
Endothelin and the Cardiovascular System: The Long Journey and Where We Are Going


{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
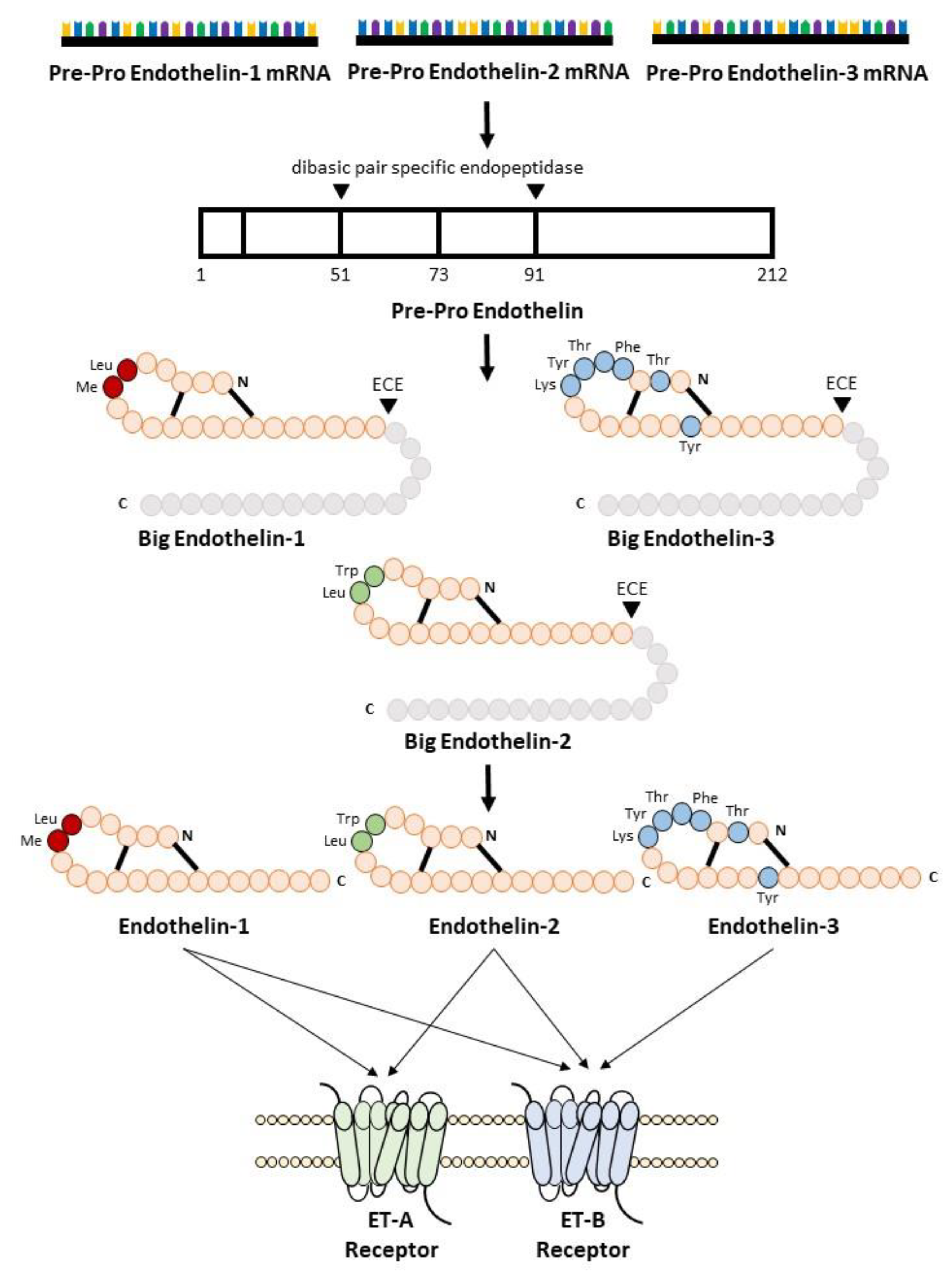
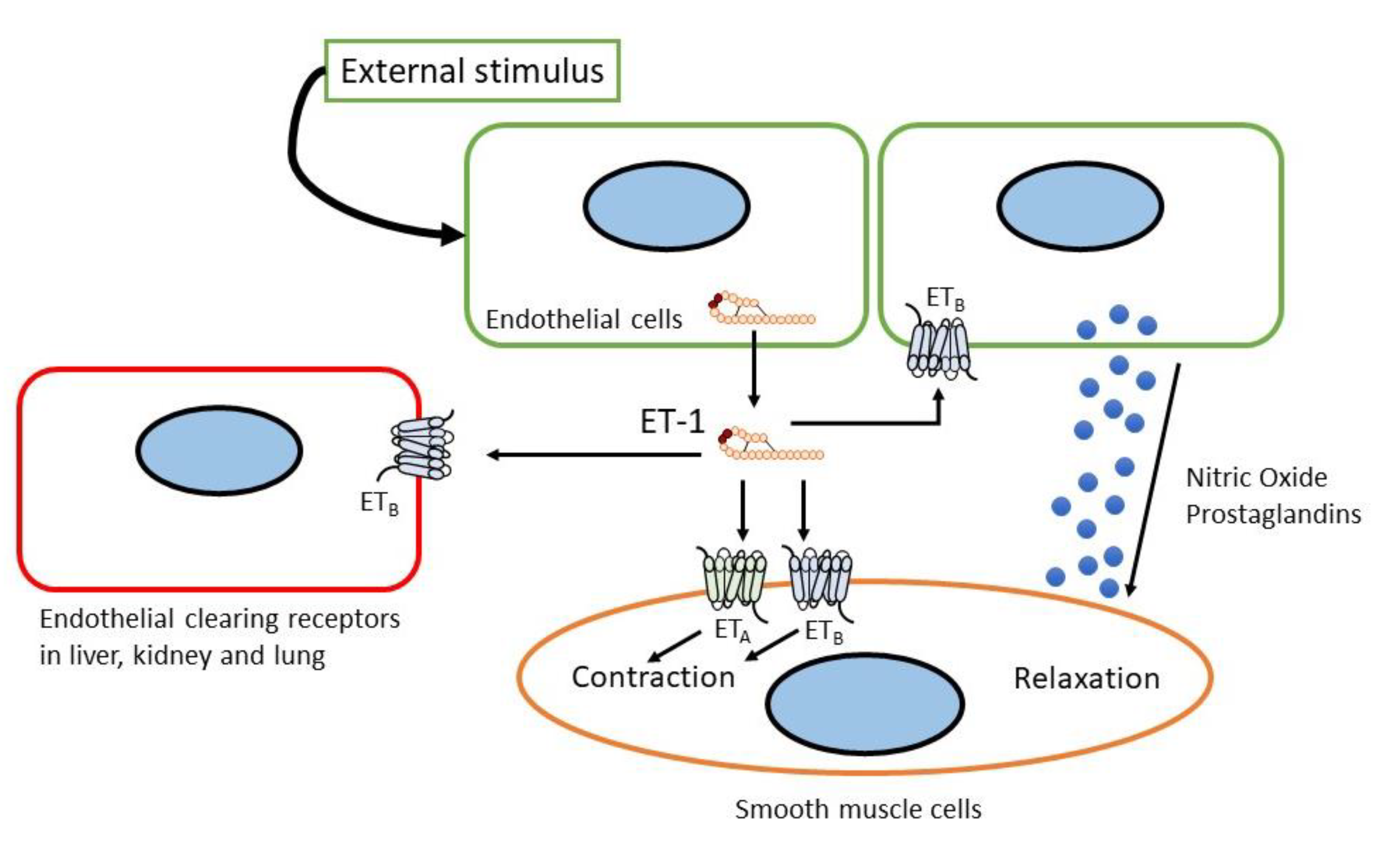
2. The Endothelin System
2.1. Biosynthesis of Endothelin
2.2. Endothelin Receptor
2.3. Endothelin Agonists and Antagonists
2.4. Genetic Mutations in Endothelin System
2.5. Phenotype of Genetic Endothelin Modification in Mice
3. Endothelin in Cardiovascular Diseases
3.1. Pulmonary Hypertension
3.1.1. Relations between Endothelin and PAH
3.1.2. Clinical Applications of Endothelin and ERAs in PH
3.2. Systemic Arterial Hypertension
3.2.1. ET in Basic Molecular Mechanism of Systemic Arterial Hypertension
3.2.2. Clinical Implications of Endothelin in Systemic Arterial Hypertension
3.3. Heart Failure
3.3.1. Endothelin and Heart Failure (HF)
3.3.2. Clinical Evidence of Endothelin in Chronic and Acute Heart Failure
3.4. Atherosclerosis, Acute Coronary Syndrome and Coronary Artery Disease
3.4.1. Endothelin and Coronary Artery Pathologies
3.4.2. Clinical Application of Endothelin in Coronary Artery Disease
3.5. Others
3.5.1. Cardiac Arrythmia
3.5.2. Antiangiogenic Treatment Adverse Effects
3.5.3. Peripheral Artery Disease
4. Future Perspectives
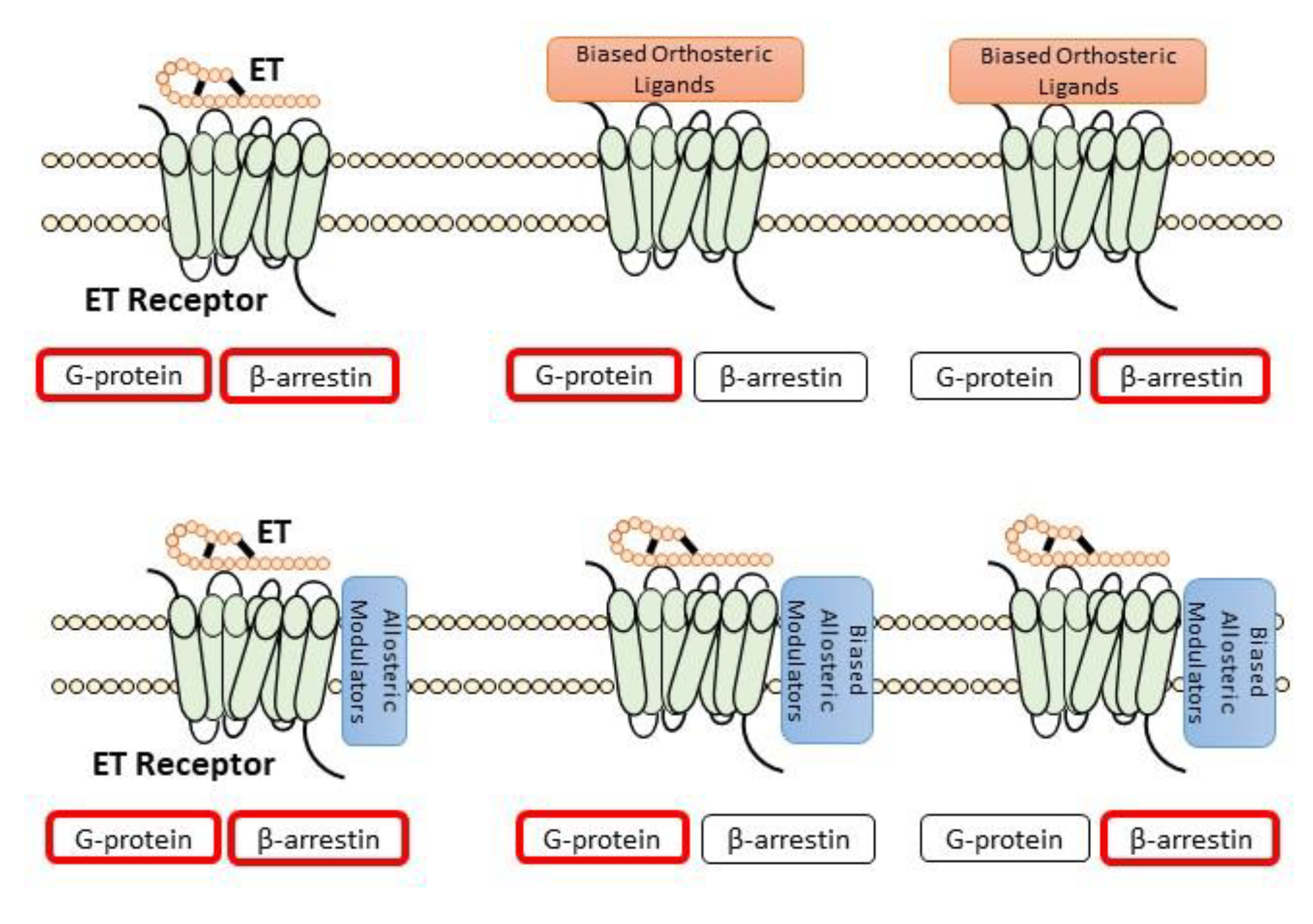
4.1. Allosteric Modulators
4.2. Peptide-Based Biased ET Receptor Signaling
4.3. Pepducins/Cell-Penetrating Peptides
4.4. Antibody against ET Receptors
4.5. ET-1 Traps
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Furchgott, R.F.; Vanhoutte, P.M. Endothelium-derived relaxing and contracting factors. FASEB J. 1989, 3, 2007–2018. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.; Yanagisawa, M. Endothelin: 30 years from discovery to therapy. Hypertension 2019, 74, 1232–1265. [Google Scholar] [CrossRef] [PubMed]
- Hickey, K.A.; Rubanyi, G.; Paul, R.J.; Highsmith, R.F. Characterization of a coronary vasoconstrictor produced by cultured endothelial cells. Am. J. Physiol. Cell Physiol. 1985, 17, C550–C556. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.F.; Robbins, R.J.; McMurtry, I.F. Endothelial cells in culture produce a vasoconstrictor substance. J. Cell. Physiol. 1987, 132, 263–270. [Google Scholar] [CrossRef]
- Yanagisawa, M.; Kurihara, H.; Kimura, S.; Tomobe, Y.; Kobayashi, M.; Mitsui, Y.; Yazaki, Y.; Goto, K.; Masaki, T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988, 332, 411–415. [Google Scholar] [CrossRef]
- Saida, K.; Mitsui, Y.; Ishida, N. A Novel Peptide, Vasoactive Intestinal Contractor, of a New (Endothelin) Peptide Family: Molecular Cloning, Expression, and Biological Activity. J. Biol. Chem. 1989, 264, 14613–14616. [Google Scholar] [CrossRef]
- Saida, K.; Hashimoto, M.; Mitsui, Y.; Ishida, N.; Uchide, T. The Prepro Vasoactive Intestinal Contractor (VIC)/Endothelin-2 Gene (EDN2): Structure, Evolution, Production, and Embryonic Expression. Genomics 2000, 64, 51–61. [Google Scholar] [CrossRef]
- Inoue, A.; Yanagisawa, M.; Kimura, S.; Kasuya, Y.; Miyauchi, T.; Goto, K.; Masaki, T. The human endothelin family: Three structurally and pharmacologically distinct isopeptides predicted by three separate genes. Proc. Natl. Acad. Sci. USA 1989, 86, 2863–2867. [Google Scholar] [CrossRef]
- Arai, H.; Hori, S.; Aramori, I.; Ohkubo, H.; Nakanishi, S. Cloning and expression of a cDNA encoding an endothelin receptor. Nature 1990, 348, 730–732. [Google Scholar] [CrossRef]
- Sakurai, T.; Yanagisawa, M.; Takuwat, Y.; Miyazakit, H.; Kimura, S.; Goto, K.; Masaki, T. Cloning of a cDNA encoding a non-isopeptide-selective subtype of the endothelin receptor. Nature 1990, 348, 732–735. [Google Scholar] [CrossRef]
- Sakamoto, A.; Yanagisawa, M.; Sakurai, T.; Takuwa, Y.; Yanagisawa, H.; Masaki, T. Cloning and functional expression of human cDNA for the ETB endothelin receptor. Biochem. Biophys. Res. Commun. 1991, 178, 656–663. [Google Scholar] [CrossRef]
- Saito, Y.; Mizuno, T.; Itakura, M.; Suzuki, Y.; Ito, T.; Hagiwara, H.; Hirose, S. Primary structure of bovine endothelin ETB receptor and identification of signal peptidase and metal proteinase cleavage sites. J. Biol. Chem. 1991, 266, 23433–23437. [Google Scholar] [CrossRef]
- Davenport, A.P.; Hyndman, K.A.; Dhaun, N.; Southan, C.; Kohan, D.E.; Pollock, J.S.; Pollock, D.M.; Webb, D.J.; Maguire, J.J. Endothelin. Pharmacol. Rev. 2016, 68, 357–418. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Emoto, N.; Giaid, A.; Slaughter, C.; Kaw, S.; deWit, D.; Yanagisawa, M. ECE-1: A membrane-bound metalloprotease that catalyzes the proteolytic activation of big endothelin-1. Cell 1994, 78, 473–485. [Google Scholar] [CrossRef]
- Takahashi, M.; Matsushita, Y.; Iijima, Y.; Tanzawa, K. Purification and characterization of endothelin-converting enzyme from rat lung. J. Biol. Chem. 1993, 268, 21394–21398. [Google Scholar] [CrossRef]
- Emoto, N.; Yanagisawa, M. Endothelin-converting Enzyme-2 Is a Membrane-bound, Phosphoramidon-sensitive Metalloprotease with Acidic pH Optimum (∗). J. Biol. Chem. 1995, 270, 15262–15268. [Google Scholar] [CrossRef]
- McPherson, A.; Larson, S.B. The X-ray crystal structure of human endothelin 1, a polypeptide hormone regulator of blood pressure. Acta Crystallogr. Sect. F 2019, 75, 47–53. [Google Scholar] [CrossRef]
- Stanfield, R.L. Never too late for endothelin. Acta Crystallogr. Sect. F 2019, 75, 45–46. [Google Scholar] [CrossRef]
- Kloog, Y.; Ambar, I.; Sokolovsky, M.; Kochva, E.; Wollberg, Z.; Bdolah, A. Sarafotoxin, a Novel Vasoconstrictor Peptide: Phosphoinositide Hydrolysis in Rat Heart and Brain. Science 1988, 242, 268–270. [Google Scholar] [CrossRef]
- Takasaki, C.; Yanagisawa, M.; Kimura, S.; Goto, K.; Masaki, T. Similarity of endothelin to snake venom toxin. Nature 1988, 335, 303. [Google Scholar] [CrossRef]
- Abd-Elsalam, M.A. Bosentan, a selective and more potent antagonist for Atractaspis envenomation than the specific antivenom. Toxicon 2011, 57, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Barton, M. Aging and endothelin: Determinants of disease. Life Sci. 2014, 118, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Stow, L.R.; Jacobs, M.E.; Wingo, C.S.; Cain, B.D. Endothelin-1 gene regulation. FASEB J. 2011, 25, 16–28. [Google Scholar] [CrossRef] [PubMed]
- von Brandenstein, M.; Richter, C.; Fries, J.W.U. MicroRNAs: Small but amazing, and their association with endothelin. Life Sci. 2012, 91, 475–489. [Google Scholar] [CrossRef] [PubMed]
- Houde, M.; Desbiens, L.; D’Orléans-Juste, P. Chapter Five—Endothelin-1: Biosynthesis, Signaling and Vasoreactivity. Adv. Pharmacol. 2016, 77, 143–175. [Google Scholar] [CrossRef] [PubMed]
- Denault, J.-B.; Claing, A.; D’Orléans-Juste, P.; Sawamura, T.; Kido, T.; Masaki, T.; Leduc, R. Processing of proendothelin-1 by human furin convertase. FEBS Lett. 1995, 362, 276–280. [Google Scholar] [CrossRef]
- Yanagisawa, H.; Hammer, R.E.; Richardson, J.A.; Emoto, N.; Williams, S.C.; Takeda, S.; Clouthier, D.E.; Yanagisawa, M. Disruption of ECE-1 and ECE-2 reveals a role for endothelin-converting enzyme-2 in murine cardiac development. J. Clin. Investig. 2000, 105, 1373–1382. [Google Scholar] [CrossRef]
- Wypij, D.M.; Nichols, J.S.; Novak, P.J.; Lowell Stacy, D.; Berman, J.; Wiseman, J.S. Role of mast cell chymase in the extracellular processing of big-endothelin-1 to endothelin-1 in the perfused rat lung. Biochem. Pharmacol. 1992, 43, 845–853. [Google Scholar] [CrossRef]
- Maguire, J.; Davenport, A.P. Alternative Pathway to Endothelin-Converting Enzyme for the Synthesis of Endothelin in Human Blood Vessels. J. Cardiovasc. Pharmacol. 2004, 44, S27. [Google Scholar] [CrossRef]
- Houde, M.; Desbiens, L.; Schwertani, A.; Pejler, G.; Iglarz, M.; D’Orléans-Juste, P. Endothelin receptor antagonist macitentan or deletion of mouse mast cell protease 4 delays lesion development in atherosclerotic mice. Life Sci. 2016, 159, 71–75. [Google Scholar] [CrossRef]
- Houde, M.; Jamain, M.-D.; Labonté, J.; Desbiens, L.; Pejler, G.; Gurish, M.; Takai, S.; D’Orléans-Juste, P. Pivotal Role of Mouse Mast Cell Protease 4 in the Conversion and Pressor Properties of Big-Endothelin-1. J. Pharmacol. Exp. Ther. 2013, 346, 31–37. [Google Scholar] [CrossRef]
- Guo, C.; Ju, H.; Leung, D.; Massaeli, H.; Shi, M.; Rabinovitch, M. A novel vascular smooth muscle chymase is upregulated in hypertensive rats. J. Clin. Investig. 2001, 107, 703–715. [Google Scholar] [CrossRef][Green Version]
- Ju, H.; Gros, R.; You, X.; Tsang, S.; Husain, M.; Rabinovitch, M. Conditional and targeted overexpression of vascular chymase causes hypertension in transgenic mice. Proc. Natl. Acad. Sci. USA 2001, 98, 7469–7474. [Google Scholar] [CrossRef]
- Dhaun, N.; Webb, D.J. Endothelins in cardiovascular biology and therapeutics. Nat. Rev. Cardiol. 2019, 16, 491–502. [Google Scholar] [CrossRef]
- Regard, J.B.; Sato, I.T.; Coughlin, S.R. Anatomical Profiling of G Protein-Coupled Receptor Expression. Cell 2008, 135, 561–571. [Google Scholar] [CrossRef]
