
Hemophagocytic Lymphohistiocytosis Gene Variants in Multisystem Inflammatory Syndrome in Children

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Identification of Gene Variants in the Patient Cohort
2.2. DOCK8 WT and Mutant Gene Preparations
2.3. Recombinant FV Preparation and Infection of NK-92 Cells
2.4. NK-92 Cell CD107a Expression Assays (Degranulation Assays)
2.5. NK-92 Cell Cytotoxicity Assays
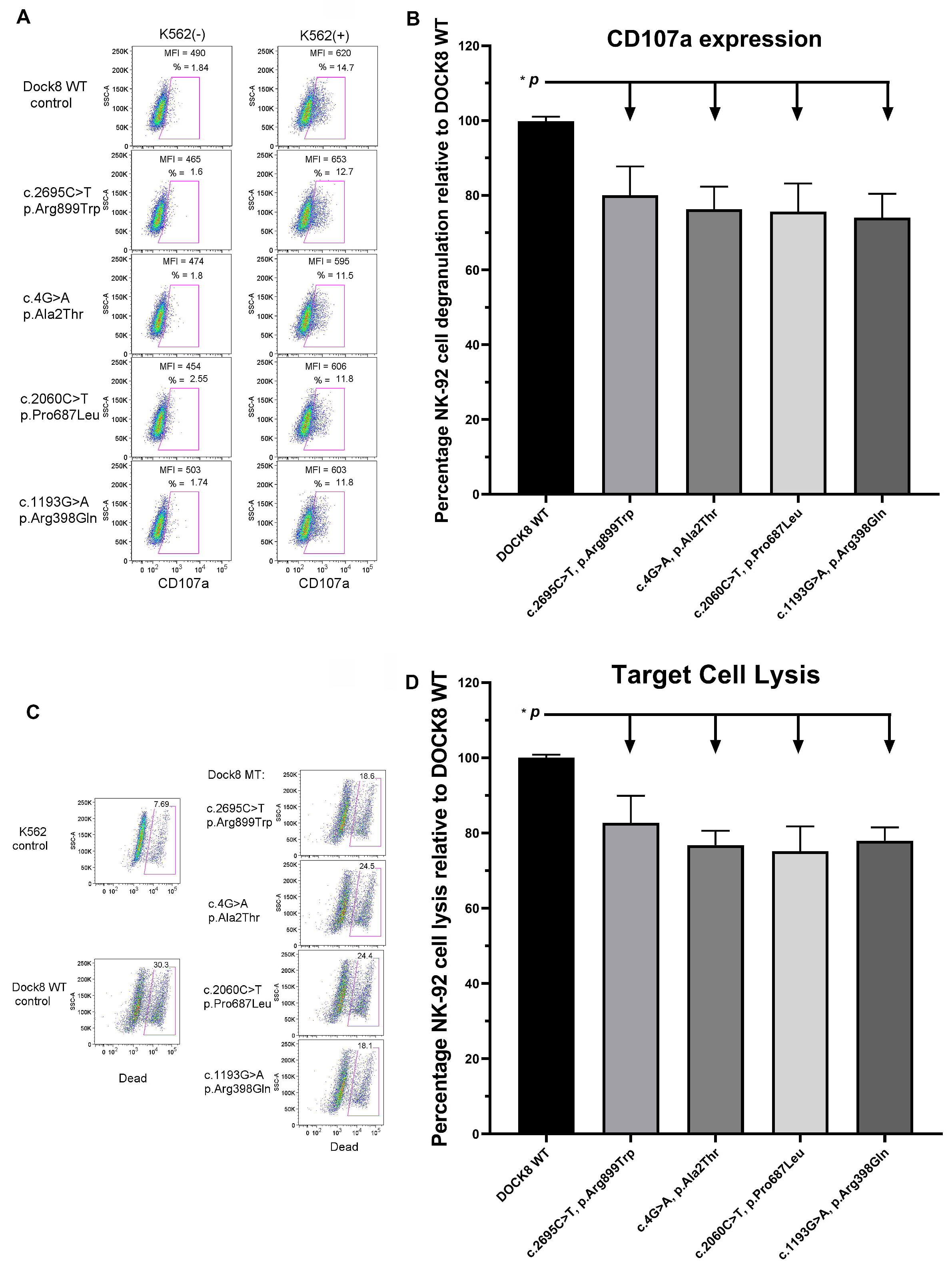
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Multisystem Inflammatory Syndrome in Children (MIS-C); Center for Disease Control and Prevention: Atlanta, GA, USA, 2020. Available online: https://www.cdc.gov/mis-c/hcp/ (accessed on 5 January 2022).
