
Fusion Genes in Prostate Cancer: A Comparison in Men of African and European Descent

 ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
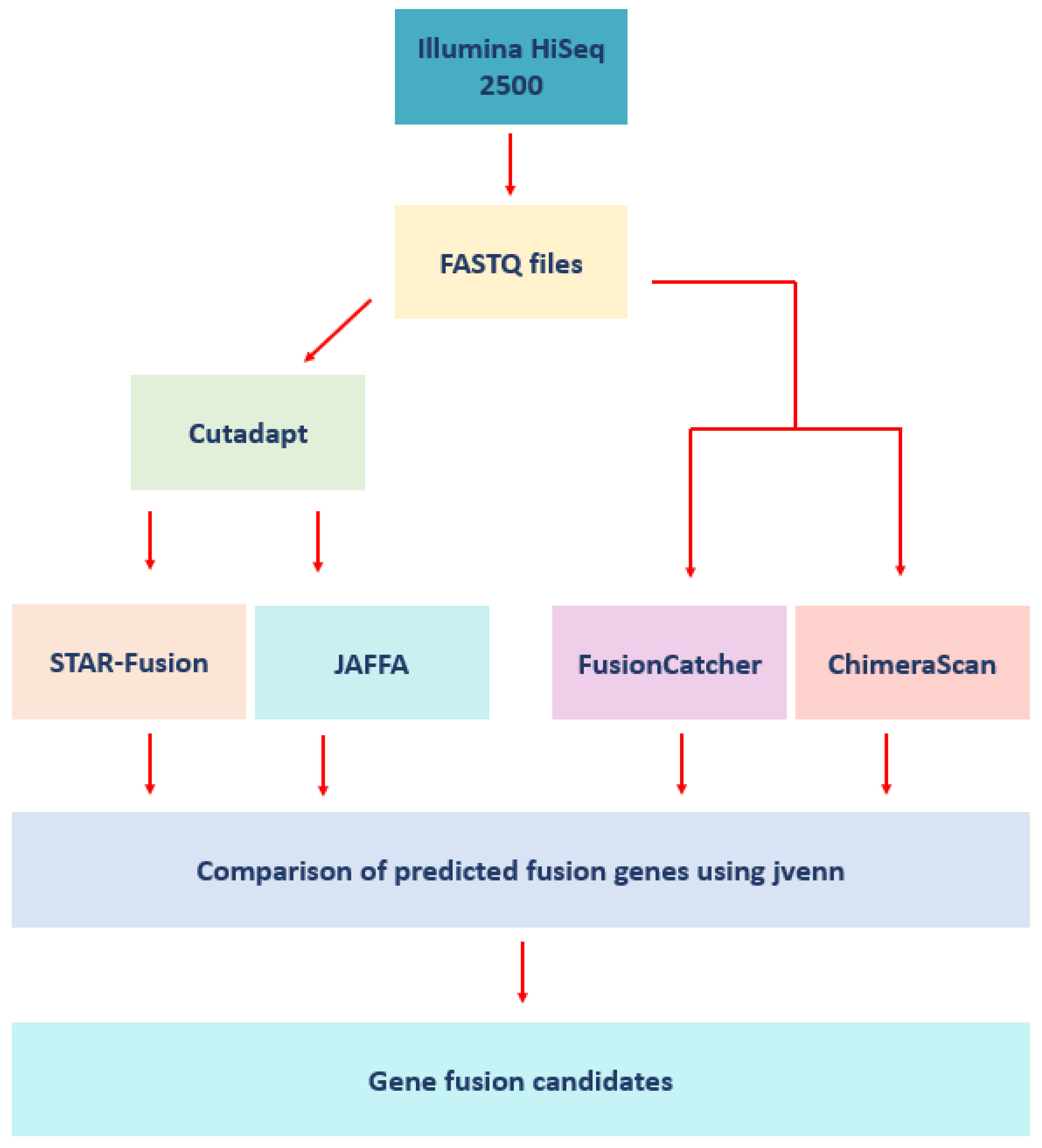
2. Materials and Methods
2.1. Human Subjects
2.2. Tissue Sample Procurement and RNA Purification
2.3. RNA Sequencing (RNA-seq) and Quality Control
2.4. Fusion Detection Software
2.5. Benchmarking the Fusion Detection Tools Using Prostate Cancer Cell Lines
2.6. Analysis of the Patient Samples
3. Results
3.1. Cell Line Analysis
3.2. STAR-Fusion
3.3. FusionCatcher
3.4. JAFFA
3.5. ChimeraScan
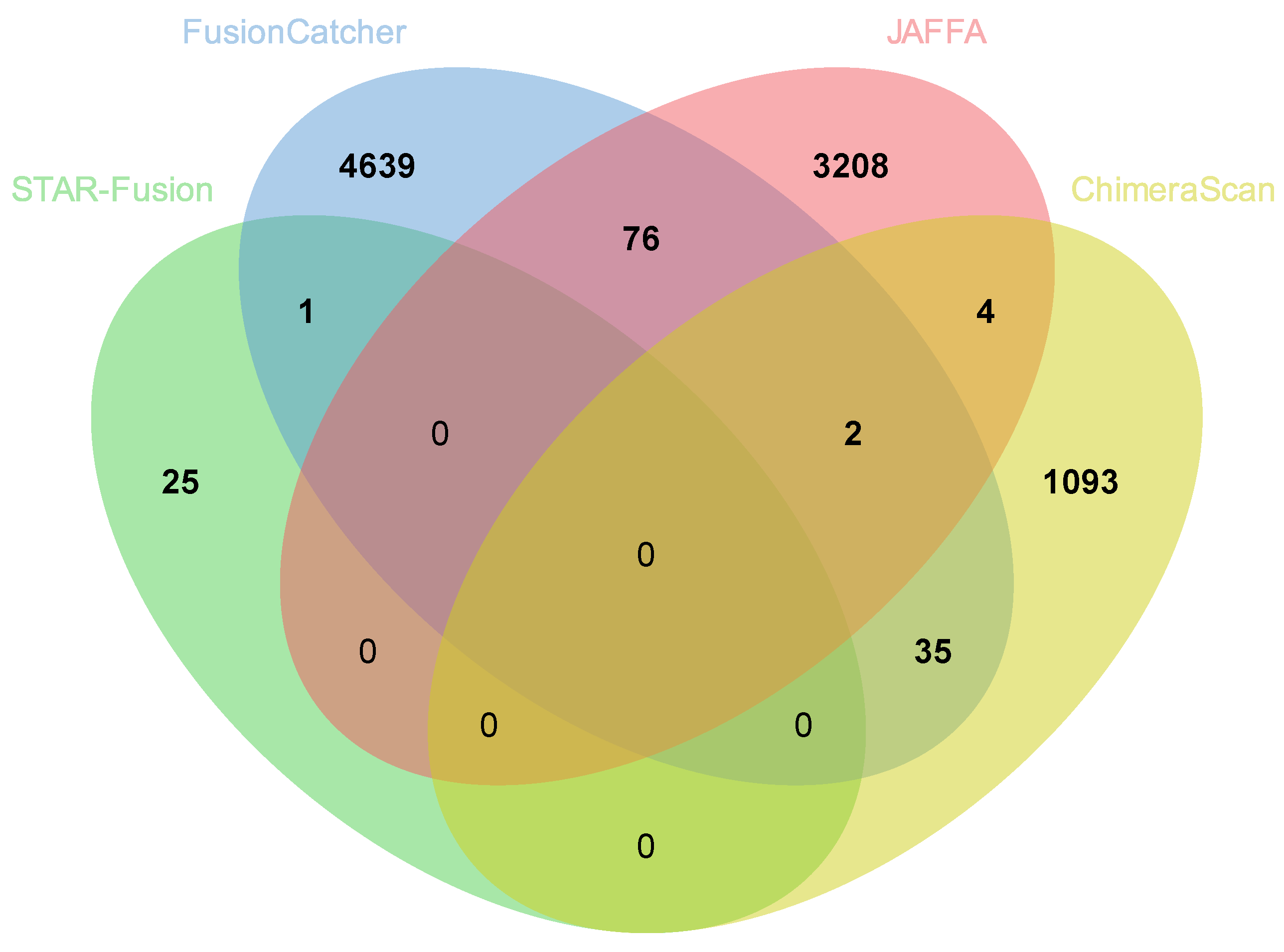
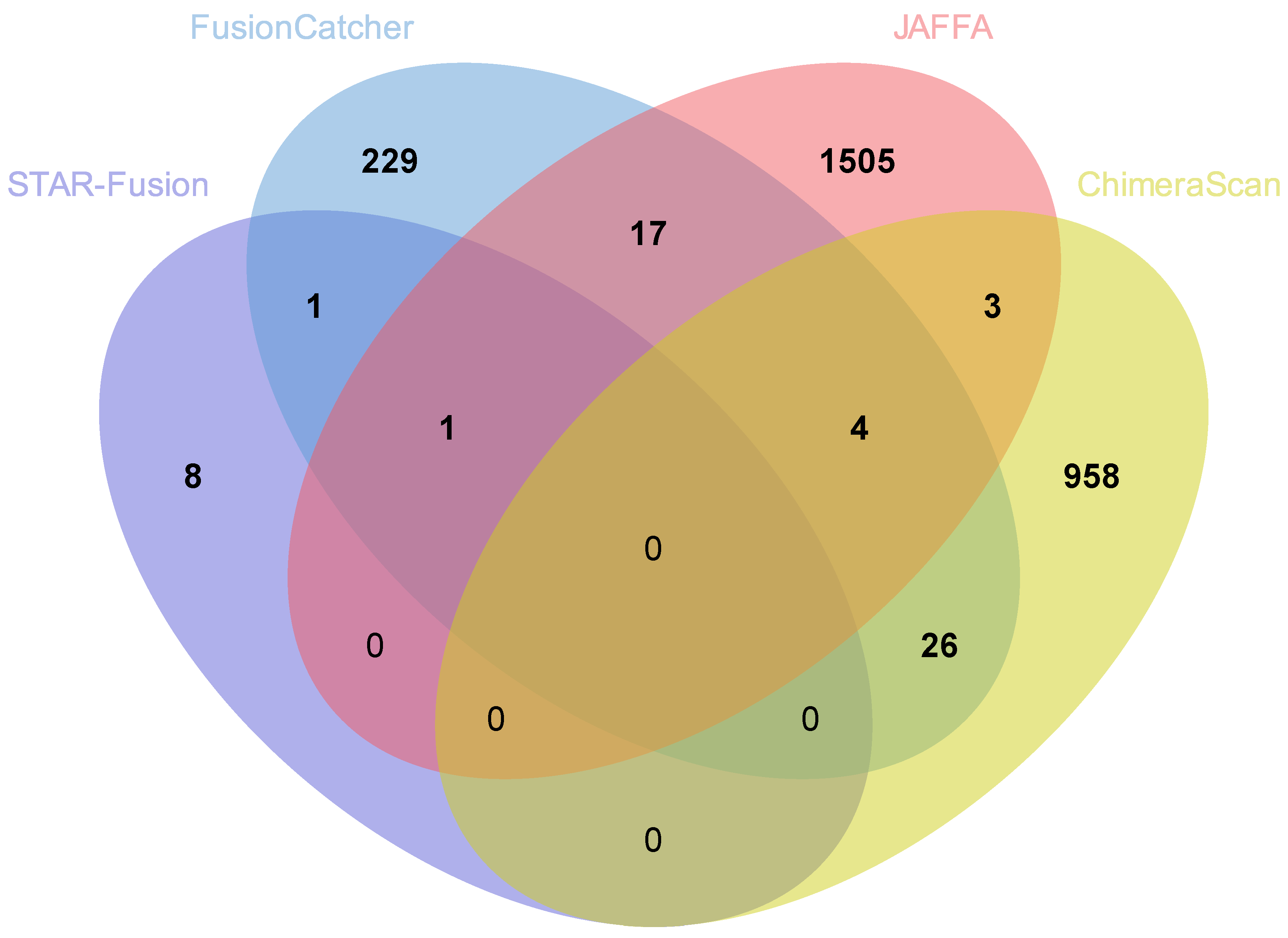
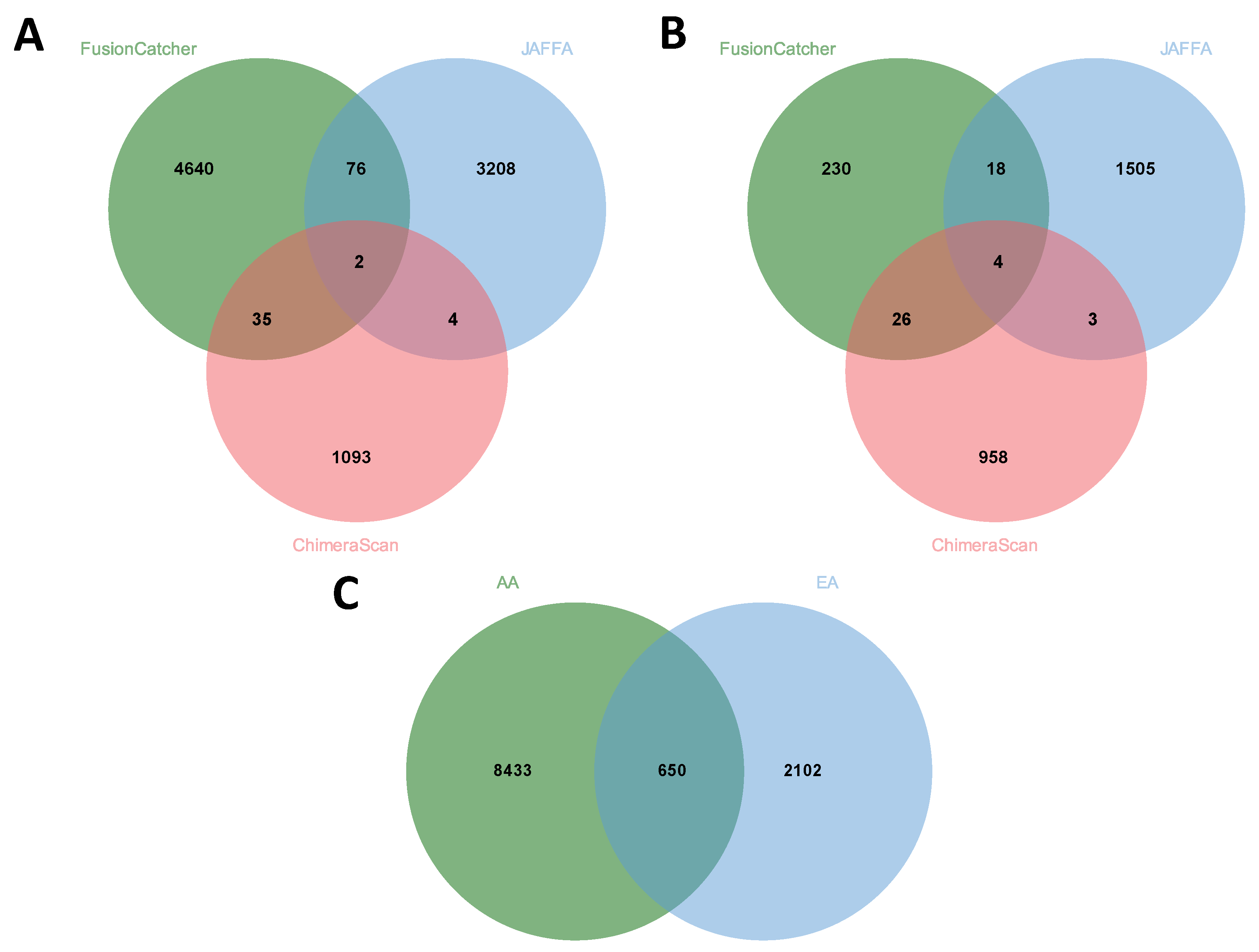
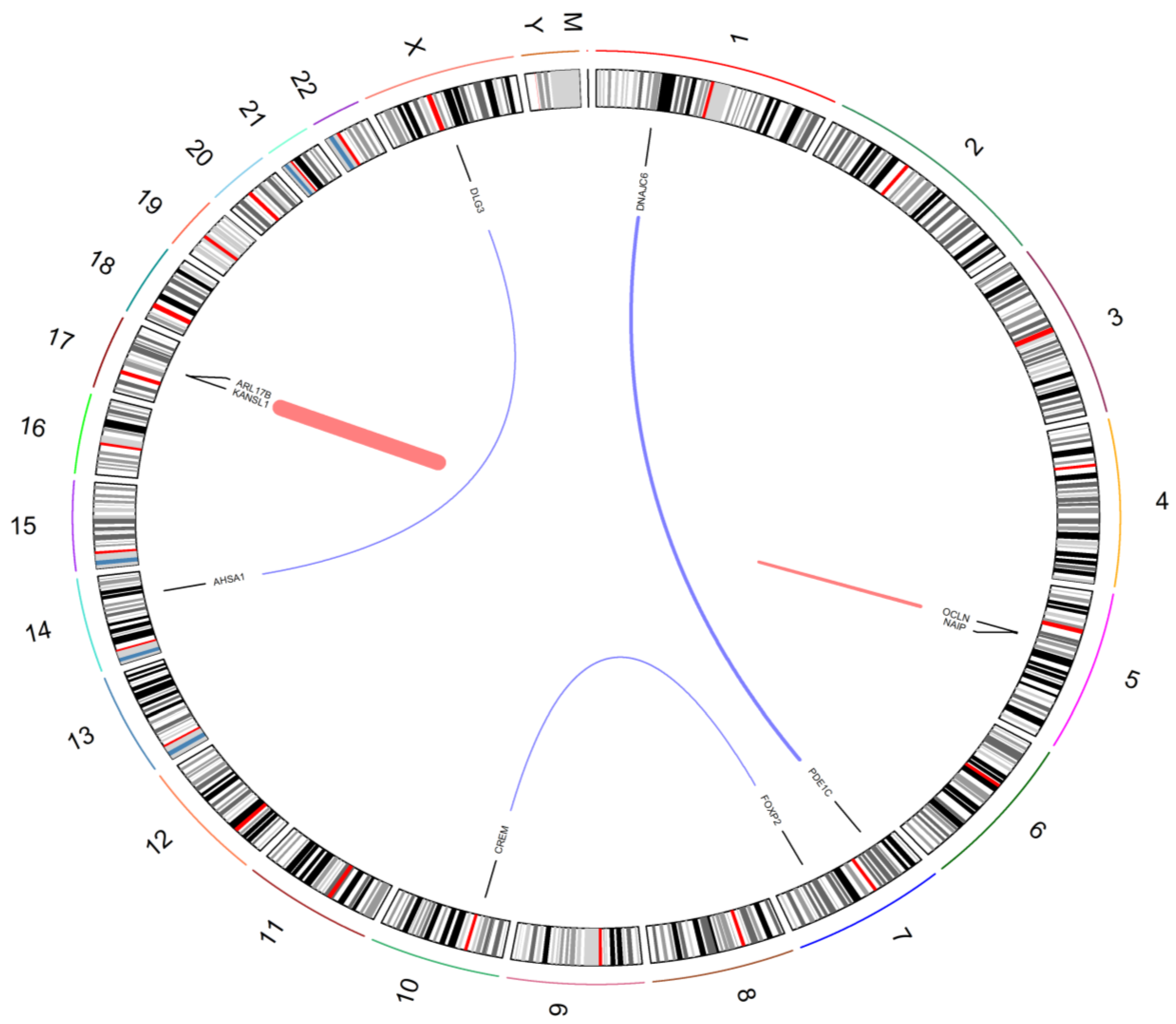
3.6. Comparison of Fusion Genes Based on Race
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wallace, T.A.; Prueitt, R.L.; Yi, M.; Howe, T.M.; Gillespie, J.W.; Yfantis, H.G.; Stephens, R.M.; Caporaso, N.E.; Loffredo, C.A.; Ambs, S. Tumor immunobiological differences in prostate cancer between African-American and European-American men. Cancer Res. 2008, 68, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.J.; Mallin, K.; Graves, A.J.; Chang, S.S.; Penson, D.F.; Resnick, M.J.; Barocas, D.A. Recent Changes in Prostate Cancer Screening Practices and Epidemiology. J. Urol. 2017, 198, 1230–1240. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Perdomo, H.A.; Chaves, M.J.; Osorio, J.C.; Sanchez, A. Association between TMPRSS2:ERG fusion gene and the prostate cancer: Systematic review and meta-analysis. Cent Eur. J. Urol. 2018, 71, 410–419. [Google Scholar] [CrossRef]