- Schneider, M.P.; Boesen, E.I.; Pollock, D.M. Contrasting Actions of Endothelin ETA and ETB Receptors in Cardiovascular Disease. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 731–759. [Google Scholar] [CrossRef]
- Dupuis, J.; Goresky, C.A.; Fournier, A. Pulmonary clearance of circulating endothelin-1 in dogs in vivo: Exclusive role of ETBreceptors. J. Appl. Physiol. 1996, 81, 1510–1515. [Google Scholar] [CrossRef]
- Dupuis, J.; Stewart, D.J.; Cernacek, P.; Gosselin, G. Human Pulmonary Circulation Is an Important Site for Both Clearance and Production of Endothelin-1. Circulation 1996, 94, 1578–1584. [Google Scholar] [CrossRef]
- Fukuroda, T.; Fujikawa, T.; Ozaki, S.; Ishikawa, K.; Yano, M.; Nishikibe, M. Clearance of Circulating Endothelin-1 by ETB Receptors in Rats. Biochem. Biophys. Res. Commun. 1994, 199, 1461–1465. [Google Scholar] [CrossRef]
- Shihoya, W.; Nishizawa, T.; Okuta, A.; Tani, K.; Dohmae, N.; Fujiyoshi, Y.; Nureki, O.; Doi, T. Activation mechanism of endothelin ETB receptor by endothelin-1. Nature 2016, 537, 363–368. [Google Scholar] [CrossRef]
- Shihoya, W.; Izume, T.; Inoue, A.; Yamashita, K.; Kadji, F.M.N.; Hirata, K.; Aoki, J.; Nishizawa, T.; Nureki, O. Crystal structures of human ETB receptor provide mechanistic insight into receptor activation and partial activation. Nat. Commun. 2018, 9, 4711. [Google Scholar] [CrossRef]
- Shihoya, W.; Nishizawa, T.; Yamashita, K.; Inoue, A.; Hirata, K.; Kadji, F.M.N.; Okuta, A.; Tani, K.; Aoki, J.; Fujiyoshi, Y.; et al. X-ray structures of endothelin ETB receptor bound to clinical antagonist bosentan and its analog. Nat. Struct. Mol. Biol. 2017, 24, 758–764. [Google Scholar] [CrossRef]
- Nagiri, C.; Shihoya, W.; Inoue, A.; Kadji, F.M.N.; Aoki, J.; Nureki, O. Crystal structure of human endothelin ETB receptor in complex with peptide inverse agonist IRL2500. Commun. Biol. 2019, 2, 236. [Google Scholar] [CrossRef]
- Izume, T.; Miyauchi, H.; Shihoya, W.; Nureki, O. Crystal structure of human endothelin ETB receptor in complex with sarafotoxin S6b. Biochem. Biophys. Res. Commun. 2020, 528, 383–388. [Google Scholar] [CrossRef]
- Haynes, W.G.; Strachan, F.E.; Webb, D.J. Endothelin ETA and ETB Receptors Cause Vasoconstriction of Human Resistance and Capacitance Vessels In Vivo. Circulation 1995, 92, 357–363. [Google Scholar] [CrossRef]
- Davenport, A.P.; Kuc, R.E.; Southan, C.; Maguire, J.J. New drugs and emerging therapeutic targets in the endothelin signaling pathway and prospects for personalized precision medicine. Physiol. Res. 2018, 67, S37–S54. [Google Scholar] [CrossRef]
- Russell, F.D.; Davenport, A.P. Characterization of the binding of endothelin ETB selective ligands in human and rat heart. Br. J. Pharmacol. 1996, 119, 631–636. [Google Scholar] [CrossRef]
- Williams, D.L.; Jones, K.L.; Pettibone, D.J.; Lis, E.V.; Clineschmidt, B.V. Sarafotoxin S6c: An agonist which distinguishes between endothelin receptor subtypes. Biochem. Biophys. Res. Commun. 1991, 175, 556–561. [Google Scholar] [CrossRef]
- Takai, M.; Umemura, I.; Yamasaki, K.; Watakabe, T.; Fujitani, Y.; Oda, K.; Urade, Y.; Inui, T.; Yamamura, T.; Okada, T. A potent and specific agonist, Suc-[Glu9,Ala11,15]-endothelin-1(8-21), IRL 1620, for the ETB receptor. Biochem. Biophys. Res. Commun. 1992, 184, 953–959. [Google Scholar] [CrossRef]
- Ihara, M.; Saeki, T.; Fukuroda, T.; Kimura, S.; Ozaki, S.; Patel, A.C.; Yano, M. A novel radioligand [125I]BQ-3020 selective for endothelin (ETB) receptors. Life Sci. 1992, 51, PL47–PL52. [Google Scholar] [CrossRef]
- Leonard, M.G.; Briyal, S.; Gulati, A. Endothelin B receptor agonist, IRL-1620, reduces neurological damage following permanent middle cerebral artery occlusion in rats. Brain Res. 2011, 1420, 48–58. [Google Scholar] [CrossRef]
- Cifuentes, E.G.; Hornick, M.G.; Havalad, S.; Donovan, R.L.; Gulati, A. Neuroprotective Effect of IRL-1620, an Endothelin B Receptor Agonist, on a Pediatric Rat Model of Middle Cerebral Artery Occlusion. Front. Pediatr. 2018, 6, 310. [Google Scholar] [CrossRef]
- Briyal, S.; Ranjan, A.K.; Hornick, M.G.; Puppala, A.K.; Luu, T.; Gulati, A. Anti-apoptotic activity of ETB receptor agonist, IRL-1620, protects neural cells in rats with cerebral ischemia. Sci. Rep. 2019, 9, 10439. [Google Scholar] [CrossRef]
- Cemazar, M.; Wilson, I.; Prise, V.E.; Bell, K.M.; Hill, S.A.; Tozer, G.M. The endothelin B (ETB) receptor agonist IRL 1620 is highly vasoconstrictive in two syngeneic rat tumour lines: Potential for selective tumour blood flow modification. Br. J. Cancer 2005, 93, 98–106. [Google Scholar] [CrossRef]
- Rajeshkumar, N.V.; Rai, A.; Gulati, A. Endothelin B receptor agonist, IRL 1620, enhances the anti-tumor efficacy of paclitaxel in breast tumor rats. Breast Cancer Res. Treat. 2005, 94, 237–247. [Google Scholar] [CrossRef]
- Gulati, A.; Sunila, E.; Kuttan, G. IRL-1620, an Endothelin-B Receptor Agonist, Enhanced Radiation Induced Reduction in Tumor Volume in Dalton’s Lymphoma Ascites Tumor Model. Arzneimittelforschung 2012, 62, 14–17. [Google Scholar] [CrossRef]
- Perreault, T.; Baribeau, J. Characterization of endothelin receptors in newborn piglet lung. Am. J. Physiol. Cell. Mol. Physiol. 1995, 268, L607–L614. [Google Scholar] [CrossRef]
- Hirata, Y.; Emori, T.; Eguchi, S.; Kanno, K.; Imai, T.; Ohta, K.; Marumo, F. Endothelin receptor subtype B mediates synthesis of nitric oxide by cultured bovine endothelial cells. J. Clin. Investig. 1993, 91, 1367–1373. [Google Scholar] [CrossRef]
- Molenaar, P.; O’Reilly, G.; Sharkey, A.; Kuc, R.E.; Harding, D.P.; Plumpton, C.; Gresham, G.A.; Davenport, A.P. Characterization and localization of endothelin receptor subtypes in the human atrioventricular conducting system and myocardium. Circ. Res. 1993, 72, 526–538. [Google Scholar] [CrossRef]
- Johnström, P.; Rudd, J.H.F.; Richards, H.K.; Fryer, T.D.; Clark, J.C.; Weissberg, P.L.; Pickard, J.D.; Davenport, A.P. Imaging Endothelin ETB Receptors Using [18F]-BQ3020: In Vitro Characterization and Positron Emission Tomography (MicroPET). Exp. Biol. Med. 2006, 231, 736–740. [Google Scholar] [CrossRef]
- Maguire, J.J.; Davenport, A.P. Endothelin Receptors and Their Antagonists. Semin. Nephrol. 2015, 35, 125–136. [Google Scholar] [CrossRef]
- Palmer, M.J. Endothelin Receptor Antagonists: Status and Learning 20 Years On. In Progress in Medicinal Chemistry; Elsevier: Amsterdam, The Netherlands, 2009; ISBN 0079-6468. [Google Scholar]
- Rubin, L.J.; Badesch, D.B.; Barst, R.J.; Galiè, N.; Black, C.M.; Keogh, A.; Pulido, T.; Frost, A.; Roux, S.; Leconte, I.; et al. Bosentan Therapy for Pulmonary Arterial Hypertension. N. Engl. J. Med. 2002, 346, 896–903. [Google Scholar] [CrossRef]
- Abman, S.H. Role of Endothelin Receptor Antagonists in the Treatment of Pulmonary Arterial Hypertension. Annu. Rev. Med. 2009, 60, 13–23. [Google Scholar] [CrossRef]
- Maguire, J.J.; Davenport, A.P. Endothelin@25—New agonists, antagonists, inhibitors and emerging research frontiers: IUPHAR Review 12. Br. J. Pharmacol. 2014, 171, 5555–5572. [Google Scholar] [CrossRef]
- Serasli, E.; Michaelidis, V.; Kosmas, A.; Antoniadou, M.; Tsara, V. Review on bosentan, a dual endothelin receptor antagonist for the treatment of pulmonary arterial hypertension. Recent Pat. Cardiovasc. Drug Discov. 2010, 5, 184–195. [Google Scholar] [CrossRef]
- Angeli, F.; Verdecchia, P.; Reboldi, G. Aprocitentan, A Dual Endothelin Receptor Antagonist Under Development for the Treatment of Resistant Hypertension. Cardiol. Ther. 2021, 10, 397–406. [Google Scholar] [CrossRef]
- Verweij, P.; Danaietash, P.; Flamion, B.; Ménard, J.; Bellet, M. Randomized dose-response study of the new dual endothelin receptor antagonist aprocitentan in hypertension. Hypertension 2020, 75, 956–965. [Google Scholar] [CrossRef]
- Ihara, M.; Fukuroda, T.; Saeki, T.; Nishikibe, M.; Kojiri, K.; Suda, H.; Yano, M. An endothelin receptor (ETA) antagonist isolated from Streptomyces misakiensis. Biochem. Biophys. Res. Commun. 1991, 178, 132–137. [Google Scholar] [CrossRef]
- Aramori, I.; Nirei, H.; Shoubo, M.; Sogabe, K.; Nakamura, K.; Kojo, H.; Notsu, Y.; Ono, T.; Nakanishi, S. Subtype selectivity of a novel endothelin antagonist, FR139317, for the two endothelin receptors in transfected Chinese hamster ovary cells. Mol. Pharmacol. 1993, 43, 127–131. [Google Scholar]
- Masuda, Y.; Sugo, T.; Kikuchi, T.; Kawata, A.; Satoh, M.; Fujisawa, Y.; Itoh, Y.; Wakimasu, M.; Ohtaki, T. Receptor binding and antagonist properties of a novel endothelin receptor antagonist, TAK-044 [cyclo[D-alpha-aspartyl-3-[(4-phenylpiperazin-1-yl) carbonyl]-L-alanyl-L-alpha-aspartyl-D-2-(2-thienyl) glycyl-L-leucyl-D-tryptophyl]disodium salt], in human end. J. Pharmacol. Exp. Ther. 1996, 279, 675–685. [Google Scholar]
- Vatter, H.; Zimmermann, M.; Weyrauch, E.; Lange, B.N.; Setzer, M.; Raabe, A.; Seifert, V. Cerebrovascular Characterization of the Novel Nonpeptide Endothelin-A Receptor Antagonist LU 208075. Clin. Neuropharmacol. 2003, 26, 73–83. [Google Scholar] [CrossRef]
