
AmazonForest: In Silico Metaprediction of Pathogenic Variants

 , , , ,
, , , ,  , and
, and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Fetch ClinVar .vcf File
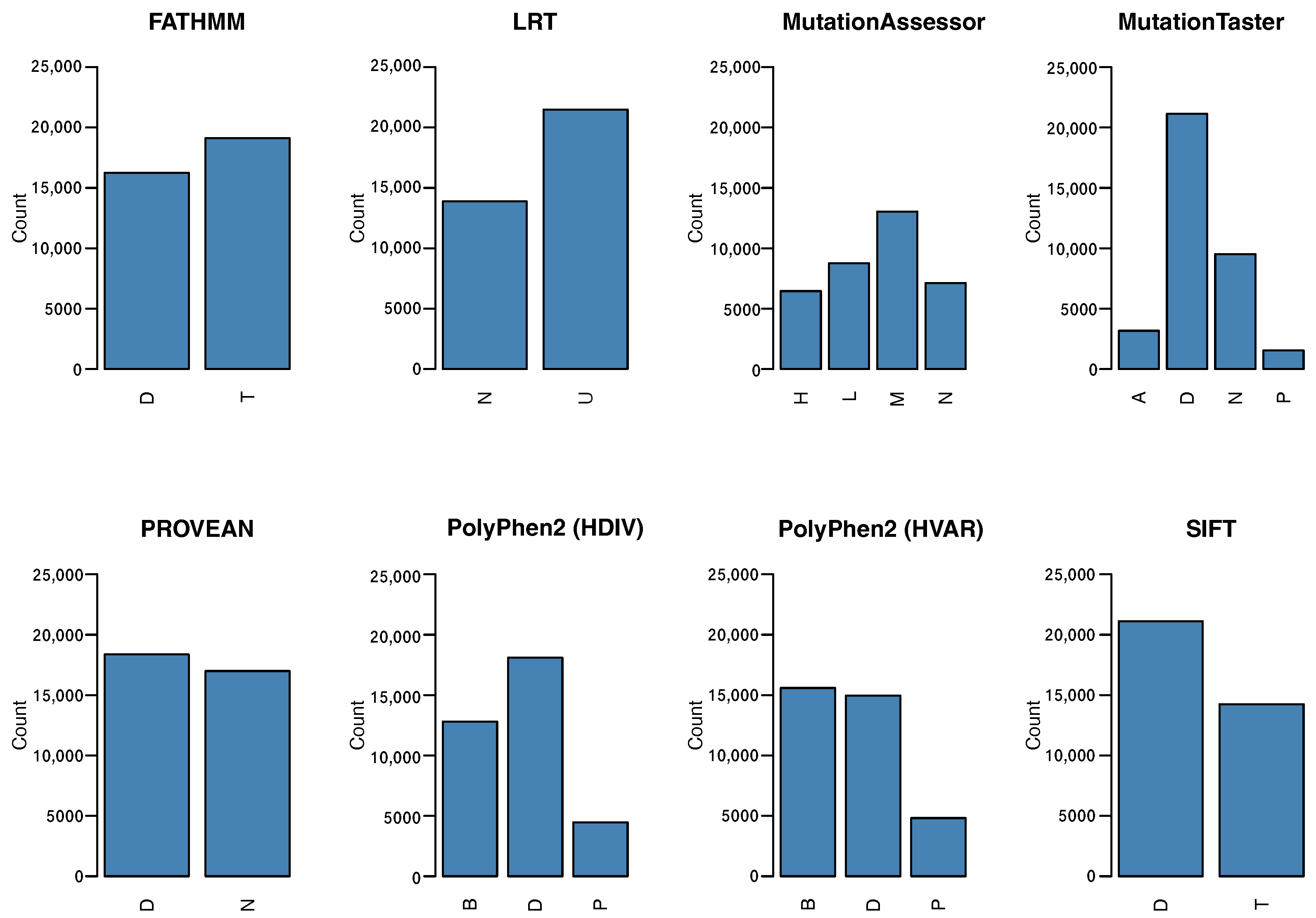
2.2. Functional Impact Variant Annotation by Single Predictors
- FATHMM predicts the functional effects of coding and noncoding variants. This predictor combines wild-type and mutated sequences in a hidden Markov model, which identifies mutations in peptide chains, showing the alignment of homologous sequences and conserved protein domains [15];
- SIFT (Sorting Intolerant From Tolerant) is a prediction tool that codes an algorithm for amino acid substitution analyses. It assumes that important positions in a protein sequence have been conserved throughout evolution, and therefore substitutions at these positions may affect protein function. The algorithm sorts changes in a polypeptide chain as tolerant or intolerant according to its evolutionary conservation [16];
- Polyphen-2 (Polymorphism Phenotyping v2) predicts the impact of amino acid substitutions on structural stability, physical interactions, and human protein function. The probability of a mutation being pathogenic depends on the extraction of sequence annotations, structural attributes, and conservation profiles in protein-coding regions [17];
- PROVEAN (Protein Variation Effect Analyzer) is a predictor that provides a generalized approach to predict the functional effects on variations in a peptide chain. These effects include SNPs, INDELs, or multiple amino acid substitutions. Prediction is performed by employing a mutation database obtained from UniProtKB/Swiss-Prot and other experimental data previously generated from mutagenesis experiments [18];
- MutationAssessor predicts the functional impact of amino acid substitutions on proteins using the evolutionary conservation of the affected amino acid in protein counterparts. Multiple Sequence Alignment is used to reflect functional specificity, represent the functional impact of a missense variant, and generate conservation scores. Variants with higher scores are more likely to be pathogenic [19];