- Riphagen, S.; Gomez, X.; Gonzalez-Martinez, C.; Wilkinson, N.; Theocharis, P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet 2020, 395, 1607–1608. [Google Scholar] [CrossRef]
- Davies, P.; Evans, C.; Kanthimathinathan, H.K.; Lillie, J.; Brierley, J.; Waters, G.; Johnson, M.; Griffiths, B.; Du Pré, P.; Mohammad, Z.; et al. Intensive care admissions of children with paediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2 (PIMS-TS) in the UK: A multicentre observational study. Lancet Child Adolesc. Health 2020, 4, 669–677. [Google Scholar] [CrossRef]
- Galeotti, C.; Bayry, J. Autoimmune and inflammatory diseases following COVID-19. Nat. Rev. Rheumatol. 2020, 16, 413–414. [Google Scholar] [CrossRef]
- Reiff, D.D.; Mannion, M.L.; Samuy, N.; Scalici, P.; Cron, R.Q. Distinguishing active pediatric COVID-19 pneumonia from MIS-C. Pediatr. Rheumatol. 2021, 19, 21. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Hostallero, D.E.; El Khili, M.R.; Fonseca, G.J.; Milette, S.; Noorah, N.; Guay-Belzile, M.; Spicer, J.; Daneshtalab, N.; Sirois, M.; et al. A network-informed analysis of SARS-CoV-2 and hemophagocytic lymphohistiocytosis genes’ interactions points to Neutrophil extracellular traps as mediators of thrombosis in COVID-19. PLoS Comput. Biol. 2021, 17, e1008810. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Liu, D.; Liu, W.; Wang, G.; Chen, L.; Cao, Y.; Wei, J.; Xiao, M.; Liu, X.; Huang, G.; et al. Germline variants in UNC13D and AP3B1 are enriched in COVID-19 patients experiencing severe cytokine storms ]. Eur. J. Hum. Genet. 2021, 29, 1312–1315. [Google Scholar] [CrossRef] [PubMed]
- Bami, S.; Vagrecha, A.; Soberman, D.; Badawi, M.; Cannone, D.; Lipton, J.M.; Cron, R.Q.; Levy, C.F. The use of anakinra in the treatment of secondary hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer 2020, 67, e28581. [Google Scholar] [CrossRef] [PubMed]
- Eloseily, E.M.; Weiser, P.; Crayne, C.B.; Haines, H.; Mannion, M.L.; Stoll, M.L.; Beukelman, T.; Atkinson, T.P.; Cron, R.Q. Benefit of Anakinra in Treating Pediatric Secondary Hemophagocytic Lymphohistiocytosis. Arthritis Rheumatol. 2020, 72, 326–334. [Google Scholar] [CrossRef]
- Trobridge, G.D. Foamy virus vectors for gene transfer. Expert Opin. Biol. Ther. 2009, 9, 1427–1436. [Google Scholar] [CrossRef]
- Zhang, M.; Bracaglia, C.; Prencipe, G.; Bemrich-Stolz, C.J.; Beukelman, T.; Dimmitt, R.A.; Chatham, W.W.; Zhang, K.; Li, H.; Walter, M.R.; et al. A Heterozygous RAB27A Mutation Associated with Delayed Cytolytic Granule Polarization and Hemophagocytic Lymphohistiocytosis. J. Immunol. 2016, 196, 2492–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulert, G.S.; Cron, R.Q. The genetics of macrophage activation syndrome. Genes Immun. 