- Wu, C.; Chan, M.F.; Stavros, F.; Raju, B.; Okun, I.; Mong, S.; Keller, K.M.; Brock, T.; Kogan, T.P.; Dixon, R.A.F. Discovery of TBC11251, a Potent, Long Acting, Orally Active Endothelin Receptor-A Selective Antagonist. J. Med. Chem. 1997, 40, 1690–1697. [Google Scholar] [CrossRef]
- Don, G.W.; Joseph, F.; Celermajer, D.S.; Corte, T.J. Ironic case of hepatic dysfunction following the global withdrawal of sitaxentan. Intern. Med. J. 2012, 42, 1351–1354. [Google Scholar] [CrossRef]
- Jarvis, M.F.; Wessale, J.L.; Zhu, C.Z.; Lynch, J.J.; Dayton, B.D.; Calzadilla, S.V.; Padley, R.J.; Opgenorth, T.J.; Kowaluk, E.A. ABT-627, an endothelin ETA receptor-selective antagonist, attenuates tactile allodynia in a diabetic rat model of neuropathic pain. Eur. J. Pharmacol. 2000, 388, 29–35. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Parving, H.-H.; Andress, D.L.; Bakris, G.; Correa-Rotter, R.; Hou, F.-F.; Kitzman, D.W.; Kohan, D.; Makino, H.; McMurray, J.J.V.; et al. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): A double-blind, randomised, placebo-controlled trial. Lancet 2019, 393, 1937–1947. [Google Scholar] [CrossRef]
- Ishikawa, K.; Ihara, M.; Noguchi, K.; Mase, T.; Mino, N.; Saeki, T.; Fukuroda, T.; Fukami, T.; Ozaki, S.; Nagase, T. Biochemical and pharmacological profile of a potent and selective endothelin B-receptor antagonist, BQ-788. Proc. Natl. Acad. Sci. USA 1994, 91, 4892–4896. [Google Scholar] [CrossRef]
- Komers, R.; Diva, U.; Inrig, J.K.; Loewen, A.; Trachtman, H.; Rote, W.E. Study Design of the Phase 3 Sparsentan Versus Irbesartan (DUPLEX) Study in Patients with Focal Segmental Glomerulosclerosis. Kidney Int. Rep. 2020, 5, 494–502. [Google Scholar] [CrossRef]
- Kumar, A.; Choudhury, M.; Batra, S.D.; Sikri, K.; Gupta, A. In vivo assessment of a single adenine mutation in 5′UTR of Endothelin-1 gene in paediatric cases with severe pulmonary hypertension: An observational study. BMC Res. Notes 2021, 14, 194. [Google Scholar] [CrossRef]
- Gordon, C.T.; Petit, F.; Kroisel, P.M.; Jakobsen, L.; Zechi-Ceide, R.M.; Oufadem, M.; Bole-Feysot, C.; Pruvost, S.; Masson, C.; Tores, F.; et al. Mutations in Endothelin 1 Cause Recessive Auriculocondylar Syndrome and Dominant Isolated Question-Mark Ears. Am. J. Hum. Genet. 2013, 93, 1118–1125. [Google Scholar] [CrossRef]
- Kiando, S.R.; Tucker, N.R.; Castro-Vega, L.-J.; Katz, A.; D’Escamard, V.; Tréard, C.; Fraher, D.; Albuisson, J.; Kadian-Dodov, D.; Ye, Z.; et al. PHACTR1 Is a Genetic Susceptibility Locus for Fibromuscular Dysplasia Supporting Its Complex Genetic Pattern of Inheritance. PLoS Genet. 2016, 12, e1006367. [Google Scholar] [CrossRef]
- Gupta, R.M.; Hadaya, J.; Trehan, A.; Zekavat, S.M.; Roselli, C.; Klarin, D.; Emdin, C.A.; Hilvering, C.R.E.; Bianchi, V.; Mueller, C.; et al. A Genetic Variant Associated with Five Vascular Diseases Is a Distal Regulator of Endothelin-1 Gene Expression. Cell 2017, 170, 522–533.e15. [Google Scholar] [CrossRef] [PubMed]
- Krystek, S.R.; Patel, P.S.; Rose, P.M.; Fisher, S.M.; Kienzle, B.K.; Lach, D.A.; Liu, E.C.; Lynch, J.S.; Novotny, J.; Webb, M.L. Mutation of peptide binding site in transmembrane region of a G protein-coupled receptor accounts for endothelin receptor subtype selectivity. J. Biol. Chem. 1994, 269, 12383–12386. [Google Scholar] [CrossRef]
- Gordon, C.T.; Weaver, K.N.; Zechi-Ceide, R.M.; Madsen, E.C.; Tavares, A.L.P.; Oufadem, M.; Kurihara, Y.; Adameyko, I.; Picard, A.; Breton, S.; et al. Mutations in the Endothelin Receptor Type A Cause Mandibulofacial Dysostosis with Alopecia. Am. J. Hum. Genet. 2015, 96, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Low, S.-K.; Takahashi, A.; Cha, P.-C.; Zembutsu, H.; Kamatani, N.; Kubo, M.; Nakamura, Y. Genome-wide association study for intracranial aneurysm in the Japanese population identifies three candidate susceptible loci and a functional genetic variant at EDNRA. Hum. Mol. Genet. 2012, 21, 2102–2110. [Google Scholar] [CrossRef]
- Yasuno, K.; Bakircioglu, M.; Low, S.-K.; Bilguvar, K.; Gaal, E.; Ruigrok, Y.M.; Niemela, M.; Hata, A.; Bijlenga, P.; Kasuya, H.; et al. Common variant near the endothelin receptor type A (EDNRA) gene is associated with intracranial aneurysm risk. Proc. Natl. Acad. Sci. USA 2011, 108, 19707–19712. [Google Scholar] [CrossRef]
- Hong, E.P.; Kim, B.J.; Jeon, J.P.; Yang, J.S.; Choi, H.J.; Kang, S.H.; Cho, Y.J. Association of Endothelin Receptor Type A with Intracranial Aneurysm in 20,609 East Asians: An Updated Meta-Analysis. World Neurosurg. 2019, 130, e804–e814. [Google Scholar] [CrossRef]
- Tzourio, C.; El Amrani, M.; Poirier, O.; Nicaud, V.; Bousser, M.-G.; Alperovitch, A. Association between migraine and endothelin type A receptor (ETA -231 A/G) gene polymorphism. Neurology 2001, 56, 1273–1277. [Google Scholar] [CrossRef]
- Miao, J.; Wang, F.; Fang, Y. Association of 231G>A polymorphism of endothelin type A receptor gene with migraine: A meta-analysis. J. Neurol. Sci. 2012, 323, 232–235. [Google Scholar] [CrossRef]
- Hofstra, R.M.W.; Osinga, J.; Tan-Sindhunata, G.; Wu, Y.; Kamsteeg, E.-J.; Stulp, R.P.; van Ravenswaaij-Arts, C.; Majoor-Krakauer, D.; Angrist, M.; Chakravarti, A.; et al. A homozygous mutation in the endothelin-3 gene associated with a combined Waardenburg type 2 and Hirschsprung phenotype (Shah-Waardenburg syndrome). Nat. Genet. 1996, 12, 445–447. [Google Scholar] [CrossRef]
- Edery, P.; Attie, T.; Amiel, J.; Pelet, A.; Eng, C.; Hofstra, R.M.W.; Martelli, H.; Bidaud, C.; Munnich, A.; Lyonnet, S. Mutation of the endothelin-3 gene in the Waardenburg-Hirschsprung disease (Shah-Waardenburg syndrome). Nat. Genet. 1996, 12, 442–444. [Google Scholar] [CrossRef]
- Pingault, V.; Bondurand, N.; Lemort, N.; Sancandi, M.; Ceccherini, I.; Hugot, J.-P.; Jouk, P.-S.; Goossens, M. A heterozygous endothelin 3 mutation in Waardenburg-Hirschsprung disease: Is there a dosage effect ofEDN3/EDNRB gene mutations on neurocristopathy phenotypes? J. Med. Genet. 2001, 38, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Auricchio, A.; Casari, G.; Staiano, A.; Ballabio, A. Endothelin-B Receptor Mutations in Patients with Isolated Hirschsprung Disease from a Non-Inbred Population. Hum. Mol. Genet. 1996, 5, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.; Amiel, J.; Claudel, S.; Lyonnet, S.; Corvol, P.; Pinet, F. Functional Characterization of Three Mutations of the Endothelin B Receptor Gene in Patients with Hirschsprung’s Disease: Evidence for Selective Loss of Gi Coupling. Mol. Med. 2001, 7, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Varga, L.; Danis, D.; Drsata, J.; Masindova, I.; Skopkova, M.; Slobodova, Z.; Chrobok, V.; Profant, M.; Gasperikova, D. Novel variants in EDNRB gene in Waardenburg syndrome type II and SOX10 gene in PCWH syndrome. Int. J. Pediatr. Otorhinolaryngol. 2021, 140, 110499. [Google Scholar] [CrossRef]
- Puffenberger, E.G.; Hosoda, K.; Washington, S.S.; Nakao, K.; deWit, D.; Yanagisawa, M.; Chakravarti, A. A missense mutation of the endothelin-B receptor gene in multigenic hirschsprung’s disease. Cell 1994, 79, 1257–1266. [Google Scholar] [CrossRef]
- Wei, F.; Ge, Y.; Li, W.; Wang, X.; Chen, B. Role of endothelin receptor type B (EDNRB) in lung adenocarcinoma. Thorac. Cancer 2020, 11, 1885–1890. [Google Scholar] [CrossRef]
- Smith, S.L.; Damato, B.E.; Scholes, A.G.M.; Nunn, J.; Field, J.K.; Heighway, J. Decreased endothelin receptor B expression in large primary uveal melanomas is associated with early clinical metastasis and short survival. Br. J. Cancer 2002, 87, 1308–1313. [Google Scholar] [CrossRef][Green Version]
- Knight, L.; Gibson, N.; Bujac, S.; Ellison, G.; Growcott, J.; Brooks, N.; Hughes, A.; Xinarianos, G.; Liloglou, T.; Field, J. Hypermethylation of endothelin receptor type B (EDNRB) is a frequent event in non-small cell lung cancer. Cancer Res. 2007, 67, 1135. [Google Scholar]
- Tanaka, T.; Sho, M.; Takayama, T.; Wakatsuki, K.; Matsumoto, S.; Migita, K.; Ito, M.; Hamada, K.; Nakajima, Y. Endothelin B receptor expression correlates with tumour angiogenesis and prognosis in oesophageal squamous cell carcinoma. Br. J. Cancer 2014, 110, 1027–1033. [Google Scholar] [CrossRef]
- Hofstra, R.M.W.; Valdenaire, O.; Arch, E.; Osinga, J.; Kroes, H.; Löffler, B.-M.; Hamosh, A.; Meijers, C.; Buys, C.H.C.M. A Loss-of-Function Mutation in the Endothelin-Converting Enzyme 1 (ECE-1) Associated with Hirschsprung Disease, Cardiac Defects, and Autonomic Dysfunction. Am. J. Hum. Genet. 1999, 64, 304–307. [Google Scholar] [CrossRef][Green Version]