- MutationTaster2 predicts functional changes in DNA sequences. It is designed to predict consequences based on amino acid substitutions, and intronic substitutions such as synonymous changes, short insertion or exclusion mutations, and variants that cover the limits of introns and exons [20];
- Likelihood Ratio Test (LRT) is a metric that evaluates the proportion of synonymous and nonsynonymous mutations in protein-coding regions. The altered proportion of mutations means that a negative selection process occurred over that region during evolution, which consequently modifies codons in peptide chains [21].
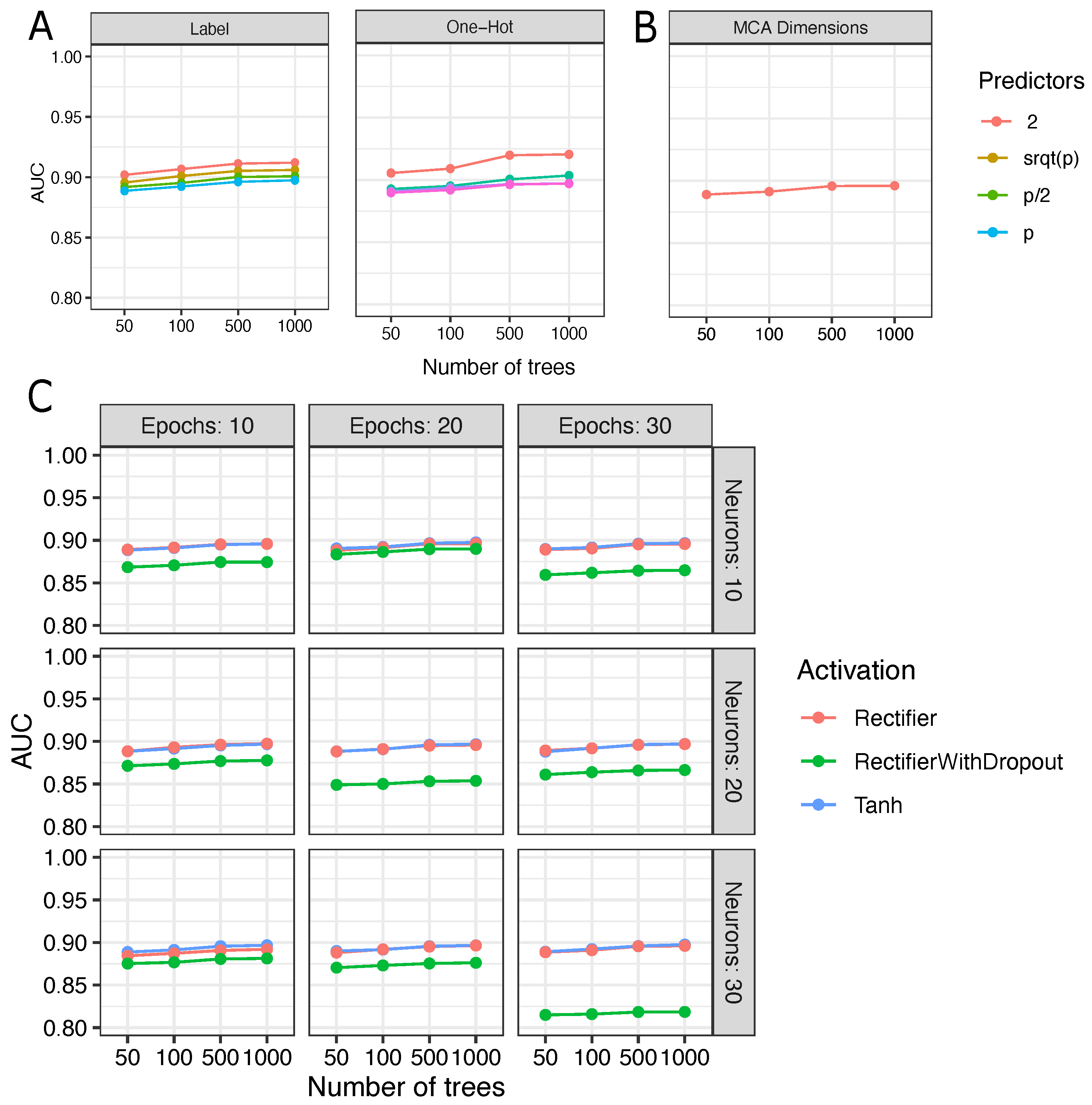
2.3. Encoding Genome-Wide Training and Test Dataset
- Label encoding is an approach that assigns numerical values from 0 to the number of classes −1 to each of the categorical values in a dataset. For example, if the column with categorical values contains five classes, then the label encoding assigns numerical values between 0 and 4;
- One hot encoding transforms categorical variables using a dummy strategy. Each variable category is transformed into a binary column. For example, given a dataset with two categories, the one hot encoder creates two new columns to store binary values, 0 or 1;
- Multiple Correspondence Analysis (MCA) is a statistical method that handles categorical variables for dimensionality reduction. MCA is an extension of simple correspondence analysis and a generalization of principal component analysis, which is appropriate for quantitative data [22]. The MCA is used to create a low-dimensional space for samples and predictor points based on a contingency table, and the dimensions are retained as eigenvalues;
- Autoencoders are unsupervised learning algorithms that aim to obtain a data representation by reconstructing the input data at the output [23]. In this study, artificial neural networks were implemented to learn representations of the ClinVar data. We used an autoencoder similar to a multilayer perceptron (MLP), with