- Chen, S.L.; Wang, S.C.; Ho, C.J.; Kao, Y.L.; Hsieh, T.Y.; Chen, W.J.; Chen, C.J.; Wu, P.R.; Ko, J.L.; Lee, H.; et al. Prostate Cancer Mortality-To-Incidence Ratios Are Associated with Cancer Care Disparities in 35 Countries. Sci. Rep. 2017, 7, 40003. [Google Scholar] [CrossRef]
- Lee, H.; Lee, D.; Park, J.H.; Song, S.H.; Jeong, I.G.; Kim, C.S.; Searson, P.C.; Lee, K.H. High throughput differential identification of TMPRSS2-ERG fusion genes in prostate cancer patient urine. Biomaterials 2017, 135, 23–29. [Google Scholar] [CrossRef]
- Ilic, D.; Djulbegovic, M.; Jung, J.H.; Hwang, E.C.; Zhou, Q.; Cleves, A.; Agoritsas, T.; Dahm, P. Prostate cancer screening with prostate-specific antigen (PSA) test: A systematic review and meta-analysis. BMJ 2018, 362, k3519. [Google Scholar] [CrossRef]
- Gulati, R.; Cheng, H.H.; Lange, P.H.; Nelson, P.S.; Etzioni, R. Screening Men at Increased Risk for Prostate Cancer Diagnosis: Model Estimates of Benefits and Harms. Cancer Epidemiol. Biomark. Prev. 2017, 26, 222–227. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Srivastava, S.K.; Khan, M.A.; Prajapati, V.K.; Singh, S.; Carter, J.E.; Singh, A.P. Racial disparities in prostate cancer: A molecular perspective. Front. Biosci. 2017, 22, 772–782. [Google Scholar] [CrossRef]
- Dai, X.; Theobard, R.; Cheng, H.; Xing, M.; Zhang, J. Fusion genes: A promising tool combating against cancer. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 149–160. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, Q.; Wu, J.; Han, P.; Song, X.F. GFusion: An Effective Algorithm to Identify Fusion Genes from Cancer RNA-Seq Data. Sci. Rep. 2017, 7, 6880. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.C.; Zhang, W. Fusion genes in solid tumors: An emerging target for cancer diagnosis and treatment. Chin. J. Cancer 2013, 32, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Latysheva, N.S.; Babu, M.M. Discovering and understanding oncogenic gene fusions through data intensive computational approaches. Nucleic Acids Res. 2016, 44, 4487–4503. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.; Jang, Y.E.; Lee, S. FusionScan: Accurate prediction of fusion genes from RNA-Seq data. Genom. Inf. 2019, 17, e26. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Y.; Zhang, J.; Hu, Q.; Zhi, F.; Zhang, S.; Mao, D.; Zhang, Y.; Liang, H. Significance of the TMPRSS2:ERG gene fusion in prostate cancer. Mol. Med. Rep. 2017, 16, 5450–5458. [Google Scholar] [CrossRef] [PubMed]
- Ayala, G.; Frolov, A.; Chatterjee, D.; He, D.; Hilsenbeck, S.; Ittmann, M. Expression of ERG protein in prostate cancer: Variability and biological correlates. Endocr. Relat. Cancer 2015, 22, 277–287. [Google Scholar] [CrossRef]
- Yuan, J.; Kensler, K.H.; Hu, Z.; Zhang, Y.; Zhang, T.; Jiang, J.; Xu, M.; Pan, Y.; Long, M.; Montone, K.T.; et al. Integrative comparison of the genomic and transcriptomic landscape between prostate cancer patients of predominantly African or European genetic ancestry. PLoS Genet. 2020, 16, e1008641. [Google Scholar] [CrossRef]