- Funke-Kaiser, H.; Reichenberger, F.; Köpke, K.; Herrmann, S.-M.; Pfeifer, J.; Orzechowski, H.-D.; Zidek, W.; Paul, M.; Brand, E. Differential binding of transcription factor E2F-2 to the endothelin-converting enzyme-1b promoter affects blood pressure regulation. Hum. Mol. Genet. 2003, 12, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, Y.; Kurihara, H.; Suzuki, H.; Kodama, T.; Maemura, K.; Nagai, R.; Oda, H.; Kuwaki, T.; Cao, W.-H.; Kamada, N.; et al. Elevated blood pressure and craniofaclal abnormalities in mice deficient in endothelin-1. Nature 1994, 368, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, Y.; Kurihara, H.; Oda, H.; Maemura, K.; Nagai, R.; Ishikawa, T.; Yazaki, Y. Aortic arch malformations and ventricular septal defect in mice deficient in endothelin-1. J. Clin. Investig. 1995, 96, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, Y.; Kurihara, H.; Maemura, K.; Kuwaki, T.; Kumada, M.; Yazaki, Y. Impaired development of the thyroid and thymus in endothelin-1 knockout mice. J. Cardiovasc. Pharmacol. 1995, 26 (Suppl. 3), S13-6. [Google Scholar] [CrossRef]
- Ieda, M.; Fukuda, K.; Hisaka, Y.; Kimura, K.; Kawaguchi, H.; Fujita, J.; Shimoda, K.; Takeshita, E.; Okano, H.; Kurihara, Y.; et al. Endothelin-1 regulates cardiac sympathetic innervation in the rodent heart by controlling nerve growth factor expression. J. Clin. Investig. 2004, 113, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Hocher, B.; Thöne-Reineke, C.; Rohmeiss, P.; Schmager, F.; Slowinski, T.; Burst, V.; Siegmund, F.; Quertermous, T.; Bauer, C.; Neumayer, H.H.; et al. Endothelin-1 transgenic mice develop glomerulosclerosis, interstitial fibrosis, and renal cysts but not hypertension. J. Clin. Investig. 1997, 99, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Hocher, B.; Rohmeiss, P.; Thöne-Reineke, C.; Schwarz, A.; Burst, V.; van der Woude, F.; Bauer, C. Theuring Apoptosis in Kidneys of Endothelin-1 Transgenic Mice. J. Cardiovasc. Pharmacol. 1998, 31, S554–S556. [Google Scholar] [CrossRef]
- Kalk, P.; Thöne-Reineke, C.; Schwarz, A.; Godes, M.; Bauer, C.; Pfab, T.; Hocher, B. Renal phenotype of Et-1 transgenic mice is modulated by androgens. Eur. J. Med. Res. 2009, 14, 55. [Google Scholar] [CrossRef]
- Shindo, T.; Kurihara, H.; Maemura, K.; Kurihara, Y.; Ueda, O.; Suzuki, H.; Kuwaki, T.; Ju, K.-H.; Wang, Y.; Ebihara, A.; et al. Renal damage and salt-dependent hypertension in aged transgenic mice overexpressing endothelin-1. J. Mol. Med. 2002, 80, 105–116. [Google Scholar] [CrossRef]
- Chang, I.; Bramall, A.N.; Baynash, A.G.; Rattner, A.; Rakheja, D.; Post, M.; Joza, S.; McKerlie, C.; Stewart, D.J.; McInnes, R.R.; et al. Endothelin-2 deficiency causes growth retardation, hypothermia, and emphysema in mice. J. Clin. Investig. 2013, 123, 2643–2653. [Google Scholar] [CrossRef]
- Liefeldt, L.; Schönfelder, G.; Böcker, W.; Hocher, B.; Talsness, C.E.; Rettig, R.; Paul, M. Transgenic rats expressing the human ET-2 gene: A model for the study of endothelin actions in vivo. J. Mol. Med. 1999, 77, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Hocher, B.; Liefeldt, L.; Thöne-Reineke, C.; Orzechowski, H.-D.; Distler, A.; Bauer, C.; Paul, M. Characterization of the Renal Phenotype of Transgenic Rats Expressing the Human Endothelin-2 Gene. Hypertension 1996, 28, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Baynash, A.G.; Hosoda, K.; Giaid, A.; Richardson, J.A.; Emoto, N.; Hammer, R.E.; Yanagisawa, M. Interaction of endothelin-3 with endothelin-B receptor is essential for development of epidermal melanocytes and enteric neurons. Cell 1994, 79, 1277–1285. [Google Scholar] [CrossRef]
- Rice, J.; Doggett, B.; Sweetser, D.A.; Yanagisawa, H.; Yanagisawa, M.; Kapur, R.P. Transgenic rescue of aganglionosis and piebaldism in lethal spotted mice. Dev. Dyn. 2000, 217, 120–132. [Google Scholar] [CrossRef]
- Clouthier, D.E.; Hosoda, K.; Richardson, J.A.; Williams, S.C.; Yanagisawa, H.; Kuwaki, T.; Kumada, M.; Hammer, R.E.; Yanagisawa, M. Cranial and cardiac neural crest defects in endothelin-A receptor-deficient mice. Development 1998, 125, 813–824. [Google Scholar] [CrossRef]
- Yanagisawa, H.; Hammer, R.E.; Richardson, J.A.; Williams, S.C.; Clouthier, D.E.; Yanagisawa, M. Role of Endothelin-1/Endothelin-A receptor-mediated signaling pathway in the aortic arch patterning in mice. J. Clin. Investig. 1998, 102, 22–33. [Google Scholar] [CrossRef]
- Hosoda, K.; Hammer, R.E.; Richardson, J.A.; Baynash, A.G.; Cheung, J.C.; Giaid, A.; Yanagisawa, M. Targeted and natural (piebald-lethal) mutations of endothelin-B receptor gene produce megacolon associated with spotted coat color in mice. Cell 1994, 79, 1267–1276. [Google Scholar] [CrossRef]
- Yanagisawa, H.; Yanagisawa, M.; Kapur, R.P.; Richardson, J.A.; Williams, S.C.; Clouthier, D.E.; de Wit, D.; Emoto, N.; Hammer, R.E. Dual genetic pathways of endothelin-mediated intercellular signaling revealed by targeted disruption of endothelin converting enzyme-1 gene. Development 1998, 125, 825–836. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endor. Eur. Respir. J. 2015, 46, 903–975. [Google Scholar] [CrossRef]
- Tuder, R.M. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res. 2017, 367, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Archer, S.L.; Dorfmuller, P.; Erzurum, S.C.; Guignabert, C.; Michelakis, E.; Rabinovitch, M.; Schermuly, R.; Stenmark, K.R.; Morrell, N.W. Relevant issues in the pathology and pathobiology of pulmonary hyerptension. J. Am. Coll. Cardiol. 2014, 62, D4–D12. [Google Scholar] [CrossRef]
- Miyauchi, T.; Yorikane, R.; Sakai, S.; Sakurai, T.; Okada, M.; Nishikibe, M.; Yano, M.; Yamaguchi, I.; Sugishita, Y.; Goto, K. Contribution of endogenous endothelin-1 to the progression of cardiopulmonary alterations in rats with monocrotaline-induced pulmonary hypertension. Circ. Res. 1993, 73, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, S.J.; Chen, Y.F.; Meng, Q.C.; Durand, J.; Oparil, S.; Elton, T.S. Enhanced endothelin-1 and endothelin receptor gene expression in chronic hypoxia. J. Appl. Physiol. 1994, 77, 1451–1459. [Google Scholar] [CrossRef]
- Kang, B.-Y.; Park, K.K.; Kleinhenz, J.M.; Murphy, T.C.; Green, D.E.; Bijli, K.M.; Yeligar, S.M.; Carthan, K.A.; Searles, C.D.; Sutliff, R.L.; et al. Peroxisome Proliferator–Activated Receptor γ and microRNA 98 in Hypoxia-Induced Endothelin-1 Signaling. Am. J. Respir. Cell Mol. Biol. 2016, 54, 136–146. [Google Scholar] [CrossRef]
- Amiri, F.; Virdis, A.; Neves, M.F.; Iglarz, M.; Seidah, N.G.; Touyz, R.M.; Reudelhuber, T.L.; Schiffrin, E.L. Endothelium-Restricted Overexpression of Human Endothelin-1 Causes Vascular Remodeling and Endothelial Dysfunction. Circulation 2004, 110, 2233–2240. [Google Scholar] [CrossRef]
- Lambers, C.; Roth, M.; Zhong, J.; Campregher, C.; Binder, P.; Burian, B.; Petkov, V.; Block, L.-H. The Interaction of Endothelin-1 and TGF-β1 Mediates Vascular Cell Remodeling. PLoS ONE 2013, 8, e73399. [Google Scholar] [CrossRef]
- Bourque, S.L.; Davidge, S.T.; Adams, M.A. The interaction between endothelin-1 and nitric oxide in the vasculature: New perspectives. Am. J. Physiol. Integr. Comp. Physiol. 2011, 300, R1288–R1295. [Google Scholar] [CrossRef]
- Galie, N. The endothelin system in pulmonary arterial hypertension. Cardiovasc. Res. 2004, 61, 227–237. [Google Scholar] [CrossRef]
- Miyagawa, K.; Emoto, N. Current state of endothelin receptor antagonism in hypertension and pulmonary hypertension. Ther. Adv. Cardiovasc. Dis. 2014, 8, 202–216. [Google Scholar] [CrossRef]
- Maruyama, H.; Sakai, S.; Dewachter, L.; Dewachter, C.; Rondelet, B.; Naeije, R.; Ieda, M. Endothelin-1 Induces Lysyl Oxidase Expression in Pulmonary Artery Smooth Muscle Cells. Can. J. Physiol. Pharmacol. 2020, 98, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Wedgwood, S.; McMullan, D.M.; Bekker, J.M.; Fineman, J.R.; Black, S.M. Role for Endothelin-1–Induced Superoxide and Peroxynitrite Production in Rebound Pulmonary Hypertension Associated with Inhaled Nitric Oxide Therapy. Circ. Res. 2001, 89, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Biasin, V.; Chwalek, K.; Wilhelm, J.; Best, J.; Marsh, L.M.; Ghanim, B.; Klepetko, W.; Fink, L.; Schermuly, R.T.; Weissmann, N.; et al. Endothelin-1 driven proliferation of pulmonary arterial smooth muscle cells is c-fos dependent. Int. J. Biochem. Cell Biol. 2014, 54, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Kumar, S.; Sharma, S.; Aggarwal, S.; Lu, Q.; Gross, C.; Rafikova, O.; Lee, S.G.; Dasarathy, S.; Hou, Y.; et al. Endothelin-1 Induces a Glycolytic Switch in Pulmonary Arterial Endothelial Cells via the Mitochondrial Translocation of Endothelial Nitric Oxide Synthase. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1084–1095. [Google Scholar] [CrossRef] [PubMed]
- Giaid, A.; Yanagisawa, M.; Langleben, D.; Michel, R.P.; Levy, R.; Shennib, H.; Kimura, S.; Masaki, T.; Duguid, W.P.; Stewart, D.J. Expression of Endothelin-1 in the Lungs of Patients with Pulmonary Hypertension. N. Engl. J. Med. 1993, 328, 1732–1739. [Google Scholar] [CrossRef]
- Cacoub, P. Endothelin-1 in the lungs of patients with pulmonary hypertension. Cardiovasc. Res. 1997, 33, 196–200. [Google Scholar] [CrossRef]
- Çelik, G.; Karabiyikoglu, G. Local and Peripheral Plasma Endothelin-1 in Pulmonary Hypertension Secondary to Chronic Obstructive Pulmonary Disease. Respiration 1998, 65, 289–294. [Google Scholar] [CrossRef]