an input layer, a hidden layer with 10, 20, and 30 neurons, and an output layer with the same number of predictors. Rectifier (ReLu), Rectifier with Dropout, and Hyperbolic Tangent Function (Tanh) were used as neuron activation functions. Dropout is commonly used to reduce overfitting and can improve the results of a classifier. The function of this regularization layer is to turn off a portion of the neurons, forcing the network to readjust the weights and preventing the network from memorizing the training data [24].
2.4. Fine-Tuning of Random Forest
- (1)
- RF were trained with categorical data and one hot encoder;
- (2)
- RF were trained with two extracted MCA dimensions;
- (3)
- RF were trained with two dimensions from autoencoders based on three diferent activation functions: rectifier, rectifier with dropout and tahn. Moreover, we range the number of epochs and hidden neurons on autoencoders, which were set for 10, 20, and 30 for both parameters.
2.5. AmazonForest: Web Platform for Variant Classification
- The first is the user interface component. AmazonForest was developed as a web tool with an interface that allows performing pathogenicity prediction of SNPs or INDELs with in silico analyses employing the best metapredictor model. The simple web interface enables the user to predict pathogenicity in two ways. First, by providing genomic or dbSNP information (chromosome, chromosome position, or rsID) and second, by allowing the combination of predictor results to query pathogenicity status. The web component was developed using Python3.6 [29], Javascript (https://developer.mozilla.org/en-US/docs/Web/JavaScript/Reference), HTML5 (https://developer.mozilla.org/pt-BR/docs/Web/HTML/HTML5), and using frameworks such as Flask (v.2) (https://palletsprojects.com/p/flask/), scikit-learn [30], Pandas (v.1.1.5) [31], Numpy (V.1.19.5) [32]. All packages was acessed on 2 February 2021.
- The second is a model administrator component to assess the evolution and performance of the model. This model component enables the reproducibility of up-to-date data.