- Hardiman, G.; Savage, S.J.; Hazard, E.S.; Wilson, R.C.; Courtney, S.M.; Smith, M.T.; Hollis, B.W.; Halbert, C.H.; Gattoni-Celli, S. Systems analysis of the prostate transcriptome in African-American men compared with European-American men. Pharmacogenomics 2016, 17, 1129–1143. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 1–3. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics, Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Nicorici, D.; Şatalan, M.; Edgren, H.; Kangaspeska, S.; Murumägi, A.; Kallioniemi, O.; Virtanen, S.; Kilkku, O. FusionCatcher—A tool for finding somatic fusion genes in paired-end RNA-sequencing data. bioRxiv 2014. [Google Scholar] [CrossRef]
- Davidson, N.M.; Majewski, I.J.; Oshlack, A. JAFFA: High sensitivity transcriptome-focused fusion gene detection. Genome Med. 2015, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Iyer, M.K.; Chinnaiyan, A.M.; Maher, C.A. ChimeraScan: A tool for identifying chimeric transcription in sequencing data. Bioinformatics 2011, 27, 2903–2904. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Vo, A.D.; Qin, F.; Li, H. Comparative assessment of methods for the fusion transcripts detection from RNA-Seq data. Sci. Rep. 2016, 6, 21597. [Google Scholar] [CrossRef] [PubMed]
- Lagstad, S.; Zhao, S.; Hoff, A.M.; Johannessen, B.; Lingjaerde, O.C.; Skotheim, R.I. chimeraviz: A tool for visualizing chimeric RNA. Bioinformatics 2017, 33, 2954–2956. [Google Scholar] [CrossRef] [PubMed]
- Bardou, P.; Mariette, J.; Escudie, F.; Djemiel, C.; Klopp, C. jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef]
- Mitelman, F.; Johansson, B.; Mertens, F. Mitelman Database Chromosome Aberrations and Gene Fusions in Cancer. Available online: https://mitelmandatabase.isb-cgc.org (accessed on 10 March 2020).
