223-3p carried by cancer-associated fibroblast microvesicles targets SORBS1 to modulate the progression of gastric cancer. Cancer Cell Int. 2022, 22, 96. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nag, A.; Mandal, C.C. A comprehensive review on miR-200c, A promising cancer biomarker with therapeutic potential. Curr. Drug Targets 2015, 16, 1381–1403. [Google Scholar] [CrossRef]
- Pichler, M.; Ress, A.L.; Winter, E.; Stiegelbauer, V.; Karbiener, M.; Schwarzenbacher, D.; Scheideler, M.; Ivan, C.; Jahn, S.W.; Kiesslich, T.; et al. MiR-200a regulates epithelial to mesenchymal transition-related gene expression and determines prognosis in colorectal cancer patients. Br. J. Cancer 2014, 110, 1614–1621. [Google Scholar] [CrossRef] [Green Version]
- Legras, A.; Pécuchet, N.; Imbeaud, S.; Pallier, K.; Didelot, A.; Roussel, H.; Gibault, L.; Fabre, E.; Pimpec-Barthes, L.; Laurent-Puig, P.; et al. Epithelial-to-mesenchymal transition and MicroRNAs in lung cancer. Cancers 2017, 9, 101. [Google Scholar] [CrossRef] [Green Version]
- Raue, R.; Frank, A.; Fuhrmann, D.C.; de la Cruz-Ojeda, P.; Rösser, S.; Bauer, R.; Cardamone, G.; Weigert, A.; Syed, S.N.; Schmid, T.; et al. MicroRNA-200c attenuates the tumor-infiltrating capacity of macrophages. Biology 2022, 11, 349. [Google Scholar] [CrossRef]
- Liu, Z.; Wen, J.; Wu, C.; Hu, C.; Wang, J.; Bao, Q.; Wang, H.; Wang, J.; Zhou, Q.; Wei, L.; et al. MicroRNA-200a induces immunosuppression by promoting PTEN-mediated PD-L1 upregulation in osteosarcoma. Aging 2020, 12, 1213–1236. [Google Scholar] [CrossRef]
- Guo, J.; Wen, N.; Yang, S.; Guan, X.; Cang, S. MiR-92a regulates oral squamous cell carcinoma (OSCC) cell growth by targeting FOXP1 expression. Biomed. Pharmacother. 2018, 104, 77–86. [Google Scholar] [CrossRef]
- Elhamamsy, A.R.; El Sharkawy, M.S.; Zanaty, A.F.; Mahrous, M.A.; Mohamed, A.E.; Abushaaban, E.A. Circulating miR-92a, miR-143 and miR-342 in plasma are novel potential biomarkers for acute myeloid leukemia. Int. J. Mol. Cell. Med. 2017, 6, 77–86. [Google Scholar]
- Wang, J.; Li, Y.; Ding, M.; Zhang, H.; Xu, X.; Tang, J. Molecular mechanisms and clinical applications of miR-22 in regulating malignant progression in human cancer (review). Int. J. Oncol. 2017, 50, 345–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monin, L.; Gaffen, S.L. Interleukin 17 family cytokines: Signaling mechanisms, biological activities, and therapeutic implications. Cold Spring Harb. Perspect. Biol. 2018, 10, a028522. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, X.; Herjan, T.; Li, X. The role of interleukin-17 in tumor development and progression. J. Exp. Med. 2019, 217, e20190297. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ming, X.; Li, W.; Bi, M.; Yan, B.; Wang, X.; Yang, P.; Yang, B. The microRNA-155 mediates hepatitis B virus replication by reinforcing SOCS1 signalling-induced autophagy. Cell Biochem. Funct. 2020, 38, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Waters, J.P.; Pober, J.S.; Bradley, J.R. Tumour necrosis factor in infectious disease. J. Pathol. 2013, 230, 132–147. [Google Scholar] [CrossRef]