2020, 21, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Canna, S.W.; De Jesus, A.A.; Gouni, S.; Brooks, S.R.; Marrero, B.; Liu, Y.; DiMattia, M.A.; Zaal, K.J.; Sanchez, G.A.M.; Kim, H.; et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet. 2014, 46, 1140–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufort, E.M.; Koumans, E.H.; Chow, E.J.; Rosenthal, E.M.; Muse, A.; Rowlands, J.; Barranco, M.A.; Maxted, A.M.; Rosenberg, E.S.; Easton, D.; et al. Multisystem Inflammatory Syndrome in Children in New York State. N. Engl. J. Med. 2020, 383, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Capone, C.A.; Subramony, A.; Sweberg, T.; Schneider, J.; Shah, S.; Rubin, L.; Schleien, C.; Epstein, S.; Johnson, J.C.; Kessel, A.; et al. Characteristics, Cardiac Involvement, and Outcomes of Multisystem Inflammatory Syndrome of Childhood Associated with severe acute respiratory syndrome coronavirus 2 Infection. J. Pediatr. 2020, 224, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Consiglio, C.R.; Cotugno, N.; Sardh, F.; Pou, C.; Amodio, D.; Rodriguez, L.; Tan, Z.; Zicari, S.; Ruggiero, A.; Pascucci, G.R.; et al. The Immunology of Multisystem Inflammatory Syndrome in Children with COVID-19. Cell 2020, 183, 968–981.e7. [Google Scholar] [CrossRef]
- Lee, P.Y.; Day-Lewis, M.; Henderson, L.A.; Friedman, K.G.; Lo, J.; Roberts, J.E.; Lo, M.S.; Platt, C.D.; Chou, J.; Hoyt, K.J.; et al. Distinct clinical and immunological features of SARS-CoV-2-induced multisystem inflammatory syndrome in children. J. Clin. Investig. 2020, 130, 5942–5950. [Google Scholar] [CrossRef]
- Prilutskiy, A.; Kritselis, M.; Shevtsov, A.; Yambayev, I.; Vadlamudi, C.; Zhao, Q.; Kataria, Y.; Sarosiek, S.R.; Lerner, A.; Sloan, J.M.; et al. SARS-CoV-2 Infection-Associated Hemophagocytic Lymphohistiocytosis. Am. J. Clin. Pathol. 2020, 154, 466–474. [Google Scholar] [CrossRef]
- Vagrecha, A.; Patel, H.B.; Mamdouhi, T.; Acharya, S.; Appiah-Kubi, A.; Aygun, B.; Vlachos, A.; Wolfe, L.C.; Lipton, J.M.; Cron, R.Q.; et al. Effect of COVID-19 on anakinra-induced remission in homozygous STX11 hemophagocytosis lymphohistiocytosis. Pediatr. Blood Cancer. 2021, 68, e28897. [Google Scholar] [CrossRef]
- Chou, J.; Platt, C.D.; Habiballah, S.; Nguyen, A.A.; Elkins, M.; Weeks, S.; Peters, Z.; Day-Lewis, M.; Novak, T.; Armant, M.; et al. Mechanisms underlying genetic susceptibility to multisystem inflammatory syndrome in children (MIS-C). J. Allergy Clin. Immunol. 2021, 148, 732–738.e1. [Google Scholar] [CrossRef]
- Schulert, G.S.; Zhang, M.; Fall, N.; Husami, A.; Kissell, D.; Hanosh, A.; Zhang, K.; Davis, K.; Jentzen, J.M.; Napolitano, L.; et al. Whole-Exome Sequencing Reveals Mutations in Genes Linked to Hemophagocytic Lymphohistiocytosis and Macrophage Activation Syndrome in Fatal Cases of H1N1 Influenza. J. Infect. Dis. 2016, 213, 1180–1188. [Google Scholar] [CrossRef]