- Hu, X.; Wang, Q.; Tang, M.; Barthel, F.; Amin, S.; Yoshihara, K.; Lang, F.M.; Martinez-Ledesma, E.; Lee, S.H.; Zheng, S.; et al. TumorFusions: An integrative resource for cancer-associated transcript fusions. Nucleic Acids Res. 2018, 46, D1144–D1149. [Google Scholar] [CrossRef]
- Torres-García, W.; Zheng, S.; Sivachenko, A.; Vegesna, R.; Wang, Q.; Yao, R.; Berger, M.F.; Weinstein, J.N.; Getz, G.; Verhaak, R.G. PRADA: Pipeline for RNA sequencing data analysis. Bioinformatics 2014, 30, 2224–2226. [Google Scholar] [CrossRef]
- Kumar-Sinha, C.; Tomlins, S.A.; Chinnaiyan, A.M. Recurrent gene fusions in prostate cancer. Nat. Rev. Cancer 2008, 8, 497–511. [Google Scholar] [CrossRef]
- Annala, M.J.; Parker, B.C.; Zhang, W.; Nykter, M. Fusion genes and their discovery using high throughput sequencing. Cancer Lett. 2013, 340, 192–200. [Google Scholar] [CrossRef]
- Heyer, E.E.; Deveson, I.W.; Wooi, D.; Selinger, C.I.; Lyons, R.J.; Hayes, V.M.; O’Toole, S.A.; Ballinger, M.L.; Gill, D.; Thomas, D.M.; et al. Diagnosis of fusion genes using targeted RNA sequencing. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, N.; Liu, J.; Wu, Z.; Dong, D. FusionCancer: A database of cancer fusion genes derived from RNA-seq data. Diagn. Pathol. 2015, 10, 131. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Li, X.; Li, H. Gene fusions and chimeric RNAs, and their implications in cancer. Genes Dis. 2019, 6, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Xie, Z.; Li, H. Intergenically Spliced Chimeric RNAs in Cancer. Trends Cancer 2016, 2, 475–484. [Google Scholar] [CrossRef]
- Spratt, D.E.; Chan, T.; Waldron, L.; Speers, C.; Feng, F.Y.; Ogunwobi, O.O.; Osborne, J.R. Racial/Ethnic Disparities in Genomic Sequencing. JAMA Oncol. 2016, 2, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Freedland, S.J.; Amling, C.L.; Dorey, F.; Kane, C.J.; Presti, J.C., Jr.; Terris, M.K.; Aronson, W.J. Race as an outcome predictor after radical prostatectomy: Results from the Shared Equal Access Regional Cancer Hospital (SEARCH) database. Urology 2002, 60, 670–674. [Google Scholar] [CrossRef]
- Freedland, S.J.; Sutter, M.E.; Naitoh, J.; Dorey, F.; Csathy, G.S.; Aronson, W.J. Clinical characteristics in black and white men with prostate cancer in an equal access medical center. Urology 2000, 55, 387–390. [Google Scholar] [CrossRef]
- Woods, V.D.; Montgomery, S.B.; Belliard, J.C.; Ramirez-Johnson, J.; Wilson, C.M. Culture, black men, and prostate cancer: What is reality? Cancer Control. 2004, 11, 388–396. [Google Scholar] [CrossRef]
- Tan, S.H.; Petrovics, G.; Srivastava, S. Prostate Cancer Genomics: Recent Advances and the Prevailing Underrepresentation from Racial and Ethnic Minorities. Int. J. Mol. Sci. 2018, 19, 1255. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, W.; Huang, S.; He, Z.; Cheng, Y.; Wang, J.; Lam, T.W.; Peng, Z.; Yiu, S.M. SOAPfusion: A robust and effective computational fusion discovery tool for RNA-seq reads. Bioinformatics 2013, 29, 2971–2978. [Google Scholar] [CrossRef]
- Zhang, J.; White, N.M.; Schmidt, H.K.; Fulton, R.S.; Tomlinson, C.; Warren, W.C.; Wilson, R.K.; Maher, C.A. INTEGRATE: Gene fusion discovery using whole genome and transcriptome data. Genome Res. 2016, 26, 108–118. [Google Scholar] [CrossRef]
- Benelli, M.; Pescucci, C.; Marseglia, G.; Severgnini, M.; Torricelli, F.; Magi, A. Discovering chimeric transcripts in paired-end RNA-seq data by using EricScript. Bioinformatics 2012, 28, 3232–3239. [Google Scholar] [CrossRef] [PubMed]
- Chiu, R.; Nip, K.M.; Birol, I. Fusion-Bloom: Fusion detection in assembled transcriptomes. Bioinformatics 2020, 36, 2256–2257. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Qiao, Y.; Asangani, I.A.; Ateeq, B.; Poliakov, A.; Cieslik, M.; Pitchiaya, S.; Chakravarthi, B.; Cao, X.; Jing, X.; et al. Development of Peptidomimetic Inhibitors of the ERG Gene Fusion Product in Prostate Cancer. Cancer Cell 2017, 31, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Demichelis, F.; Rubin, M.A. TMPRSS2-ETS fusion prostate cancer: Biological and clinical implications. J. Clin. Pathol. 2007, 60, 1185–1186. [Google Scholar] [CrossRef]
- Zhou, C.K.; Young, D.; Yeboah, E.D.; Coburn, S.B.; Tettey, Y.; Biritwum, R.B.; Adjei, A.A.; Tay, E.; Niwa, S.; Truelove, A.; et al. TMPRSS2:ERG Gene Fusions in Prostate Cancer of West African Men and a Meta-Analysis of Racial Differences. Am. J. Epidemiol. 2017, 186, 1352–1361. [Google Scholar] [CrossRef]
- Blackburn, J.; Vecchiarelli, S.; Heyer, E.E.; Patrick, S.M.; Lyons, R.J.; Jaratlerdsiri, W.; van Zyl, S.; Bornman, M.S.R.; Mercer, T.R.; Hayes, V.M. TMPRSS2-ERG fusions linked to prostate cancer racial health disparities: A focus on Africa. Prostate 2019, 79, 1191–1196. [Google Scholar] [CrossRef]
- Ramanand, S.G.; Mani, R.S. Genetic, Environmental, and Nuclear Factors Governing Genomic Rearrangements. Adv. Exp. Med. Biol. 2019, 1210, 57–66. [Google Scholar] [CrossRef]