3. Results
3.1. Training and Test Data Records
3.2. Fine-Tuning and Selection of Metaprediction Model
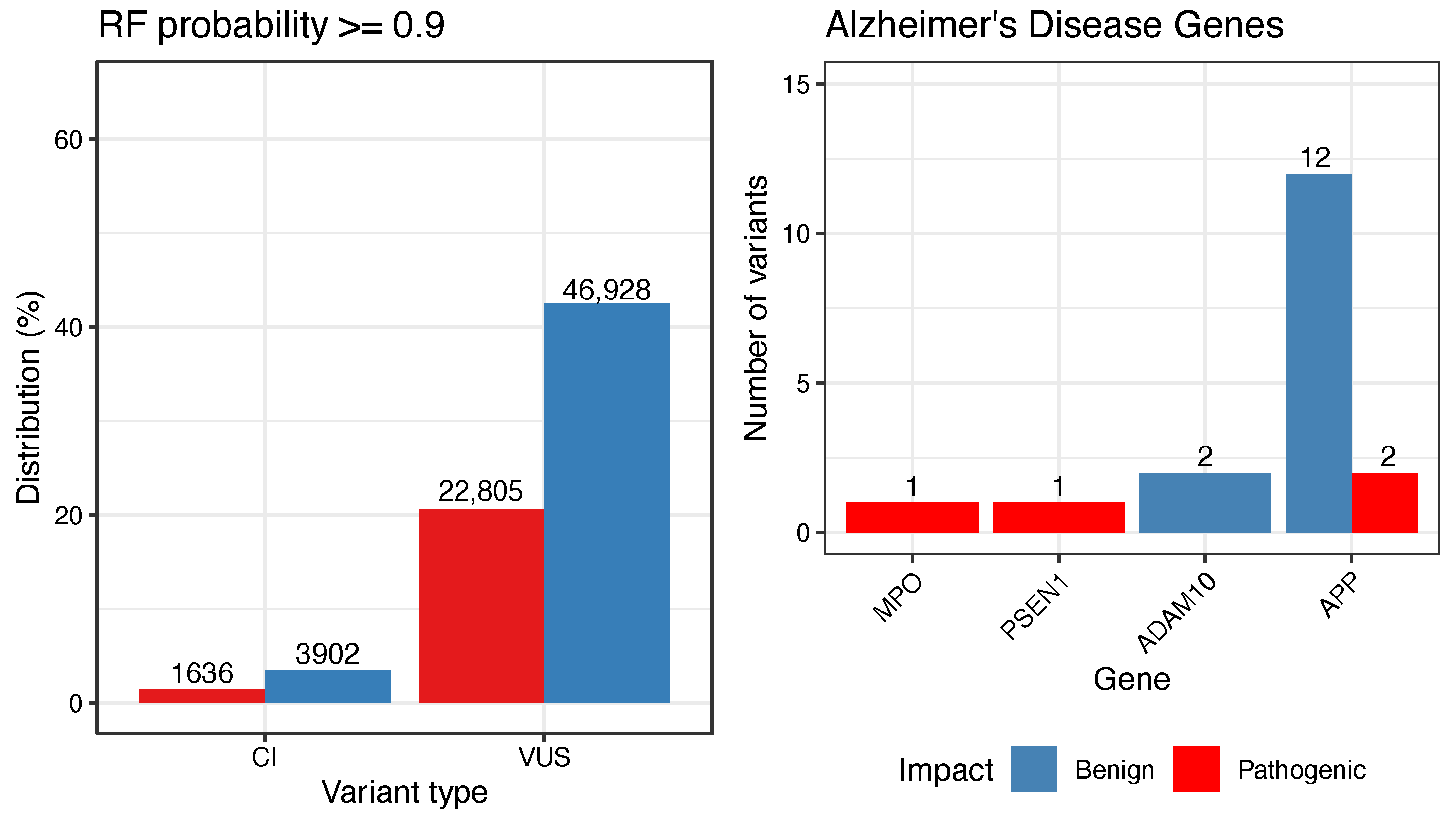
3.3. Reclassification of VUS and CI Variants
3.4. Case Study: Alzheimer’s Disease-Related Genes
4. Discussion
5. Conclusions
6. Software Availability
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NGS | Next Generation Sequencing |
| GWAS | Genome Wide Association Studies |
| SNP | Single Nucleotide Polymorphism |
| ML | Machine Learning |
| VUS | Variants of uncertain significance |
| CI | Conflit of interpretation |
| RF | Random Forest |
| ROC | Receiver Operating Curve |
| AUC | Area Under Curve |
References
- MacArthur, J.; Bowler, E.; Cerezo, M.; Gil, L.; Hall, P.; Hastings, E.; Junkins, H.; McMahon, A.; Milano, A.; Morales, J.; et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017, 45, D896–D901. [Google Scholar] [CrossRef] [PubMed]
- Araújo, G.S.; Lima, L.H.C.; Schneider, S.; Leal, T.P.; da Silva, A.P.C.; Vaz de Melo, P.O.; Tarazona-Santos, E.; Scliar, M.O.; Rodrigues, M.R. Integrating, summarizing and visualizing GWAS-hits and human diversity with DANCE (Disease-ANCEstry networks). Bioinformatics 2016, 32, 1247–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, N.; Zhou, H.; Fan, H.; Yuan, Y. Single nucleotide polymorphisms and cancer susceptibility. Oncotarget 2017, 8, 110635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unoki, H.; Takahashi, A.; Kawaguchi, T.; Hara, K.; Horikoshi, M.; Andersen, G.; Ng, D.P.; Holmkvist, J.; Borch-Johnsen, K.; Jørgensen, T.; et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat. Genet. 2008, 40, 1098–1102. [Google Scholar] [CrossRef]
- Araújo, G.S.; Souza, M.R.; Oliveira, J.R.M.; Costa, I.G. Random Forest and Gene Networks for Association of SNPs to Alzheimer’s Disease. In Brazilian Symposium on Bioinformatics; Springer: Cham, Switzerland, 2013; pp. 104–115. [Google Scholar]