- Conti, I.; Varano, G.; Simioni, C.; Laface, I.; Milani, D.; Rimondi, E.; Neri, L.M. miRNAs as influencers of cell-cell communication in tumor microenvironment. Cells 2020, 9, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Okada, M.; Kitahara, M.; Kishimoto, S.; Matsuda, T.; Hirano, T.; Kishimoto, T. IL-6/BSF-2 functions as a killer helper factor in the in vitro induction of cytotoxic T cells. J. Immunol. 1988, 141, 1543–1549. [Google Scholar]
- Chen, X.; Wang, L.; Qu, J.; Guan, N.; Li, J. Predicting miRNA-disease association based on inductive matrix completion. Bioinformatics 2018, 34, 4256–4265. [Google Scholar] [CrossRef]
- Chen, X.; Yin, J.; Qu, J.; Huang, L. MDHGI: Matrix decomposition and heterogeneous graph inference for miRNA-disease association prediction. PLoS Comput. Biol. 2018, 14, e1006418. [Google Scholar] [CrossRef]
- Chen, X.; Zhu, C.; Yin, J. Ensemble of decision tree reveals potential miRNA-disease associations. PLoS Comput. Biol. 2019, 15, e1007209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Sun, L.; Zhao, Y. NCMCMDA: miRNA-disease association prediction through neighborhood constraint matrix completion. Brief. Bioinform. 2021, 22, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, T.; Zhao, Y.; Wang, C.; Zhu, C. Deep-belief network for predicting potential miRNA-disease associations. Brief. Bioinform. 2021, 22, bbaa186. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, T.; Huang, L.; Chen, X. Prediction of potential miRNA-disease associations based on stacked autoencoder. Brief. Bioinform. 2022, 23, bbac021. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Feng, D.; Li, M.; Zhou, J.; Li, X.; Zhao, D.; Hao, B.; Li, D.; Ding, K. Transcriptomic analysis of mRNA-lncRNA-miRNA interactions in hepatocellular carcinoma. Sci. Rep. 2019, 9, 16096. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, X.; Zheng, Y.; Chen, W.; Zheng, Y.; Li, G.; Lou, W.; Wang, X. Construction of an mRNA-miRNA-lncRNA network prognostic for triple-negative breast cancer. Aging 2021, 13, 1153–1175. [Google Scholar] [CrossRef]
- Ghasemi, T.; Khalaj-Kondori, M.; Hosseinpour Feizi, M.A.; Asadi, P. lncRNA-miRNA-mRNA interaction network for colorectal cancer; an in silico analysis. Comput. Biol. Chem. 2020, 89, 107370. [Google Scholar] [CrossRef]
- Gao, L.; Li, X.; Nie, X.; Guo, Q.; Liu, Q.; Qi, Y.; Liu, J.; Lin, B. Construction of novel mRNA-miRNA-lncRNA regulatory networks associated with prognosis of ovarian cancer. J. Cancer. 2020, 11, 7057–7072. [Google Scholar] [CrossRef]
- Yang, M.; Lu, H.; Liu, J.; Wu, S.; Kim, P.; Zhou, X. lncRNAfunc: A knowledgebase of lncRNA function in human cancer. Nucleic Acids Res. 2022, 50, D1295–D1306. [Google Scholar] [CrossRef]
- Wei, G.; Dong, Y.; He, Z.; Qiu, H.; Wu, Y.; Chen, Y. Identification of hub genes and construction of an mRNA-miRNA-lncRNA network of gastric carcinoma using integrated bioinformatics analysis. PLoS ONE 2021, 16, e0261728. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Type | Abbreviation | Number of Samples |
|---|---|---|
| Breast invasive carcinoma | BRCA | 1202 |
| Kidney renal clear cell carcinoma | KIRC | 592 |
| Uterine corpus endometrial carcinoma | UCEC | 575 |
| Thyroid carcinoma | THCA | 573 |