- Mertz, K.D.; Setlur, S.R.; Dhanasekaran, S.M.; Demichelis, F.; Perner, S.; Tomlins, S.; Tchinda, J.; Laxman, B.; Vessella, R.L.; Beroukhim, R.; et al. Molecular characterization of TMPRSS2-ERG gene fusion in the NCI-H660 prostate cancer cell line: A new perspective for an old model. Neoplasia 2007, 9, 200–206. [Google Scholar] [CrossRef]
- Mounir, Z.; Lin, F.; Lin, V.G.; Korn, J.M.; Yu, Y.; Valdez, R.; Aina, O.H.; Buchwalter, G.; Jaffe, A.B.; Korpal, M.; et al. TMPRSS2:ERG blocks neuroendocrine and luminal cell differentiation to maintain prostate cancer proliferation. Oncogene 2015, 34, 3815–3825. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, N.; Liu, D.; Jin, Y. Recurrent Fusion Genes in Leukemia: An Attractive Target for Diagnosis and Treatment. Curr. Genom. 2017, 18, 378–384. [Google Scholar] [CrossRef]
- Haas, B.J.; Dobin, A.; Stransky, N.; Li, B.; Yang, X.; Tickle, T.; Bankapur, A.; Ganote, C.; Doak, T.G.; Pochet, N.; et al. STAR-Fusion: Fast and accurate fusion transcript detection from RNA-Seq. bioRxiv 2017. [Google Scholar] [CrossRef]
- Liu, S.; Tsai, W.H.; Ding, Y.; Chen, R.; Fang, Z.; Huo, Z.G.; Kim, S.; Ma, T.Z.; Chang, T.Y.; Priedigkeit, N.M.; et al. Comprehensive evaluation of fusion transcript detection algorithms and a meta-caller to combine top performing methods in paired-end RNA-seq data. Nucleic Acids Res. 2016, 44, e47. [Google Scholar] [CrossRef] [PubMed]
- Greger, L.; Su, J.; Rung, J.; Ferreira, P.G.; Geuvadis, c.; Lappalainen, T.; Dermitzakis, E.T.; Brazma, A. Tandem RNA Chimeras Contribute to Transcriptome Diversity in Human Population and Are Associated with Intronic Genetic Variants. PLoS ONE 2014, 9, e104567. [Google Scholar] [CrossRef] [PubMed]
- Courseaux, A.; Richard, F.; Grosgeorge, J.; Ortola, C.; Viale, A.; Turc-Carel, C.; Dutrillaux, B.; Gaudray, P.; Nahon, J.L. Segmental duplications in euchromatic regions of human chromosome 5: A source of evolutionary instability and transcriptional innovation. Genome Res. 2003, 13, 369–381. [Google Scholar] [CrossRef]
- Moller, K.; Kluth, M.; Ahmed, M.; Burkhardt, L.; Moller-Koop, C.; Buscheck, F.; Weidemann, S.; Tsourlakis, M.C.; Minner, S.; Heinzer, H.; et al. Chromosome 5 harbors two independent deletion hotspots at 5q13 and 5q21 that characterize biologically different subsets of aggressive prostate cancer. Int. J. Cancer 2021, 148, 748–758. [Google Scholar] [CrossRef]
- Faderl, S.; Gidel, C.; Kantarjian, H.M.; Manshouri, T.; Keating, M.; Albitar, M. Loss of heterozygosity on chromosome 5 in adults with acute lymphoblastic leukemia. Leuk. Res. 2001, 25, 39–43. [Google Scholar] [CrossRef]
- Wu, X.F.; Zhao, Y.; Kemp, B.L.; Amos, C.I.; Siciliano, M.J.; Spitz, M.R. Chromosome 5 aberrations and genetic predisposition to lung cancer. Int. J. Cancer 1998, 79, 490–493. [Google Scholar] [CrossRef]
- Johannsdottir, H.K.; Jonsson, G.; Johannesdottir, G.; Agnarsson, B.A.; Eerola, H.; Arason, A.; Heikkila, P.; Egilsson, V.; Olsson, H.; Johannsson, O.T.; et al. Chromosome 5 imbalance mapping in breast tumors from BRCA1 and BRCA2 mutation carriers and sporadic breast tumors. Int. J. Cancer 2006, 119, 1052–1060. [Google Scholar] [CrossRef]
- Chiu, H.H.L.; Yong, T.M.K.; Wang, J.; Wang, Y.W.; Vessella, R.L.; Ueda, T.; Wang, Y.Z.; Sadar, M.D. Induction of neuronal apoptosis inhibitory protein expression in response to androgen deprivation in prostate cancer. Cancer Lett. 2010, 292, 176–185. [Google Scholar] [CrossRef][Green Version]
- Bendriem, R.M.; Singh, S.; Aleem, A.A.; Antonetti, D.A.; Ross, M.E. Tight junction protein occludin regulates progenitor Self-Renewal and survival in developing cortex. Elife 2019, 8, 8. [Google Scholar] [CrossRef]
- NCBI. OCLN Occludin Homo Sapiens (Human)—Gene—National Center for Biotechnology Information. Available online: https://www.ncbi.nlm.nih.gov/gene/100506658 (accessed on 12 March 2020).
- Micci, F.; Gorunova, L.; Agostini, A.; Johannessen, L.E.; Brunetti, M.; Davidson, B.; Heim, S.; Panagopoulos, I. Cytogenetic and molecular profile of endometrial stromal sarcoma. Gene Chromosom. Cancer 2016, 55, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Iwakawa, R.; Takenaka, M.; Kohno, T.; Shimada, Y.; Totoki, Y.; Shibata, T.; Tsuta, K.; Nishikawa, R.; Noguchi, M.; Sato-Otsubo, A.; et al. Genome-wide identification of genes with amplification and/or fusion in small cell lung cancer. Genes Chromosome. Cancer 2013, 52, 802–816. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, K.; Wang, Q.; Torres-Garcia, W.; Zheng, S.; Vegesna, R.; Kim, H.; Verhaak, R.G. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene 2015, 34, 4845–4854. [Google Scholar] [CrossRef] [PubMed]
- NCBI. DNAJC6 DnaJ heat shock protein family (Hsp40) member C6 [ Homo sapiens (human)—Gene—National Center for Biotechnology Information. Available online: https://www.ncbi.nlm.nih.gov/gene/9829 (accessed on 5 March 2020).
- NIH. PDE1C Gene. Available online: https://ghr.nlm.nih.gov/gene/PDE1C (accessed on 5 March 2020).