- Souza, M.; Araujo, G.; Costa, I.; Oliveira, J.; Initiative, A.D.N. Combined genome-wide CSF Aβ-42’s associations and simple network properties highlight new risk factors for Alzheimer’s disease. J. Mol. Neurosci. 2016, 58, 120–128. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2013, 42, D980–D985. [Google Scholar] [CrossRef] [Green Version]
- Alzu’bi, A.A.; Zhou, L.; Watzlaf, V.J. Genetic variations and precision medicine. Perspect. Health Inf. Manag. 2019, 16, 1a. [Google Scholar]
- Ranganathan Ganakammal, S.; Alexov, E. An Ensemble Approach to Predict the Pathogenicity of Synonymous Variants. Genes 2020, 11, 1102. [Google Scholar] [CrossRef]
- Hassan, M.S.; Shaalan, A.; Dessouky, M.; Abdelnaiem, A.E.; ElHefnawi, M. Evaluation of computational techniques for predicting non-synonymous single nucleotide variants pathogenicity. Genomics 2019, 111, 869–882. [Google Scholar] [CrossRef]
- Jaravine, V.; Balmford, J.; Metzger, P.; Boerries, M.; Binder, H.; Boeker, M. Annotation of Human Exome Gene Variants with Consensus Pathogenicity. Genes 2020, 11, 1076. [Google Scholar] [CrossRef]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do Nascimento, P.M.; Medeiros, I.G.; Falcão, R.M.; Stransky, B.; de Souza, J.E.S. A decision tree to improve identification of pathogenic mutations in clinical practice. BMC Med. Inform. Decis. Mak. 2020, 20, 52. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.; Edwards, K.J.; Day, I.N.; Gaunt, T.R. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Reva, B.; Antipin, Y.; Sander, C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 2007, 8, R232. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef] [Green Version]
- Abdi, H.; Williams, L.J. Principal component analysis. In Wiley Interdisciplinary Reviews: Computational Statistics 2.4; Wiley: Hoboken, NJ, USA, 2010; pp. 433–459. [Google Scholar]
- Team, H. Deep Learning, Neural Networks and Autoenconders. 2022. Available online: https://docs.h2o.ai/h2o/latest-stable/h2o-docs/data-science/deep-learning.html?highlight=autoencoder (accessed on 2 December 2021).