| Head and neck squamous cell carcinoma | HNSC | 569 |
| Lung adenocarcinoma | LUAD | 564 |
| Prostate adenocarcinoma | PRAD | 551 |
| Brain lower-grade glioma | LGG | 530 |
| Lung squamous cell carcinoma | LUSC | 523 |
| Ovarian serous cystadenocarcinoma | OV | 498 |
| Stomach adenocarcinoma * | STAD | 477 |
| Colon adenocarcinoma | COAD | 461 |
| Skin cutaneous melanoma | SKCM | 452 |
| Bladder urothelial carcinoma | BLCA | 432 |
| Liver hepatocellular carcinoma | LIHC | 425 |
| Kidney renal papillary cell carcinoma | KIRP | 326 |
| Cervical squamous cell carcinoma and Endocervical adenocarcinoma | CESC | 312 |
| Tumor Type | Abbreviation | Number of Samples |
|---|---|---|
| Sarcoma | SARC | 263 |
| Esophageal carcinoma | ESCA | 198 |
| Acute myeloid leukemia | LAML | 188 |
| Pheochromocytoma and Paraganglioma | PCPG | 187 |
| Pancreatic adenocarcinoma | PAAD | 183 |
| Rectum adenocarcinoma | READ | 165 |
| Testicular germ cell tumors | TGCT | 156 |
| Thymoma | THYM | 126 |
| Kidney chromophobe | KICH | 91 |
| Mesothelioma | MESO | 87 |
| Adrenocortical carcinoma | ACC | 80 |
| Uveal melanoma | UVM | 80 |
| Uterine carcinosarcoma | UCS | 57 |
| Lymphoid neoplasm diffuse large B-cell lymphoma | DLBC | 47 |
| Cholangiocarcinoma | CHOL | 45 |
| Glioblastoma multiforme | GBM | 5 |
| Top | miRNA | Feature-Importance Score | PCC |
|---|---|---|---|
| 1 | hsa-mir-155 | 0.2309 | −0.6037 |
| 2 | hsa-mir-4772 | 0.0899 | −0.5834 |
| 3 | hsa-mir-142 | 0.0575 | −0.5111 |
| 4 | hsa-mir-150 | 0.0330 | −0.5744 |
| 5 | hsa-mir-223 | 0.0225 | −0.5003 |
| 6 | hsa-mir-200c | 0.0211 | 0.0398 |
| 7 | hsa-mir-141 | 0.0159 | −0.0197 |
| 8 | hsa-mir-200b | 0.0153 | 0.0048 |
| 9 | hsa-mir-92a-1 | 0.0134 | 0.1800 |
| 10 | hsa-mir-22 | 0.0120 | −0.3558 |
| KEGG Pathways | Adjusted p-Value |
|---|---|
| Interleukin (IL)-17 signaling pathway | 0.0014 |
| Pathways in cancer | 0.0014 |
| Hepatitis B | 0.0014 |
| T-cell receptor signaling pathway | 0.0015 |
| Tumor necrosis factor (TNF) signaling pathway | 0.0021 |
| Osteoclast differentiation | 0.0043 |
| Mitogen-activated protein kinase (MAPK) signaling pathway | 0.0052 |
| Toll-like receptor signaling pathway | 0.0052 |
| Lipid and atherosclerosis | 0.0052 |
| Pancreatic cancer | 0.0053 |
| Signaling pathways regulating pluripotency of stem cells | 0.0053 |
| B-cell receptor signaling pathway | 0.0064 |
| Colorectal cancer | 0.0082 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nam, D.-Y.; Rhee, J.-K. Assessment of MicroRNAs Associated with Tumor Purity by Random Forest Regression. Biology 2022, 11, 787. https://doi.org/10.3390/biology11050787
Nam D-Y, Rhee J-K. Assessment of MicroRNAs Associated with Tumor Purity by Random Forest Regression. Biology. 2022; 11(5):787. https://doi.org/10.3390/biology11050787
Chicago/Turabian StyleNam, Dong-Yeon, and Je-Keun Rhee. 2022. "Assessment of MicroRNAs Associated with Tumor Purity by Random Forest Regression" Biology 11, no. 5: 787. https://doi.org/10.3390/biology11050787
APA StyleNam, D.-Y., & Rhee, J.-K. (2022). Assessment of MicroRNAs Associated with Tumor Purity by Random Forest Regression. Biology, 11(5), 787. https://doi.org/10.3390/biology11050787