- Chowdhury, M.A.; Moukarbel, G.V.; Gupta, R.; Frank, S.M.; Anderson, A.M.; Liu, L.C.; Khouri, S.J. Endothelin 1 Is Associated with Heart Failure Hospitalization and Long-Term Mortality in Patients with Heart Failure with Preserved Ejection Fraction and Pulmonary Hypertension. Cardiology 2019, 143, 124–133. [Google Scholar] [CrossRef]
- Reesink, H.J.; Meijer, R.C.; Lutter, R.; Boomsma, F.; Jansen, H.M.; Kloek, J.J.; Bresser, P. Hemodynamic and Clinical Correlates of Endothelin-1 in Chronic Thromboembolic Pulmonary Hypertension. Circ. J. 2006, 70, 1058–1063. [Google Scholar] [CrossRef]
- Rubens, C.; Ewert, R.; Halank, M.; Wensel, R.; Orzechowski, H.-D.; Schultheiss, H.-P.; Hoeffken, G. Big Endothelin-1 and Endothelin-1 Plasma Levels Are Correlated with the Severity of Primary Pulmonary Hypertension. Chest 2001, 120, 1562–1569. [Google Scholar] [CrossRef]
- Correale, M.; Ferraretti, A.; Monaco, I.; Grazioli, D.; Di Biase, M.; Brunetti, N.D. Endothelin-receptor antagonists in the management of pulmonary arterial hypertension: Where do we stand? Vasc. Health Risk Manag. 2018, 14, 253–264. [Google Scholar] [CrossRef]
- Traiger, G.L. Pulmonary arterial hypertension. Crit. Care Nurs. Q. 2007, 30, 20–43. [Google Scholar] [CrossRef] [PubMed]
- Degano, B.; Yaici, A.; Le Pavec, J.; Savale, L.; Jais, X.; Camara, B.; Humbert, M.; Simonneau, G.; Sitbon, O. Long-term effects of bosentan in patients with HIV-associated pulmonary arterial hypertension. Eur. Respir. J. 2009, 33, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Savale, L.; Magnier, R.; Le Pavec, J.; Jaïs, X.; Montani, D.; O’Callaghan, D.S.; Humbert, M.; Dingemanse, J.; Simonneau, G.; Sitbon, O. Efficacy, safety and pharmacokinetics of bosentan in portopulmonary hypertension. Eur. Respir. J. 2013, 41, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M. Bosentan therapy for portopulmonary hypertension. Eur. Respir. J. 2005, 25, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Ivy, D.; Wilson, N. Tale of 2 Endothelin Receptor Antagonists in Eisenmenger Syndrome. Circulation 2019, 139, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Galieè, N.; Beghetti, M.; Gatzoulis, M.A.; Granton, J.; Berger, R.M.F.; Lauer, A.; Chiossi, E.; Landzberg, M. Bosentan Therapy in Patients with Eisenmenger Syndrome. Circulation 2006, 114, 48–54. [Google Scholar] [CrossRef]
- Pulido, T.; Adzerikho, I.; Channick, R.N.; Delcroix, M.; Galiè, N.; Ghofrani, H.-A.; Jansa, P.; Jing, Z.-C.; Le Brun, F.-O.; Mehta, S.; et al. Macitentan and Morbidity and Mortality in Pulmonary Arterial Hypertension. N. Engl. J. Med. 2013, 369, 809–818. [Google Scholar] [CrossRef]
- Vonk Noordegraaf, A.; Channick, R.; Cottreel, E.; Kiely, D.G.; Marcus, J.T.; Martin, N.; Moiseeva, O.; Peacock, A.; Swift, A.J.; Tawakol, A.; et al. The REPAIR Study. JACC Cardiovasc. Imaging 2022, 15, 240–253. [Google Scholar] [CrossRef]
- Galiè, N.; Olschewski, H.; Oudiz, R.J.; Torres, F.; Frost, A.; Ghofrani, H.A.; Badesch, D.B.; McGoon, M.D.; McLaughlin, V.V.; Roecker, E.B.; et al. Ambrisentan for the Treatment of Pulmonary Arterial Hypertension. Circulation 2008, 117, 3010–3019. [Google Scholar] [CrossRef]
- Galiè, N.; Barberà, J.A.; Frost, A.E.; Ghofrani, H.-A.; Hoeper, M.M.; McLaughlin, V.V.; Peacock, A.J.; Simonneau, G.; Vachiery, J.-L.; Grünig, E.; et al. Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension. N. Engl. J. Med. 2015, 373, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.M.; Sitbon, O.; Doelberg, M.; Feldman, J.; Gibbs, J.S.R.; Grünig, E.; Hoeper, M.M.; Martin, N.; Mathai, S.C.; McLaughlin, V.V.; et al. Three- Versus Two-Drug Therapy for Patients with Newly Diagnosed Pulmonary Arterial Hypertension. J. Am. Coll. Cardiol. 2021, 78, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Gatzoulis, M.A.; Landzberg, M.; Beghetti, M.; Berger, R.M.; Efficace, M.; Gesang, S.; He, J.; Papadakis, K.; Pulido, T.; Galiè, N. Evaluation of Macitentan in Patients with Eisenmenger Syndrome. Circulation 2019, 139, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.R.; Merino, A.; Haefeli, W.E.; Miranda, C.; Prats, M.; Bancu, I.; Bailón, L.; Moltó, J. Ambrisentan use in a HIV-1 infected patient with end-stage renal disease and pulmonary hypertension: Minimal removal by hemodialysis—A case report. BMC Nephrol. 2020, 21, 24. [Google Scholar] [CrossRef]
- Sitbon, O.; Bosch, J.; Cottreel, E.; Csonka, D.; de Groote, P.; Hoeper, M.M.; Kim, N.H.; Martin, N.; Savale, L.; Krowka, M. Macitentan for the treatment of portopulmonary hypertension (PORTICO): A multicentre, randomised, double-blind, placebo-controlled, phase 4 trial. Lancet Respir. Med. 2019, 7, 594–604. [Google Scholar] [CrossRef]
- Stolz, D.; Rasch, H.; Linka, A.; Di Valentino, M.; Meyer, A.; Brutsche, M.; Tamm, M. A randomised, controlled trial of bosentan in severe COPD. Eur. Respir. J. 2008, 32, 619–628. [Google Scholar] [CrossRef]
- King, T.E.; Brown, K.K.; Raghu, G.; du Bois, R.M.; Lynch, D.A.; Martinez, F.; Valeyre, D.; Leconte, I.; Morganti, A.; Roux, S.; et al. BUILD-3: A Randomized, Controlled Trial of Bosentan in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 92–99. [Google Scholar] [CrossRef]
- Raghu, G. Treatment of Idiopathic Pulmonary Fibrosis with Ambrisentan. Ann. Intern. Med. 2013, 158, 641. [Google Scholar] [CrossRef]
- Soraya, A.I.; Suzuki, Y.; Morimoto, M.; Ko, C.J.; Ikeda, K.; Hirata, K.-I.; Emoto, N. Protective Effects of Endothelin-2 Expressed in Epithelial Cells on Bleomycin-Induced Pulmonary Fibrosis in Mice. Kobe J. Med. Sci. 2021, 67, E61–E70. [Google Scholar]
- Jiang, B.H.; Tardif, J.-C.; Shi, Y.; Dupuis, J. Bosentan does not improve pulmonary hypertension and lung remodeling in heart failure. Eur. Respir. J. 2011, 37, 578–586. [Google Scholar] [CrossRef]
- Denault, A.Y.; Pearl, R.G.; Michler, R.E.; Rao, V.; Tsui, S.S.L.; Seitelberger, R.; Cromie, M.; Lindberg, E.; D’Armini, A.M. Tezosentan and Right Ventricular Failure in Patients with Pulmonary Hypertension Undergoing Cardiac Surgery: The TACTICS Trial. J. Cardiothorac. Vasc. Anesth. 2013, 27, 1212–1217. [Google Scholar] [CrossRef] [PubMed]
- Vachiéry, J.-L.; Delcroix, M.; Al-Hiti, H.; Efficace, M.; Hutyra, M.; Lack, G.; Papadakis, K.; Rubin, L.J. Macitentan in pulmonary hypertension due to left ventricular dysfunction. Eur. Respir. J. 2018, 51, 1701886. [Google Scholar] [CrossRef] [PubMed]
- Jaïs, X.; D’Armini, A.M.; Jansa, P.; Torbicki, A.; Delcroix, M.; Ghofrani, H.A.; Hoeper, M.M.; Lang, I.M.; Mayer, E.; Pepke-Zaba, J.; et al. Bosentan for Treatment of Inoperable Chronic Thromboembolic Pulmonary Hypertension. J. Am. Coll. Cardiol. 2008, 52, 2127–2134. [Google Scholar] [CrossRef]
- Ghofrani, H.-A.; Simonneau, G.; D’Armini, A.M.; Fedullo, P.; Howard, L.S.; Jaïs, X.; Jenkins, D.P.; Jing, Z.-C.; Madani, M.M.; Martin, N.; et al. Macitentan for the treatment of inoperable chronic thromboembolic pulmonary hypertension (MERIT-1): Results from the multicentre, phase 2, randomised, double-blind, placebo-controlled study. Lancet Respir. Med. 2017, 5, 785–794. [Google Scholar] [CrossRef]
- Sakurai, S.; Ukyo, Y. Results of Macitentan in Japanese Patients with Chronic Thromboembolic Pulmonary Hypertension—A Prospective, Multicenter, Open-Label, Single-Arm, Phase 3 Study. Circ. Rep. 2021, 3, CR-21-0034. [Google Scholar] [CrossRef] [PubMed]
- Southwood, M.; MacKenzie Ross, R.V.; Kuc, R.E.; Hagan, G.; Sheares, K.K.; Jenkins, D.P.; Goddard, M.; Davenport, A.P.; Pepke-Zaba, J. Endothelin ET A receptors predominate in chronic thromboembolic pulmonary hypertension. Life Sci. 2016, 159, 104–110. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kisanuki, Y.Y.; Emoto, N.; Ohuchi, T.; Widyantoro, B.; Yagi, K.; Nakayama, K.; Kedzierski, R.M.; Hammer, R.E.; Yanagisawa, H.; Williams, S.C.; et al. Low Blood Pressure in Endothelial Cell–Specific Endothelin 1 Knockout Mice. Hypertension 2010, 56, 121–128. [Google Scholar] [CrossRef] [PubMed]
- McEniery, C.M.; Qasem, A.; Schmitt, M.; Avolio, A.P.; Cockcroft, J.R.; Wilkinson, I.B. Endothelin-1 regulates arterial pulse wave velocity in vivo. J. Am. Coll. Cardiol. 2003, 42, 1975–1981. [Google Scholar] [CrossRef]
- Wilkinson, I.B.; Franklin, S.S.; Cockcroft, J.R. Nitric Oxide and the Regulation of Large Artery Stiffness. Hypertension 2004, 44, 112–116. [Google Scholar] [CrossRef]
- du Plooy, C.S.; Mels, C.M.C.; Huisman, H.W.; Kruger, R. The Association of Endothelin-1 with Markers of Arterial Stiffness in Black South African Women: The SABPA Study. J. Amino Acids 2015, 2015, 481517. [Google Scholar] [CrossRef]
- Kostov, K. The Causal Relationship between Endothelin-1 and Hypertension: Focusing on Endothelial Dysfunction, Arterial Stiffness, Vascular Remodeling, and Blood Pressure Regulation. Life 2021, 11, 986. [Google Scholar] [CrossRef] [PubMed]
- Fryer, R.M.; Rakestraw, P.A.; Banfor, P.N.; Cox, B.F.; Opgenorth, T.J.; Reinhart, G.A. Blood pressure regulation by ET A and ET B receptors in conscious, telemetry-instrumented mice and role of ET A in hypertension produced by selective ET B blockade. Am. J. Physiol. Circ. Physiol. 2006, 290, H2554–H2559. [Google Scholar] [CrossRef] [PubMed]