- Hinton, G.E.; Srivastava, N.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R.R. Improving neural networks by preventing co-adaptation of feature detectors. arXiv 2012, arXiv:1207.0580. [Google Scholar]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Strobl, C.; Boulesteix, A.L.; Zeileis, A.; Hothorn, T. Bias in random forest variable importance measures: Illustrations, sources and a solution. BMC Bioinform. 2007, 8, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, B.A.; Hubbard, A.E.; Cutler, A.; Barcellos, L.F. An application of Random Forests to a genome-wide association dataset: Methodological considerations & new findings. BMC Genet. 2010, 11, 49. [Google Scholar]
- Ojala, M.; Garriga, G.C. Permutation tests for studying classifier performance. J. Mach. Learn. Res. 2010, 11, 1833–1863. [Google Scholar]
- Van Rossum, G.; Drake, F.L. Python 3 Reference Manual; CreateSpace: Scotts Valley, CA, USA, 2009. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- McKinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 10–16 July 2010; pp. 56–61. [Google Scholar]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef]
- Brito, L.M.; Ribeiro-dos Santos, Â.; Vidal, A.F.; de Araújo, G.S. Differential expression and mirna–gene interactions in early and late mild cognitive impairment. Biology 2020, 9, 251. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Alasmari, F.; Alshammari, M.A.; Alasmari, A.F.; Alanazi, W.A.; Alhazzani, K. Neuroinflammatory cytokines induce amyloid beta neurotoxicity through modulating amyloid precursor protein levels/metabolism. BioMed Res. Int. 2018, 1–8. [Google Scholar] [CrossRef]
- Miller, L.M.; Wang, Q.; Telivala, T.P.; Smith, R.J.; Lanzirotti, A.; Miklossy, J. Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with β-amyloid deposits in Alzheimer’s disease. J. Struct. Biol. 2006, 155, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Zhang, Y. The association of MPO gene promoter polymorphisms with Alzheimer’s disease risk in Chinese Han population. Oncotarget 2017, 8, 107870. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, W.F.; Rhees, J.; Maciejewski, D.; Paladino, T.; Sieburg, H.; Maki, R.A.; Masliah, E. Myeloperoxidase polymorphism is associated with gender specific risk for Alzheimer’s disease. Exp. Neurol. 1999, 155, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Lanoiselée, H.M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksanen, M.; Petersen, A.J.; Naumenko, N.; Puttonen, K.; Lehtonen, Š.; Olivé, M.G.; Shakirzyanova, A.; Leskelä, S.; Sarajärvi, T.; Viitanen, M.; et al. PSEN1 mutant iPSC-derived model reveals severe astrocyte pathology in Alzheimer’s disease. Stem Cell Rep. 2017, 9, 1885–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arber, C.; Lovejoy, C.; Harris, L.; Willumsen, N.; Alatza, A.; Casey, J.M.; Lines, G.; Kerins, C.; Mueller, A.K.; Zetterberg, H.; et al. Familial Alzheimer’s disease mutations in PSEN1 lead to premature human stem cell neurogenesis. Cell Rep. 2021, 34, 108615. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.Z.; Sun, S.; Tan, C.C.; Yu, J.T.; Tan, L. The role of ADAM10 in Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 58, 303–322. [Google Scholar] [CrossRef]
- Manzine, P.R.; Ettcheto, M.; Cano, A.; Busquets, O.; Marcello, E.; Pelucchi, S.; Di Luca, M.; Endres, K.; Olloquequi, J.; Camins, A.; et al. ADAM10 in Alzheimer’s disease: Pharmacological modulation by natural compounds and its role as a peripheral marker. Biomed. Pharmacother. 2019, 113, 108661. [Google Scholar] [CrossRef]
- Spilman, P.; Bredesen, D.; John, V. Enhancement of sAPPalpha as a Therapeutic Strategy for Alzheimer’s and other Neurodegenerative Diseases. J. Alzheimer’s Neurodegener. Dis. 2015, 1, 1–10. [Google Scholar]
- Au, T.C. Random forests, decision trees, and categorical predictors: The “absent levels” problem. J. Mach. Learn. Res. 2018, 19, 1737–1766. [Google Scholar]
- Mitchell, T.M. Generative and discriminative classifiers: Naive bayes and logistic regression. Mach. Learn. 2010, 1–17. [Google Scholar]
- IJCAI 2001 workshop on empirical methods in artificial intelligence. Mach. Learn. 2001, 3, 41–46.