- de Alexandre, R.B.; Horvath, A.D.; Szarek, E.; Manning, A.D.; Leal, L.F.; Kardauke, F.; Epstein, J.A.; Carraro, D.M.; Soares, F.A.; Apanasovich, T.V.; et al. Phosphodiesterase sequence variants may predispose to prostate cancer. Endocr. Relat. Cancer 2015, 22, 519–530. [Google Scholar] [CrossRef]
- Mangangcha, I.R.; Malik, M.Z.; Kucuk, O.; Ali, S.; Singh, R.K.B. Identification of key regulators in prostate cancer from gene expression datasets of patients. Sci. Rep. 2019, 9, 16420. [Google Scholar] [CrossRef]
- Shao, J.; Wang, L.; Zhong, C.; Qi, R.; Li, Y. AHSA1 regulates proliferation, apoptosis, migration, and invasion of osteosarcoma. Biomed. Pharmacother 2016, 77, 45–51. [Google Scholar] [CrossRef]
- Lin, W.H.; Asmann, Y.W.; Anastasiadis, P.Z. Expression of polarity genes in human cancer. Cancer Inform. 2015, 14, 15–28. [Google Scholar] [CrossRef]
- GenesCards. THBS1 Gene. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=THBS1 (accessed on 10 April 2020).
- Li, Q.; Ahuja, N.; Burger, P.C.; Issa, J.-P.J. Methylation and silencing of the Thrombospondin-1 promoter in human cancer. Oncogene 1999, 18, 3284–3289. [Google Scholar] [CrossRef]
- NIH. KANSL1 Gene. Available online: https://ghr.nlm.nih.gov/gene/KANSL1 (accessed on 5 March 2020).
- GenesCards. ARL17B Gene. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=ARL17B (accessed on 5 March 2020).
- Babiceanu, M.; Qin, F.; Xie, Z.; Jia, Y.; Lopez, K.; Janus, N.; Facemire, L.; Kumar, S.; Pang, Y.; Qi, Y.; et al. Recurrent chimeric fusion RNAs in non-cancer tissues and cells. Nucleic Acids Res. 2016, 44, 2859–2872. [Google Scholar] [CrossRef]
- Wen, H.X.; Li, Y.J.; Malek, S.N.; Kim, Y.C.; Xu, J.; Chen, P.X.; Xiao, F.X.; Huang, X.; Zhou, X.Z.; Xuan, Z.Y.; et al. New Fusion Transcripts Identified in Normal Karyotype Acute Myeloid Leukemia. PLoS ONE 2012, 7, e51203. [Google Scholar] [CrossRef]
- Dupain, C.; Harttrampf, A.C.; Boursin, Y.; Lebeurrier, M.; Rondof, W.; Robert-Siegwald, G.; Khoueiry, P.; Geoerger, B.; Massaad-Massade, L. Discovery of New Fusion Transcripts in a Cohort of Pediatric Solid Cancers at Relapse and Relevance for Personalized Medicine. Mol. Ther. 2019, 27, 200–218. [Google Scholar] [CrossRef] [PubMed]
- NIH. FOXP2 Gene. Available online: https://ghr.nlm.nih.gov/gene/FOXP2 (accessed on 5 March 2020).
- Herrero, M.J.; Gitton, Y. The untold stories of the speech gene, the FOXP2 cancer gene. Genes Cancer 2018, 9, 11–38. [Google Scholar] [CrossRef]
- Yehia, G.; Razavi, R.; Memin, E.; Schlotter, F.; Molina, C.A. The Expression of Inducible cAMP Early Repressor (ICER) Is Altered in Prostate Cancer Cells and Reverses the Transformed Phenotype of the LNCaP Prostate Tumor Cell Line. Cancer Res. 2001, 61, 6055–6059. [Google Scholar] [PubMed]
- Krzyszczyk, P.; Acevedo, A.; Davidoff, E.J.; Timmins, L.M.; Marrero-Berrios, I.; Patel, M.; White, C.; Lowe, C.; Sherba, J.J.; Hartmanshenn, C.; et al. The growing role of precision and personalized medicine for cancer treatment. Technol. (Singap. World Sci.) 2018, 6, 79–100. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| European American (n = 17) | African American (n = 10) | |
|---|---|---|
| Age, years, mean (SD) | 61.4 (4.78) | 63.4 (5.21) |
| Radical Prostatectomy Gleason Score, n (%) | ||
| 3 + 3 | 4 (23.5) | 2 (20.0) |
| 3 + 4 | 10 (58.8) | 7 (70.0) |
| 4 + 3 | 1 (5.88) | 0 (0.00) |
| 4 + 3 + 5 | 2 (11.8) | 1 (10.0) |
| Tumor, n (%) | ||
| Confined | 15 (88.2) | 7 (70.0) |