- Bagnall, A.J.; Kelland, N.F.; Gulliver-Sloan, F.; Davenport, A.P.; Gray, G.A.; Yanagisawa, M.; Webb, D.J.; Kotelevtsev, Y.V. Deletion of Endothelial Cell Endothelin B Receptors Does Not Affect Blood Pressure or Sensitivity to Salt. Hypertension 2006, 48, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Boesen, E.I.; Pollock, D.M. Cooperative role of ET A and ET B receptors in mediating the diuretic response to intramedullary hyperosmotic NaCl infusion. Am. J. Physiol. Physiol. 2010, 299, F1424–F1432. [Google Scholar] [CrossRef]
- Krum, H.; Viskoper, R.J.; Lacourciere, Y.; Budde, M.; Charlon, V. The Effect of an Endothelin-Receptor Antagonist, Bosentan, on Blood Pressure in Patients with Essential Hypertension. N. Engl. J. Med. 1998, 338, 784–791. [Google Scholar] [CrossRef]
- Nakov, R.; Pfarr, E.; Eberle, S.; HEAT Investigators. Darusentan: An effective endothelinA receptor antagonist for treatment of hypertension. Am. J. Hypertens. 2002, 15, 583–589. [Google Scholar] [CrossRef]
- Webb, D.J. DORADO: Opportunity Postponed. Hypertension 2010, 56, 806–807. [Google Scholar] [CrossRef]
- Weber, M.A.; Black, H.; Bakris, G.; Krum, H.; Linas, S.; Weiss, R.; Linseman, J.V.; Wiens, B.L.; Warren, M.S.; Lindholm, L.H. A selective endothelin-receptor antagonist to reduce blood pressure in patients with treatment-resistant hypertension: A randomised, double-blind, placebo-controlled trial. Lancet 2009, 374, 1423–1431. [Google Scholar] [CrossRef]
- Bakris, G.L.; Lindholm, L.H.; Black, H.R.; Krum, H.; Linas, S.; Linseman, J.V.; Arterburn, S.; Sager, P.; Weber, M. Divergent Results Using Clinic and Ambulatory Blood Pressures. Hypertension 2010, 56, 824–830. [Google Scholar] [CrossRef]
- Trachtman, H.; Nelson, P.; Adler, S.; Campbell, K.N.; Chaudhuri, A.; Derebail, V.K.; Gambaro, G.; Gesualdo, L.; Gipson, D.S.; Hogan, J.; et al. DUET: A Phase 2 Study Evaluating the Efficacy and Safety of Sparsentan in Patients with FSGS. J. Am. Soc. Nephrol. 2018, 29, 2745–2754. [Google Scholar] [CrossRef]
- Nicoletti, A.; Michel, J. Cardiac fibrosis and inflammation: Interaction with hemodynamic and hormonal factors. Cardiovasc. Res. 1999, 41, 532–543. [Google Scholar] [CrossRef]
- Widyantoro, B.; Emoto, N.; Nakayama, K.; Anggrahini, D.W.; Adiarto, S.; Iwasa, N.; Yagi, K.; Miyagawa, K.; Rikitake, Y.; Suzuki, T.; et al. Endothelial Cell–Derived Endothelin-1 Promotes Cardiac Fibrosis in Diabetic Hearts Through Stimulation of Endothelial-to-Mesenchymal Transition. Circulation 2010, 121, 2407–2418. [Google Scholar] [CrossRef] [PubMed]
- Shin, A.N.; Dasgupta, C.; Zhang, G.; Seal, K.; Zhang, L. Proteomic Analysis of Endothelin-1 Targets in the Regulation of Cardiomyocyte Proliferation. Curr. Top. Med. Chem. 2017, 17, 1788–1802. [Google Scholar] [CrossRef] [PubMed]
- Pikkarainen, S.; Tokola, H.; Majalahti-Palviainen, T.; Kerkelä, R.; Hautala, N.; Bhalla, S.S.; Charron, F.; Nemer, M.; Vuolteenaho, O.; Ruskoaho, H. GATA-4 is a nuclear mediator of mechanical stretch-activated hypertrophic program. J. Biol. Chem. 2003, 278, 23807–23816. [Google Scholar] [CrossRef]
- Thibault, G.; Doubell, A.F.; Garcia, R.; Larivière, R.; Schiffrin, E.L. Endothelin-stimulated secretion of natriuretic peptides by rat atrial myocytes is mediated by endothelin A receptors. Circ. Res. 1994, 74, 460–470. [Google Scholar] [CrossRef]
- Konstam, M.A.; DeNofrio, D. Endothelin Expression and the Progression of Heart Failure. Circulation 2004, 109, 143–145. [Google Scholar] [CrossRef]
- Abukar, Y.; May, C.N.; Ramchandra, R. Role of endothelin-1 in mediating changes in cardiac sympathetic nerve activity in heart failure. Am. J. Physiol. Integr. Comp. Physiol. 2016, 310, R94–R99. [Google Scholar] [CrossRef]
- Yang, L.L.; Gros, R.; Kabir, M.G.; Sadi, A.; Gotlieb, A.I.; Husain, M.; Stewart, D.J. Conditional Cardiac Overexpression of Endothelin-1 Induces Inflammation and Dilated Cardiomyopathy in Mice. Circulation 2004, 109, 255–261. [Google Scholar] [CrossRef]
- Gu, M.; Shao, N.Y.; Sa, S.; Li, D.; Termglinchan, V.; Ameen, M.; Karakikes, I.; Sosa, G.; Grubert, F.; Lee, J.; et al. Patient-Specific iPSC-Derived Endothelial Cells Uncover Pathways that Protect against Pulmonary Hypertension in BMPR2 Mutation Carriers. Cell Stem Cell 2017, 20, 490–504.e5. [Google Scholar] [CrossRef]
- Heiden, S.; Vignon-Zellweger, N.; Masuda, S.; Yagi, K.; Nakayama, K.; Yanagisawa, M.; Emoto, N. Vascular endothelium derived endothelin-1 is required for normal heart function after chronic pressure overload in mice. PLoS ONE 2014, 9, e88730. [Google Scholar] [CrossRef]
- Borgeson, D.D.; Grantham, J.A.; Williamson, E.E.; Luchner, A.; Redfield, M.M.; Opgenorth, T.J.; Burnett, J.C. Chronic Oral Endothelin Type A Receptor Antagonism in Experimental Heart Failure. Hypertension 1998, 31, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Wada, A.; Tsutamoto, T.; Ohnishi, M.; Sawaki, M.; Fukai, D.; Maeda, Y.; Kinoshita, M. Effects of a Specific Endothelin-Converting Enzyme Inhibitor on Cardiac, Renal, and Neurohumoral Functions in Congestive Heart Failure. Circulation 1999, 99, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Tonnessen, T.; Christensen, G.; Oie, E.; Holt, E.; Kjekshus, H.; Smiseth, O.; Sejersted, O.; Attramadal, H. Increased cardiac expression of endothelin-1 mRNA in ischemic heart failure in rats. Cardiovasc. Res. 1997, 33, 601–610. [Google Scholar] [CrossRef]
- Sakai, S.; Miyauchi, T.; Kobayashi, M.; Yamaguchi, I.; Goto, K.; Sugishita, Y. Inhibition of myocardial endothelin pathway improves long-term survival in heart failure. Nature 1996, 384, 353–355. [Google Scholar] [CrossRef]
- Fraccarollo, D.; Hu, K.; Galuppo, P.; Gaudron, P.; Ertl, G. Chronic Endothelin Receptor Blockade Attenuates Progressive Ventricular Dilation and Improves Cardiac Function in Rats with Myocardial Infarction. Circulation 1997, 96, 3963–3973. [Google Scholar] [CrossRef]
- Vignon-zellweger, N.; Heiden, S.; Miyauchi, T.; Emoto, N. Endothelin and endothelin receptors in the renal and cardiovascular systems. Life Sci. 2012, 91, 490–500. [Google Scholar] [CrossRef]
- Gottlieb, S.S.; Harris, K.; Todd, J.; Estis, J.; Christenson, R.H.; Torres, V.; Whittaker, K.; Rebuck, H.; Wawrzyniak, A.; Krantz, D.S. Prognostic significance of active and modified forms of endothelin 1 in patients with heart failure with reduced ejection fraction. Clin. Biochem. 2015, 48, 292–296. [Google Scholar] [CrossRef]
- He, X.; Guo, H.; Xu, J. Endothelin 1: A Potential Prognostic Biomarker for Heart Failure with Preserved Ejection Fraction and Pulmonary Hypertension? Cardiology 2020, 145, 262. [Google Scholar] [CrossRef]
- Pek, S.L.T.; Lim, S.C.; Ang, K.; Kwan, P.Y.; Tang, W.E.; Sum, C.F.; Tavintharan, S. Endothelin-1 predicts incident diabetic peripheral neuropathy in Type 2 Diabetes: A cohort study. Eur. J. Endocrinol. 2020, 182, 429–438. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, Y.; Zou, Y.; Wang, D.; Zhu, L.; Tian, T.; Wang, J.; Bao, J.; Hui, R.; Kang, L.; et al. Plasma level of big endothelin-1 predicts the prognosis in patients with hypertrophic cardiomyopathy. Int. J. Cardiol. 2017, 243, 283–289. [Google Scholar] [CrossRef]
- Zymliński, R.; Sierpiński, R.; Metra, M.; Cotter, G.; Sokolski, M.; Siwołowski, P.; Garus, M.; Gajewski, P.; Tryba, J.; Samorek, M.; et al. Elevated plasma endothelin-1 is related to low natriuresis, clinical signs of congestion, and poor outcome in acute heart failure. ESC Heart Fail. 2020, 7, 3536–3544. [Google Scholar] [CrossRef] [PubMed]
- Mo, R.; Yang, Y.; Yu, L.; Tan, H.; Zhu, J. Elevated Plasma Big Endothelin-1 at Admission Is Associated with Poor Short-Term Outcomes in Patients with Acute Decompensated Heart Failure. Front. Cardiovasc. Med. 2021, 8, 101. [Google Scholar] [CrossRef] [PubMed]
- Gombos, T.; Förhécz, Z.; Pozsonyi, Z.; Wallentin, S.; Papassotiriou, J.; Kunde, J.; Morgenthaler, N.G.; Jánoskuti, L.; Prohászka, Z. Adrenomedullin and endothelin-1 are related to inflammation in chronic heart failure. Inflamm. Res. 2009, 58, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Sikkeland, L.I.B.; Dahl, C.P.; Ueland, T.; Andreassen, A.K.; Gude, E.; Edvardsen, T.; Holm, T.; Yndestad, A.; Gullestad, L.; Kongerud, J.; et al. Increased Levels of Inflammatory Cytokines and Endothelin-1 in Alveolar Macrophages from Patients with Chronic Heart Failure. PLoS ONE 2012, 7, e36815. [Google Scholar] [CrossRef]
- Ward, R.; Ergul, A. Relationship of endothelin-1 and NLRP3 inflammasome activation in HT22 hippocampal cells in diabetes. Life Sci. 2016, 159, 97–103. [Google Scholar] [CrossRef]
- Dow, C.A.; Templeton, D.L.; Lincenberg, G.M.; Greiner, J.J.; Stauffer, B.L.; DeSouza, C.A. Elevations in C-reactive protein and endothelin-1 system activity in humans. Life Sci. 2016, 159, 66–70. [Google Scholar] [CrossRef][Green Version]
- Abbate, A.; Toldo, S.; Marchetti, C.; Kron, J.; Van Tassell, B.W.; Dinarello, C.A. Interleukin-1 and the Inflammasome as Therapeutic Targets in Cardiovascular Disease. Circ. Res. 2020, 126, 1260–1280. [Google Scholar] [CrossRef]