- Risma, K.A.; Frayer, R.W.; Filipovich, A.H.; Sumegi, J. Aberrant maturation of mutant perforin underlies the clinical diversity of hemophagocytic lymphohistiocytosis. J. Clin. Investig. 2006, 116, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Behrens, E.M.; Atkinson, T.P.; Shakoory, B.; Grom, A.A.; Cron, R.Q. Genetic defects in cytolysis in macrophage activation syndrome. Curr. Rheumatol. Rep. 2014, 16, 439. [Google Scholar] [CrossRef] [PubMed]
- Spessott, W.A.; Sanmillan, M.L.; McCormick, M.E.; Patel, N.; Villanueva, J.; Zhang, K.; Nichols, K.E.; Giraudo, C.G. Hemophagocytic lymphohistiocytosis caused by dominant-negative mutations in STXBP2 that inhibit SNARE-mediated membrane fusion. Blood 2015, 125, 1566–1577. [Google Scholar] [CrossRef] [Green Version]
- Schulert, G.S.; Zhang, M.; Husami, A.; Fall, N.; Brunner, H.; Zhang, K.; Cron, R.Q.; Grom, A.A. Brief Report: Novel UNC13D Intronic Variant Disrupting an NF-κB Enhancer in a Patient With Recurrent Macrophage Activation Syndrome and Systemic Juvenile Idiopathic Arthritis. Arthritis Rheumatol. 2018, 70, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Jordan, M.B.; Marsh, R.A.; Johnson, J.A.; Kissell, D.; Meller, J.; Villanueva, J.; Risma, K.A.; Wei, Q.; Klein, P.S.; et al. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial HLH. Blood 2011, 118, 5794–5798. [Google Scholar] [CrossRef] [Green Version]
- Strippoli, R.; Caiello, I.; De Benedetti, F. Reaching the threshold: A multilayer pathogenesis of macrophage activation syndrome. J. Rheumatol. 2013, 40, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Chinn, I.K.; Eckstein, O.S.; Peckham-Gregory, E.C.; Goldberg, B.R.; Forbes, L.R.; Nicholas, S.K.; Mace, E.M.; Vogel, T.P.; Abhyankar, H.A.; Diaz, M.I.; et al. Genetic and mechanistic diversity in pediatric hemophagocytic lymphohistiocytosis. Blood 2018, 132, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Biggs, C.M.; Keles, S.; Chatila, T.A. DOCK8 deficiency: Insights into pathophysiology, clinical features and management. Clin. Immunol. 2017, 181, 75–82. [Google Scholar] [CrossRef]
- Cron, R.Q.; Zhang, M.; Cron, R.R.; Absher, D.; Atkinson, P.; Chatham, W. Characterization of DOCK8 as a novel gene associated with hemophagocytic lymphohistiocytosis (abstract). J. Immunol. 2020, 204 (Suppl. 1), 29. [Google Scholar]