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Category of Genetic Variants in CinVar | Original Dataset | Training Dataset | Reclassification Dataset |
|---|---|---|---|
| Benign | 266,145 | 18,891 | - |
| Pathogenic | 130,739 | 16,471 | - |
| With conflit of interpretation | 42,609 | - | 7193 |
| With uncertain significance | 349,926 | - | 93,612 |
| Chromosome | Position | Gene | Protein | Protein Change | dbSNP ID | ClinVar Significance | AmazonForest Prediction |
|---|---|---|---|---|---|---|---|
| 21 | 26000066 | APP | NM_000484.4 | c.982CT (p.Arg328Trp) | VUS | Pathogenic | |
| 21 | 26090000 | APP | NM_000484.4 | c.298CT (p.Arg100Trp) | rs200347552 | VUS | Pathogenic |
| 17 | 58278000 | MPO | NM_000250.2 | c.1031GA (p.Gly344Asp) | VUS | Pathogenic | |
| 14 | 73173702 | PSEN1 | NM_000021.4 | c.475TC (p.Tyr159His) | VUS | Pathogenic | |
| Chromosome | Position | Gene | Protein | Protein Change | dbSNP ID | ClinVar Significance | named-content content-type="color: #FFFFFF">AmazonForest Prediction |
| 15 | 58665141 | ADAM10 | NM_001110.4 | c.541AG (p.Arg181Gly) | rs145518263 | VUS | Benign |
| 15 | 58665172 | ADAM10 | NM_001110.4 | c.510GC (p.Gln170His) | rs61751103 | VUS | Benign |
| 21 | 25997360 | APP | NM_000484.4 | c.1090CT (p.Leu364Phe) | rs749453173 | VUS | Benign |
| 21 | 25997413 | APP | NM_000484.4 | c.1037CA (p.Ser346Tyr) | VUS | Benign | |
| 21 | 26000018 | APP | NM_000484.4 | c.1030GA (p.Ala344Thr) | rs201045185 | VUS | Benign |
| 21 | 26000167 | APP | NM_000484.4 | c.881AG (p.Gln294Arg) | VUS | Benign | |
| 21 | 26021902 | APP | NM_000484.4 | c.803GA (p.Arg268Lys) | rs1601237753 | VUS | Benign |
| 21 | 26021954 | APP | NM_000484.4 | c.751GA (p.Gly251Ser | VUS | Benign | |
| 21 | 26021978 | APP | NM_000484.4 | c.727GA (p.Asp243Asn) | VUS | Benign | |
| 21 | 26022001 | APP | NM_000484.4 | c.704CT (p.Ala235Val) | CI | Benign | |
| 21 | 26022031 | APP | NM_000484.4 | c.674TC (p.Val225Ala) | rs746313873 | VUS | Benign |
| 21 | 26051060 | APP | NM_000484.4 | c.602CT (p.Ala201Val) | rs149995579 | VUS | Benign |
| 21 | 26051088 | APP | NM_000484.4 | c.574GA (p.Glu192Lys) | VUS | Benign | |
| 21 | 26170574 | APP | NM_000484.4 | c.47GA (p.Arg16Gln) | VUS | Benign |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palheta, H.G.A.; Gonçalves, W.G.; Brito, L.M.; dos Santos, A.R.; dos Reis Matsumoto, M.; Ribeiro-dos-Santos, Â.; de Araújo, G.S. AmazonForest: In Silico Metaprediction of Pathogenic Variants. Biology 2022, 11, 538. https://doi.org/10.3390/biology11040538
Palheta HGA, Gonçalves WG, Brito LM, dos Santos AR, dos Reis Matsumoto M, Ribeiro-dos-Santos Â, de Araújo GS. AmazonForest: In Silico Metaprediction of Pathogenic Variants. Biology. 2022; 11(4):538. https://doi.org/10.3390/biology11040538
Chicago/Turabian StylePalheta, Helber Gonzales Almeida, Wanderson Gonçalves Gonçalves, Leonardo Miranda Brito, Arthur Ribeiro dos Santos, Marlon dos Reis Matsumoto, Ândrea Ribeiro-dos-Santos, and Gilderlanio Santana de Araújo. 2022. "AmazonForest: In Silico Metaprediction of Pathogenic Variants" Biology 11, no. 4: 538. https://doi.org/10.3390/biology11040538
APA StylePalheta, H. G. A., Gonçalves, W. G., Brito, L. M., dos Santos, A. R., dos Reis Matsumoto, M., Ribeiro-dos-Santos, Â., & de Araújo, G. S. (2022). AmazonForest: In Silico Metaprediction of Pathogenic Variants. Biology, 11(4), 538. https://doi.org/10.3390/biology11040538