- Świątkiewicz, I.; Magielski, P.; Kubica, J. C-Reactive Protein as a Risk Marker for Post-Infarct Heart Failure over a Multi-Year Period. Int. J. Mol. Sci. 2021, 22, 3169. [Google Scholar] [CrossRef]
- Mylona, P.; Cleland, J.G.F. Update of REACH-1 and MERIT-HF clinical trials in heart failure. Eur. J. Heart Fail. 1999, 1, 197–200. [Google Scholar] [CrossRef]
- Packer, M.; McMurray, J.J.V.; Krum, H.; Kiowski, W.; Massie, B.M.; Caspi, A.; Pratt, C.M.; Petrie, M.C.; DeMets, D.; Kobrin, I.; et al. Long-Term Effect of Endothelin Receptor Antagonism with Bosentan on the Morbidity and Mortality of Patients with Severe Chronic Heart Failure. JACC Heart Fail. 2017, 5, 317–326. [Google Scholar] [CrossRef]
- Anand, I.; McMurray, J.; Cohn, J.N.; Konstam, M.A.; Notter, T.; Quitzau, K.; Ruschitzka, F.; Lüscher, T.F. Long-term effects of darusentan on left-ventricular remodelling and clinical outcomes in the Endothelin A Receptor Antagonist Trial in Heart Failure (EARTH): Randomised, double-blind, placebo-controlled trial. Lancet 2004, 364, 347–354. [Google Scholar] [CrossRef]
- Waijer, S.W.; Gansevoort, R.T.; Bakris, G.L.; Correa-Rotter, R.; Hou, F.-F.; Kohan, D.E.; Kitzman, D.W.; Makino, H.; McMurray, J.J.V.; Perkovic, V.; et al. The Effect of Atrasentan on Kidney and Heart Failure Outcomes by Baseline Albuminuria and Kidney Function. Clin. J. Am. Soc. Nephrol. 2021, 16, 1824–1832. [Google Scholar] [CrossRef] [PubMed]
- Coletta, A.P.; Cleland, J.G.F. Clinical trials update: Highlights of the scientific sessions of the XXIII Congress of the European Society of Cardiology—WARIS II, ESCAMI, PAFAC, RITZ-1 and TIME. Eur. J. Heart Fail. 2001, 3, 747–750. [Google Scholar] [CrossRef]
- Louis, A.; Cleland, J.G.F.; Crabbe, S.; Ford, S.; Thackray, S.; Houghton, T.; Clark, A. Clinical Trials Update: CAPRICORN, COPERNICUS, MIRACLE, STAF, RITZ-2, RECOVER and RENAISSANCE and cachexia and cholesterol in heart failure. Highlights of the Scientific Sessions of the American College of Cardiology, 2001. Eur. J. Heart Fail. 2001, 3, 381–387. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Teerlink, J.R.; Cotter, G.; Bourge, R.C.; Cleland, J.G.F.; Jondeau, G.; Krum, H.; Metra, M.; O’Connor, C.M.; Parker, J.D.; et al. Effects of Tezosentan on Symptoms and Clinical Outcomes in Patients with Acute Heart Failure. JAMA 2007, 298, 2009. [Google Scholar] [CrossRef]
- Sutton, G.; Pugh, D.; Dhaun, N. Developments in the Role of Endothelin-1 in Atherosclerosis: A Potential Therapeutic Target? Am. J. Hypertens. 2019, 32, 813–815. [Google Scholar] [CrossRef]
- Böhm, F.; Pernow, J. The importance of endothelin-1 for vascular dysfunction in cardiovascular disease. Cardiovasc. Res. 2007, 76, 8–18. [Google Scholar] [CrossRef]
- Jaguszewski, M.; Osipova, J.; Ghadri, J.-R.; Napp, L.C.; Widera, C.; Franke, J.; Fijalkowski, M.; Nowak, R.; Fijalkowska, M.; Volkmann, I.; et al. A signature of circulating microRNAs differentiates takotsubo cardiomyopathy from acute myocardial infarction. Eur. Heart J. 2014, 35, 999–1006. [Google Scholar] [CrossRef]
- Campia, U.; Tesauro, M.; Di Daniele, N.; Cardillo, C. The vascular endothelin system in obesity and type 2 diabetes: Pathophysiology and therapeutic implications. Life Sci. 2014, 118, 149–155. [Google Scholar] [CrossRef]
- Pernow, J.; Shemyakin, A.; Böhm, F. New perspectives on endothelin-1 in atherosclerosis and diabetes mellitus. Life Sci. 2012, 91, 507–516. [Google Scholar] [CrossRef]
- Pernow, J.; Wang, Q. Endothelin in myocardial ischaemia and reperfusion. Cardiovasc. Res. 1997, 33, 518–526. [Google Scholar] [CrossRef][Green Version]
- Ryu, S.M.; Kim, H.J.; Cho, K.R.; Jo, W.-M. Myocardial Protective Effect of Tezosentan, an Endothelin Receptor Antagonist, for Ischemia-Reperfusion Injury in Experimental Heart Failure Models. J. Korean Med. Sci. 2009, 24, 782. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, Y.-J.; Wang, X.; Xu, L.; Yang, X.-C.; Zhao, W.-S. PKC-Mediated Endothelin-1 Expression in Endothelial Cell Promotes Macrophage Activation in Atherogenesis. Am. J. Hypertens. 2019, 32, 880–889. [Google Scholar] [CrossRef]
- Molet, S.; Furukawa, K.; Maghazechi, A.; Hamid, Q.; Giaid, A. Chemokine- and cytokine-induced expression of endothelin 1 and endothelin-converting enzyme 1 in endothelial cells. J. Allergy Clin. Immunol. 2000, 105, 333–338. [Google Scholar] [CrossRef]
- Li, M.W.; Mian, M.O.R.; Barhoumi, T.; Rehman, A.; Mann, K.; Paradis, P.; Schiffrin, E.L. Endothelin-1 Overexpression Exacerbates Atherosclerosis and Induces Aortic Aneurysms in Apolipoprotein E Knockout Mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2306–2315. [Google Scholar] [CrossRef] [PubMed]
- Świątkiewicz, I.; Magielski, P.; Kubica, J.; Zadourian, A.; DeMaria, A.N.; Taub, P.R. Enhanced Inflammation is a Marker for Risk of Post-Infarct Ventricular Dysfunction and Heart Failure. Int. J. Mol. Sci. 2020, 21, 807. [Google Scholar] [CrossRef] [PubMed]
- Olivier, A.; Girerd, N.; Michel, J.; Ketelslegers, J.; Fay, R.; Vincent, J.; Bramlage, P.; Pitt, B.; Zannad, F.; Rossignol, P. Combined baseline and one-month changes in big endothelin-1 and brain natriuretic peptide plasma concentrations predict clinical outcomes in patients with left ventricular dysfunction after acute myocardial infarction: Insights from the Eplerenone Post-Ac. Int. J. Cardiol. 2017, 241, 344–350. [Google Scholar] [CrossRef]
- Øie, E.; Yndestad, A.; Robins, S.P.; Bjørnerheim, R.; Åsberg, A.; Attramadal, H. Early intervention with a potent endothelin-A/endothelin-B receptor antagonist aggravates left ventricular remodeling after myocardial infarction in rats. Basic Res. Cardiol. 2002, 97, 239–247. [Google Scholar] [CrossRef]
- Solomon, S.D.; Pfeffer, M.A. Renin-Angiotensin System and Cardiac Rupture After Myocardial Infarction. Circulation 2002, 106, 2167–2169. [Google Scholar] [CrossRef]
- Fraccarollo, D.; Widder, J.D.; Galuppo, P.; Thum, T.; Tsikas, D.; Hoffmann, M.; Ruetten, H.; Ertl, G.; Bauersachs, J. Improvement in Left Ventricular Remodeling by the Endothelial Nitric Oxide Synthase Enhancer AVE9488 After Experimental Myocardial Infarction. Circulation 2008, 118, 818–827. [Google Scholar] [CrossRef]
- Tzanidis, A.; Lim, S.; Hannan, R.D.; See, F.; Ugoni, A.M.; Krum, H. Combined Angiotensin and Endothelin Receptor Blockade Attenuates Adverse Cardiac Remodeling Post-Myocardial Infarction in the Rat: Possible Role of Transforming Growth Factor β1. J. Mol. Cell. Cardiol. 2001, 33, 969–981. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Tian, J.; Jiang, L.; Xu, L.; Liu, J.; Zhao, X.; Feng, X.; Wang, D.; Zhang, Y.; Sun, K.; et al. Prognostic Value of Plasma Big Endothelin-1 Level among Patients with Three-Vessel Disease: A Cohort Study. J. Atheroscler. Thromb. 2019, 26, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, M.-H.; Guo, Y.-L.; Zhu, C.-G.; Xu, R.-X.; Dong, Q.; Li, J.-J. Plasma Big Endothelin-1 Level and the Severity of New-onset Stable Coronary Artery Disease. J. Atheroscler. Thromb. 2015, 22, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Maitland, A.; Weisel, R.D.; Fedak, P.W.M.; Li, S.-H.; Mickle, D.A.G.; Li, R.-K.; Ko, L.; Rao, V. Increased endothelin-1 production in diabetic patients after cardioplegic arrest and reperfusion impairs coronary vascular reactivity: Reversal by means of endothelin antagonism. J. Thorac. Cardiovasc. Surg. 2002, 123, 1114–1119. [Google Scholar] [CrossRef][Green Version]
- Tsutamoto, T. Relationship between transcardiac gradient of endothelin-1 and left ventricular remodelling in patients with first anterior myocardial infarction. Eur. Heart J. 2003, 24, 346–355. [Google Scholar] [CrossRef]
- Ihling, C.; Szombathy, T.; Bohrmann, B.; Brockhaus, M.; Schaefer, H.E.; Loeffler, B.M. Coexpression of Endothelin-Converting Enzyme-1 and Endothelin-1 in Different Stages of Human Atherosclerosis. Circulation 2001, 104, 864–869. [Google Scholar] [CrossRef]
- Ruschitzka, F.; Moehrlen, U.; Quaschning, T.; Lachat, M.; Noll, G.; Shaw, S.; Yang, Z.; Teupser, D.; Subkowski, T.; Turina, M.I.; et al. Tissue Endothelin-Converting Enzyme Activity Correlates with Cardiovascular Risk Factors in Coronary Artery Disease. Circulation 2000, 102, 1086–1092. [Google Scholar] [CrossRef]
- Böhm, F.; Jensen, J.; Svane, B.; Settergren, M.; Pernow, J. Intracoronary endothelin receptor blockade improves endothelial function in patients with coronary artery disease. Can. J. Physiol. Pharmacol. 2008, 86, 745–751. [Google Scholar] [CrossRef]
- Rafnsson, A.; Shemyakin, A.; Pernow, J. Selective endothelin ETA and dual ETA/ETB receptor blockade improve endothelium-dependent vasodilatation in patients with type 2 diabetes and coronary artery disease. Life Sci. 2014, 118, 435–439. [Google Scholar] [CrossRef]
- Liou, K.; Jepson, N.; Buckley, N.; Chen, V.; Thomas, S.; Russell, E.A.; Ooi, S.-Y. Design and Rationale for the Endothelin-1 Receptor Antagonism in the Prevention of Microvascular Injury in Patients with non-ST Elevation Acute Coronary Syndrome Undergoing Percutaneous Coronary Intervention (ENDORA-PCI) Trial. Cardiovasc. Drugs Ther. 2016, 30, 169–175. [Google Scholar] [CrossRef]