- Mizesko, M.C.; Banerjee, P.P.; Monaco-Shawver, L.; Mace, E.M.; Bernal, W.E.; Sawalle-Belohradsky, J.; Belohradsky, B.H.; Heinz, V.; Freeman, A.F.; Sullivan, K.E.; et al. Defective actin accumulation impairs human natural killer cell function in patients with dedicator of cytokinesis 8 deficiency. J. Allergy Clin. Immunol. 2013, 131, 840–848. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Dove, C.G.; Hor, J.L.; Murdock, H.M.; Strauss-Albee, D.M.; Garcia, J.A.; Mandl, J.N.; Grodick, R.A.; Jing, H.; Chandler-Brown, D.B.; et al. DOCK8 regulates lymphocyte shape integrity for skin antiviral immunity. J. Exp. Med. 2014, 211, 2549–2566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, M.T.; Coppola, S.; Krumbach, O.H.; Prencipe, G.; Insalaco, A.; Cifaldi, C.; Brigida, I.; Zara, E.; Scala, S.; Di Cesare, S.; et al. A novel disorder involving dyshematopoiesis, inflammation, and HLH due to aberrant CDC42 function. J. Exp. Med. 2019, 216, 2778–2799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anft, M.; Netter, P.; Urlaub, D.; Prager, I.; Schaffner, S.; Watzl, C. NK cell detachment from target cells is regulated by successful cytotoxicity and influences cytokine production. Cell. Mol. Immunol. 2020, 17, 347–355. [Google Scholar] [CrossRef]
- Jenkins, M.R.; Rudd-Schmidt, J.A.; Lopez, J.A.; Ramsbottom, K.M.; Mannering, S.I.; Andrews, D.M.; Voskoboinik, I.; Trapani, J.A. Failed CTL/NK cell killing and cytokine hypersecretion are directly linked through prolonged synapse time. J. Exp. Med. 2015, 212, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Canna, S.W.; Cron, R.Q. Highways to hell: Mechanism-based management of cytokine storm syndromes. J. Allergy Clin. Immunol. 2020, 146, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Schulert, G.S.; Blum, S.A.; Cron, R.Q. Host genetics of pediatric SARS-CoV-2 COVID-19 and multisystem inflammatory syndrome in children. Curr. Opin. Pediatr. 2021, 33, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.M.; Lewin, A.S.; Ahmed, C.M. SOCS, Intrinsic Virulence Factors, and Treatment of COVID-19. Front. Immunol. 2020, 11, 582102. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.Y.; Platt, C.D.; Weeks, S.; Grace, R.F.; Maher, G.; Gauthier, K.; Devana, S.; Vitali, S.; Randolph, A.G.; McDonald, D.R.; et al. Immune dysregulation and multisystem inflammatory syndrome in children (MIS-C) in individuals with haploinsufficiency of SOCS1. J. Allergy Clin. Immunol. 2020, 146, 1194–1200.e1. [Google Scholar] [CrossRef]

{kind=link}
| Parameter (Mean/SD) | GROUP 1 MIS-C Group | GROUP 2 Non-MIS-C Group | GROUP 1a MIS-C with No VUS | GROUP 1b MIS-C with a VUS Other Than DOCK8/pHLH | GROUP 1c MIS-C with Either DOCK8 or pHLH VUS | GROUP 1c.1 (Subgroup of 1c) MIS-C with Only DOCK8 VUS |
|---|---|---|---|---|---|---|
| No. of patients | n = 39 | n = 6 | n = 10 | n = 19 | n = 10 | n = 4 |
| Age (yrs) | 9.0 ± 4.2 | 5.5 ± 3.1 | 10.5 ± 3.1 | 9.19 ± 4.42 | 7.4 ± 4.5 | 8.0 ± 6.3 |
| Gender | Male 62% | Male 66.7% | Male 55.6% | Male 66% | Male 60% | Male 75% |