| Extension | 2 (11.8) | 3 (30.0) |
| Nodal Involvement, n (%) | ||
| N0 | 13 (76.5) | 7 (70.0) |
| NX | 4 (23.5) | 2 (20.0) |
| Program | STAR-Fusion | FusionCatcher | JAFFA | ChimeraScan |
|---|---|---|---|---|
| Data type | Single and paired end | Single and paired end | Single and paired end | Paired end only |
| Preprocessing (Y/N) | Y | N | Y | N |
| Samples analyzed | 27 samples | 17 samples | 27 samples | 17 samples |
| 10 AA | 10 AA | 10 AA | 10 AA | |
| 17 EA | 7 EA | 17 EA | 7 EA | |
| Genome build | GRCh38 | GRCh38 | GRCh38 | GRCh37 |
| Program | STAR-Fusion | FusionCatcher | JAFFA | ChimeraScan |
|---|---|---|---|---|
| Release year | 2017 | 2012 | 2015 | 2011 |
| Installation | Python script | Java script | C++ script | |
| Alignment tools | STAR | Bowtie, Bowtie2, Liftover, STAR, Velvet, FaToTwoBit, SAMtools, Seqtk, Numpy, Biopython, Picard, Parallel | Bpipe, Velvet, Oases, SAMtools, Bowtie2, BLAT, Dedupe, Reformat | Bowtie |
| Reference | Transcriptome | Genome and transcriptome | Transcriptome | Genome and transcriptome |
| Reads format | Single end (suggested reads > 100 bp) and paired end | Single end (reads > 130 bp) and paired end | Single end (any length) and paired end | Paired end |
| Class | Read mapping | Read mapping | Read mapping | Read mapping |
| Input data | RNA-Seq | RNA-Seq | RNA-Seq | RNA-Seq |
| Human Assembly | GRCh38 | GRCh38 | GRCh38 | GRCh37 |
| Number of Different Fusion Genes Detected by Each Tool | |||||
|---|---|---|---|---|---|
| Group | STAR-Fusion | FusionCatcher | JAFFA | ChimeraScan | Total (Nonredundant) Fusions |
| AA | 26 | 4753 | 3290 | 1134 | 9203 |
| EA | 10 | 278 | 1530 | 991 | 2809 |
| Total | 36 | 5031 | 4820 | 2125 | |
| Fusion Gene | Race | Patient ID | Tools |
|---|---|---|---|
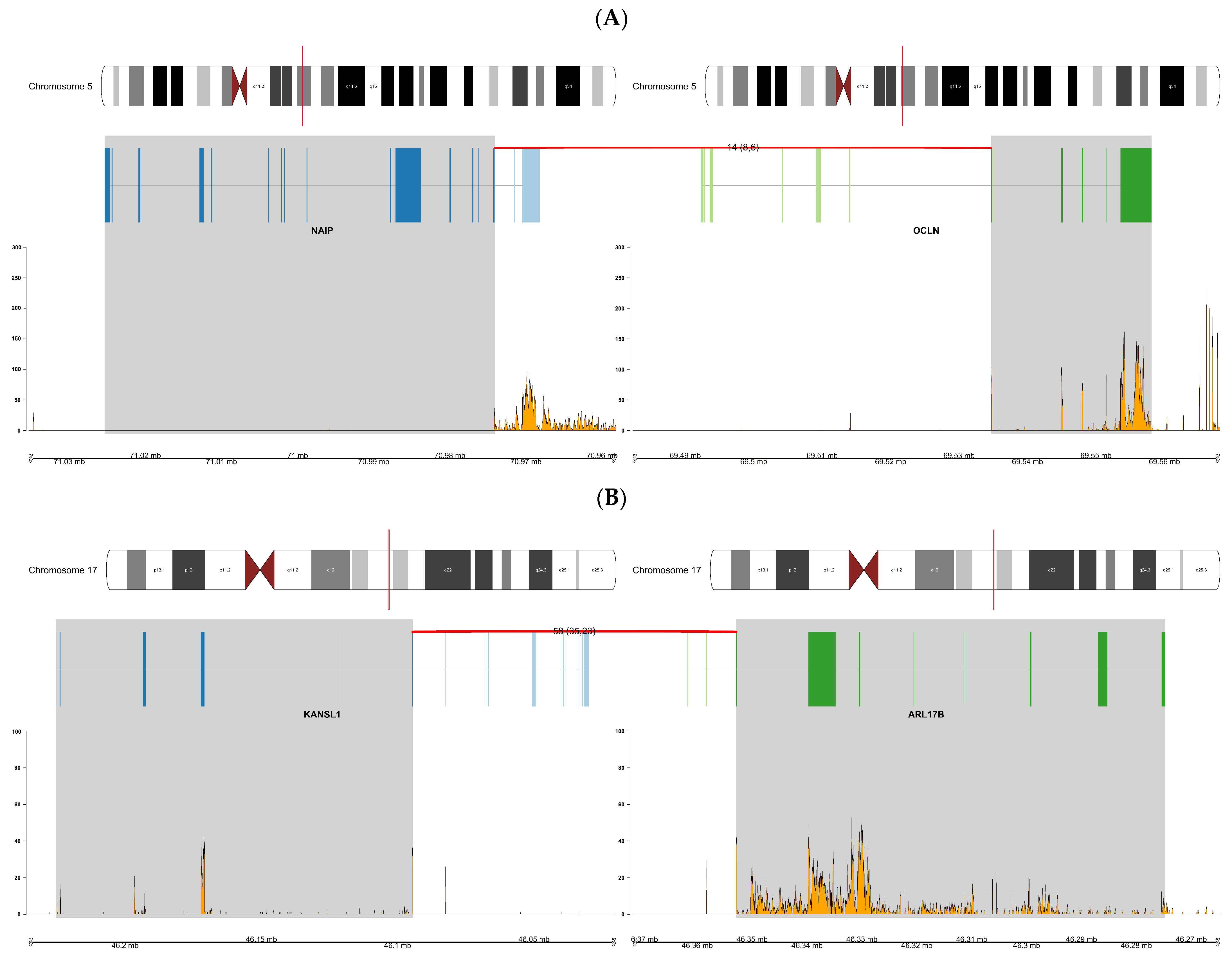
| NAIP:OCLN | AA | AA 20, 22, 24 | ChimeraScan, FusionCatcher, JAFFA |
| NAIP:OCLN | EA | EA 15 | ChimeraScan, FusionCatcher, JAFFA |
| PDE1C:DNAJC6 | AA | AA 24 | ChimeraScan, FusionCatcher, JAFFA |
| KANSL1:ARL17B | EA | EA 5, 6, 15 | ChimeraScan, FusionCatcher, JAFFA |
| FOXP2:CREM | EA | EA 1 | ChimeraScan, FusionCatcher, JAFFA |
| AHSA1:DLG3 | EA | EA 15 | STAR-Fusion, FusionCatcher, JAFFA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgan, R.; Keeley, D.; Hazard, E.S.; Allott, E.H.; Wolf, B.; Savage, S.J.; Hughes Halbert, C.; Gattoni-Celli, S.; Hardiman, G. Fusion Genes in Prostate Cancer: A Comparison in Men of African and European Descent. Biology 2022, 11, 625. https://doi.org/10.3390/biology11050625
Morgan R, Keeley D, Hazard ES, Allott EH, Wolf B, Savage SJ, Hughes Halbert C, Gattoni-Celli S, Hardiman G. Fusion Genes in Prostate Cancer: A Comparison in Men of African and European Descent. Biology. 2022; 11(5):625. https://doi.org/10.3390/biology11050625
Chicago/Turabian StyleMorgan, Rebecca, Dulcie Keeley, E. Starr Hazard, Emma H. Allott, Bethany Wolf, Stephen J. Savage, Chanita Hughes Halbert, Sebastiano Gattoni-Celli, and Gary Hardiman. 2022. "Fusion Genes in Prostate Cancer: A Comparison in Men of African and European Descent" Biology 11, no. 5: 625. https://doi.org/10.3390/biology11050625
APA StyleMorgan, R., Keeley, D., Hazard, E. S., Allott, E. H., Wolf, B., Savage, S. J., Hughes Halbert, C., Gattoni-Celli, S., & Hardiman, G. (2022). Fusion Genes in Prostate Cancer: A Comparison in Men of African and European Descent. Biology, 11(5), 625. https://doi.org/10.3390/biology11050625