- O’Connor, C.M.; Gattis, W.A.; Adams, K.F.; Hasselblad, V.; Chandler, B.; Frey, A.; Kobrin, I.; Rainisio, M.; Shah, M.R.; Teerlink, J.; et al. Tezosentan in patients with acuteheart failure and acute coronary syndromes. J. Am. Coll. Cardiol. 2003, 41, 1452–1457. [Google Scholar] [CrossRef]
- Garjani, A.; Wainwright, C.L.; Zeitlin, I.J.; Wilson, C.; Slee, S.-J. Effects of Endothelin-1 and the ETA-Receptor Antagonist, BQ123, on Ischemic Arrhythmias in Anesthetized Rats. J. Cardiovasc. Pharmacol. 1995, 25, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Hathaway, C.K.; Grant, R.; Hagaman, J.R.; Hiller, S.; Li, F.; Xu, L.; Chang, A.S.; Madden, V.J.; Bagnell, C.R.; Rojas, M.; et al. Endothelin-1 critically influences cardiac function via superoxide-MMP9 cascade. Proc. Natl. Acad. Sci. USA 2015, 112, 5141–5146. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, Y.; Ashihara, T.; Tsutamoto, T.; Ito, M.; Horie, M. Endothelin-1 as a predictor of atrial fibrillation recurrence after pulmonary vein isolation. Heart Rhythm 2009, 6, 725–730. [Google Scholar] [CrossRef]
- Mayyas, F.; Niebauer, M.; Zurick, A.; Barnard, J.; Gillinov, A.M.; Chung, M.K.; Van Wagoner, D.R. Association of Left Atrial Endothelin-1 with Atrial Rhythm, Size, and Fibrosis in Patients with Structural Heart Disease. Circ. Arrhythmia Electrophysiol. 2010, 3, 369–379. [Google Scholar] [CrossRef]
- Wu, S.; Yang, Y.; Zhu, J.; Ren, J.; Wang, J.; Zhang, H.; Shao, X. The association between plasma big endothelin-1 levels at admission and long-term outcomes in patients with atrial fibrillation. Atherosclerosis 2018, 272, 1–7. [Google Scholar] [CrossRef]
- Zheng, Y.; Lan, C.; Wang, N.; Xu, X.; Hu, T.; Wu, Q.; Xie, X.; Wang, Z.; Zhang, Y.; Li, C. Significant Association of rs2147555 Genetic Polymorphism in the EDNRB Gene with Hirschsprung Disease in Southern Chinese Children. Biomed Res. Int. 2020, 2020, 5956412. [Google Scholar] [CrossRef]
- Sharif, I.; Crockett, T.R.; Kane, K.A.; Wainwright, C.L. The effects of endothelin-1 on ischaemia-induced ventricular arrhythmias in rat isolated hearts. Eur. J. Pharmacol. 2001, 427, 235–242. [Google Scholar] [CrossRef]
- Zirlik, K.; Duyster, J. Anti-Angiogenics: Current Situation and Future Perspectives. Oncol. Res. Treat. 2018, 41, 166–171. [Google Scholar] [CrossRef]
- Iglarz, M.; Silvestre, J.-S.; Duriez, M.; Henrion, D.; Lévy, B.I. Chronic Blockade of Endothelin Receptors Improves Ischemia-Induced Angiogenesis in Rat Hindlimbs Through Activation of Vascular Endothelial Growth Factor–NO Pathway. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1598–1603. [Google Scholar] [CrossRef][Green Version]
- Pandey, A.K.; Singhi, E.K.; Arroyo, J.P.; Ikizler, T.A.; Gould, E.R.; Brown, J.; Beckman, J.A.; Harrison, D.G.; Moslehi, J. Mechanisms of VEGF (Vascular Endothelial Growth Factor) Inhibitor–Associated Hypertension and Vascular Disease. Hypertension 2018, 71, e1–e8. [Google Scholar] [CrossRef]
- Tsui, J.; Dashwood, M. A Role for Endothelin-1 in Peripheral Vascular Disease. Curr. Vasc. Pharmacol. 2005, 3, 325–332. [Google Scholar] [CrossRef] [PubMed]
- De Haro, J.; Bleda, S.; Gonzalez-Hidalgo, C.; Michel, I.; Acin, F. Long-Term Effects of Bosentan on Cardiovascular Events in Hispanic Patients with Intermittent Claudication: Four-Year Follow-up of the CLAU Trial. Am. J. Cardiovasc. Drugs 2019, 19, 203–209. [Google Scholar] [CrossRef] [PubMed]
- De Haro, J.; Bleda, S.; Varela, C.; Esparza, L.; Acin, F. Effect of Bosentan on Claudication Distance and Endothelium-Dependent Vasodilation in Hispanic Patients with Peripheral Arterial Disease. Am. J. Cardiol. 2016, 117, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Bartfai, T.; Wang, M.-W. Positive allosteric modulators to peptide GPCRs: A promising class of drugs. Acta Pharmacol. Sin. 2013, 34, 880–885. [Google Scholar] [CrossRef]
- Gentry, P.R.; Sexton, P.M.; Christopoulos, A. Novel Allosteric Modulators of G Protein-coupled Receptors *. J. Biol. Chem. 2015, 290, 19478–19488. [Google Scholar] [CrossRef]
- Talbodec, A.; Berkane, N.; Blandin, V.; Breittmayer, J.P.; Ferrari, E.; Frelin, C.; Vigne, P. Aspirin and sodium salicylate inhibit endothelin ETA receptors by an allosteric type of mechanism. Mol. Pharmacol. 2000, 57, 797–804. [Google Scholar] [CrossRef]
- Blandin, V.; Vigne, P.; Breittmayer, J.P.; Frelin, C. Allosteric Inhibition of Endothelin ETA Receptors by 3,5-Dibromosalicylic Acid. Mol. Pharmacol. 2000, 58, 1461–1469. [Google Scholar] [CrossRef]
- Wisler, J.W.; Rockman, H.A.; Lefkowitz, R.J. Biased G protein-coupled receptor signaling: Changing the paradigm of drug discovery. Circulation 2018, 137, 2315–2317. [Google Scholar] [CrossRef]
- Pang, P.S.; Butler, J.; Collins, S.P.; Cotter, G.; Davison, B.A.; Ezekowitz, J.A.; Filippatos, G.; Levy, P.D.; Metra, M.; Ponikowski, P.; et al. Biased ligand of the angiotensin II type 1 receptor in patients with acute heart failure: A randomized, double-blind, placebo-controlled, phase IIB, dose ranging trial (BLAST-AHF). Eur. Heart J. 2017, 38, 2364–2373. [Google Scholar] [CrossRef]
- Kashihara, T.; Kawagishi, H.; Nakada, T.; Numaga-Tomita, T.; Kadota, S.; Wolf, E.E.; Du, C.K.; Shiba, Y.; Morimoto, S.; Yamada, M. β-Arrestin–Biased AT1 Agonist TRV027 Causes a Neonatal-Specific Sustained Positive Inotropic Effect without Increasing Heart Rate. JACC Basic Transl. Sci. 2020, 5, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Teoh, J.P.; Park, K.M.; Wang, Y.; Hu, Q.; Kim, S.; Wu, G.; Huang, S.; Maihle, N.; Kim, I. man Endothelin-1/Endothelin A receptor-mediated biased signaling is a new player in modulating human ovarian cancer cell tumorigenesis. Cell. Signal. 2014, 26, 2885. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, M.; Schilling, J.; Beautrait, A.; Bouvier, M.; Benovic, J.L.; Shukla, A.K. Emerging Paradigm of Intracellular Targeting of G Protein-Coupled Receptors. Trends Biochem. Sci. 2018, 43, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Sallum, C.O.; Wilson, J.L.; Rupasinghe, C.; Berg, E.; Yu, J.; Green, D.S.; Taylor, L.; Mierke, D.; Polgar, P. Enhancing and limiting endothelin-1 signaling with a cell-penetrating peptide mimicking the third intracellular loop of the ETB receptor. Chem. Biol. Drug Des. 2012, 80, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Green, D.S.; Rupasinghe, C.; Warburton, R.; Wilson, J.L.; Sallum, C.O.; Taylor, L.; Yatawara, A.; Mierke, D.; Polgar, P.; Hill, N. A Cell Permeable Peptide Targeting the Intracellular Loop 2 of Endothelin B Receptor Reduces Pulmonary Hypertension in a Hypoxic Rat Model. PLoS ONE 2013, 8, e81309. [Google Scholar] [CrossRef] [PubMed]
- Kamath, A.V. Translational pharmacokinetics and pharmacodynamics of monoclonal antibodies. Drug Discov. Today Technol. 2016, 21–22, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, X.; Zhang, H.; Yao, C.; Pan, H.; Guo, Y.; Fan, K.; Jing, S. Therapeutic monoclonal antibody antagonizing endothelin receptor a for pulmonary arterial hypertension. J. Pharmacol. Exp. Ther. 2019, 370, 54–61. [Google Scholar] [CrossRef]
- Dai, Y.; Chen, X.; Song, X.; Chen, X.; Ma, W.; Lin, J.; Wu, H.; Hu, X.; Zhou, Y.; Zhang, H.; et al. Immunotherapy of Endothelin-1 Receptor Type A for Pulmonary Arterial Hypertension. J. Am. Coll. Cardiol. 2019, 73, 2567–2580. [Google Scholar] [CrossRef]
- Dai, Y.; Qiu, Z.; Ma, W.; Li, C.; Chen, X.; Song, X.; Bai, Z.; Shi, D.; Zheng, J.; Pan, G.; et al. Long-Term Effect of a Vaccine Targeting Endothelin-1 Receptor Type A in Pulmonary Arterial Hypertension. Front. Cardiovasc. Med. 2021, 0, 570. [Google Scholar] [CrossRef]
- Jain, A.; Chen, S.; Yong, H.; Chakrabarti, S. Endothelin-1 traps potently reduce pathologic markers back to basal levels in an in vitro model of diabetes. J. Diabetes Metab. Disord. 2018, 17, 189. [Google Scholar] [CrossRef]
- Jain, A.; Mehrotra, V.; Jha, I.; Jain, A. In vivo studies demonstrate that endothelin-1 traps are a potential therapy for type I diabetes. J. Diabetes Metab. Disord. 2019, 18, 133–143. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haryono, A.; Ramadhiani, R.; Ryanto, G.R.T.; Emoto, N. Endothelin and the Cardiovascular System: The Long Journey and Where We Are Going. Biology 2022, 11, 759. https://doi.org/10.3390/biology11050759
Haryono A, Ramadhiani R, Ryanto GRT, Emoto N. Endothelin and the Cardiovascular System: The Long Journey and Where We Are Going. Biology. 2022; 11(5):759. https://doi.org/10.3390/biology11050759
Chicago/Turabian StyleHaryono, Andreas, Risa Ramadhiani, Gusty Rizky Teguh Ryanto, and Noriaki Emoto. 2022. "Endothelin and the Cardiovascular System: The Long Journey and Where We Are Going" Biology 11, no. 5: 759. https://doi.org/10.3390/biology11050759
APA StyleHaryono, A., Ramadhiani, R., Ryanto, G. R. T., & Emoto, N. (2022). Endothelin and the Cardiovascular System: The Long Journey and Where We Are Going. Biology, 11(5), 759. https://doi.org/10.3390/biology11050759