| Presenting symptoms | Fever (97%) Abd. pain (72%) Rash (53%) | Fever (100%) Abd. pain (65%) Rash (40.2%) | Fever (100%) Abd. pain (55.6%) Rash (55.6%) | Fever (95%) Abd. pain (81%) Rash (57.2%) | Fever (100%) Abd. pain (71%) Rash (42%) | Fever (100%) Abd. pain/Rash (both 33%) |
| Length of stay (days) | 6.7 ± 3.7 | 4.0 ± 3.0 | 5.4 ± 2.7 | 7.48 ± 4.26 | 5.6 ± 2.4 | 6.7 ± 3.5 |
| PICU stay | Yes (64%) | No (100%) | Yes (77.8%) | Yes (61.9%) | Yes (50%) | Yes (75%) |
| Oxygen | Yes (49%) | No (100%) | Yes (55.6%) | Yes (52.4%) | Yes (50%) | Yes (75%) |
| WBC (×103/dL) | 11.0 ± 6.3 | 12.3 ± 5.8 | 10.27 ± 5.01 | 11.8 ± 7.7 | 9.4 ± 3.8 | 8.6 ± 1.9 |
| Hb (g/dL) | 11.2 ± 1.5 | 10.78 ± 1.5 | 11.08 ± 1.58 | 11.4 ± 1.5 | 10.7 ± 1.1 | 11.0 ± 1.8 |
| Plt (×103/dL) | 187 ± 80 | 163 ± 73 | 170 ± 48 | 204 ± 96 | 159 ± 51 | 154 ± 45 |
| Fibrinogen | 787 ± 203 | 509 ± 150 | 768 ± 216 | 799 ± 200 | 756 ± 211 | 665 ± 149 |
| D-dimer | 2137 ± 1652 | 915 ± 828 | 2712 ± 1455 | 1810 ± 1684 | 2090 ± 1757 | 2363 ± 1544 |
| LDH | 351 ± 136 | 315 ± 39 | 335 ± 172 | 366 ± 136 | 330 ± 86 | 230 ± 15 |
| Ferritin | 972 ± 1177 | 447 ± 515 | 1318 ± 1520 | 958.44 ± 1178 | 531 ± 440 | 864 ± 614 |
| Procalcitonin | 11.3 ± 19.3 | 29 ± 24.2 | 10.67 ± 11.9 | 10.1 ± 24 | 11.8 ± 11.5 | 9.8 ± 5.3 |
| CRP | 191 ± 107 | 95 ± 52 | 236 ± 90 | 185.1 ± 123.7 | 150 ± 75 | 166 ± 98 |
| Echocardiography findings | WNL (30%) Cor. ch. (22%) Vent. dys. (10%) Valv. dys. (35%) | WNL (66.6%) Hypokinesia (33.3%) | WNL (22.2%) Hypokinesia (33.3%) | WNL (30%) Cor. ch. (25%) Vent. dys. (10%) Valv. dys. (40%) | WNL (56%) Cor. ch. (33%) Vent. dys. (11%) Valv. dys. (22%) | WNL (50%) Cor. ch. (50%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vagrecha, A.; Zhang, M.; Acharya, S.; Lozinsky, S.; Singer, A.; Levine, C.; Al-Ghafry, M.; Fein Levy, C.; Cron, R.Q. Hemophagocytic Lymphohistiocytosis Gene Variants in Multisystem Inflammatory Syndrome in Children. Biology 2022, 11, 417. https://doi.org/10.3390/biology11030417
Vagrecha A, Zhang M, Acharya S, Lozinsky S, Singer A, Levine C, Al-Ghafry M, Fein Levy C, Cron RQ. Hemophagocytic Lymphohistiocytosis Gene Variants in Multisystem Inflammatory Syndrome in Children. Biology. 2022; 11(3):417. https://doi.org/10.3390/biology11030417
Chicago/Turabian StyleVagrecha, Anshul, Mingce Zhang, Suchitra Acharya, Shannon Lozinsky, Aaron Singer, Chana Levine, Maha Al-Ghafry, Carolyn Fein Levy, and Randy Q. Cron. 2022. "Hemophagocytic Lymphohistiocytosis Gene Variants in Multisystem Inflammatory Syndrome in Children" Biology 11, no. 3: 417. https://doi.org/10.3390/biology11030417
APA StyleVagrecha, A., Zhang, M., Acharya, S., Lozinsky, S., Singer, A., Levine, C., Al-Ghafry, M., Fein Levy, C., & Cron, R. Q. (2022). Hemophagocytic Lymphohistiocytosis Gene Variants in Multisystem Inflammatory Syndrome in Children. Biology, 11(3), 417. https://doi.org/10.3390/biology11030417