
Integration of Multimodal Data from Disparate Sources for Identifying Disease Subtypes

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
- To the best of our knowledge, our approach is the first study that aims to discover the salient genetic knowledge of a completely missing modality through a mapping function learned by the neural network. This neural network model is able to reconstruct a lower dimensional representation of the missing information based on the correlation between shared and unshared modalities across data sources. Such mapping provides the ability to produce more accurate and consistent identification of aggressive and indolent patients for lethal cancers;
- We have discovered patient subgroups and disease subtypes that have significantly different survival patterns through an unsupervised learning approach combined with manual adjustments which was then used for labeling the samples;
- We quantitatively demonstrate that our work outperforms other baselines with partially available modalities.
2. Method
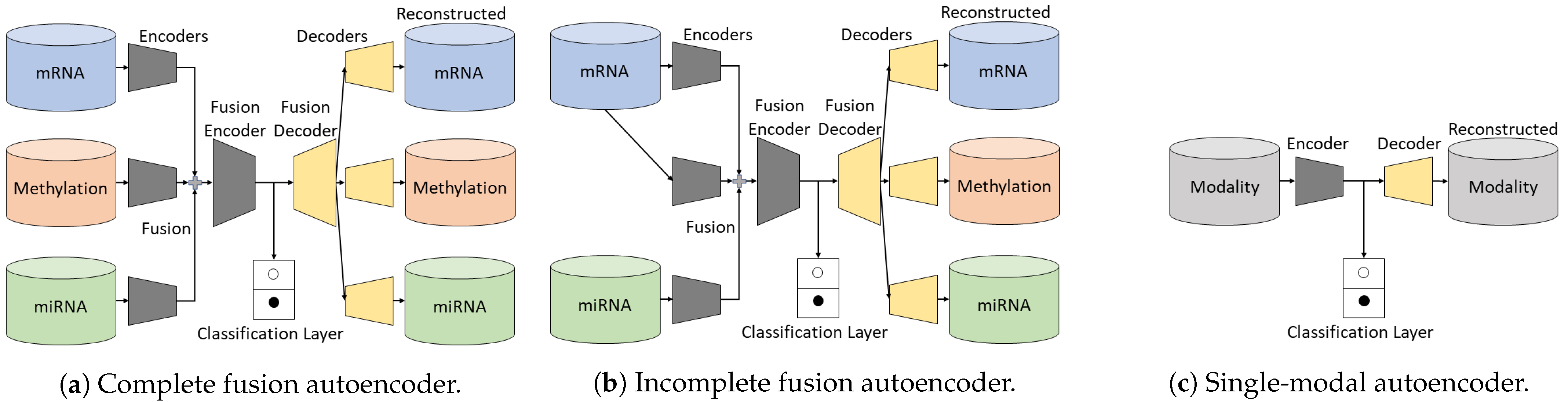
2.1. Learning Associations between Shared and Unshared Modalities
2.1.1. Complete Fusion Autoencoder
2.1.2. Incomplete Fusion Autoencoder
2.1.3. Single-Modal Autoencoder
2.1.4. Classification Layer
2.2. Joint Loss Optimization
3. Experiments
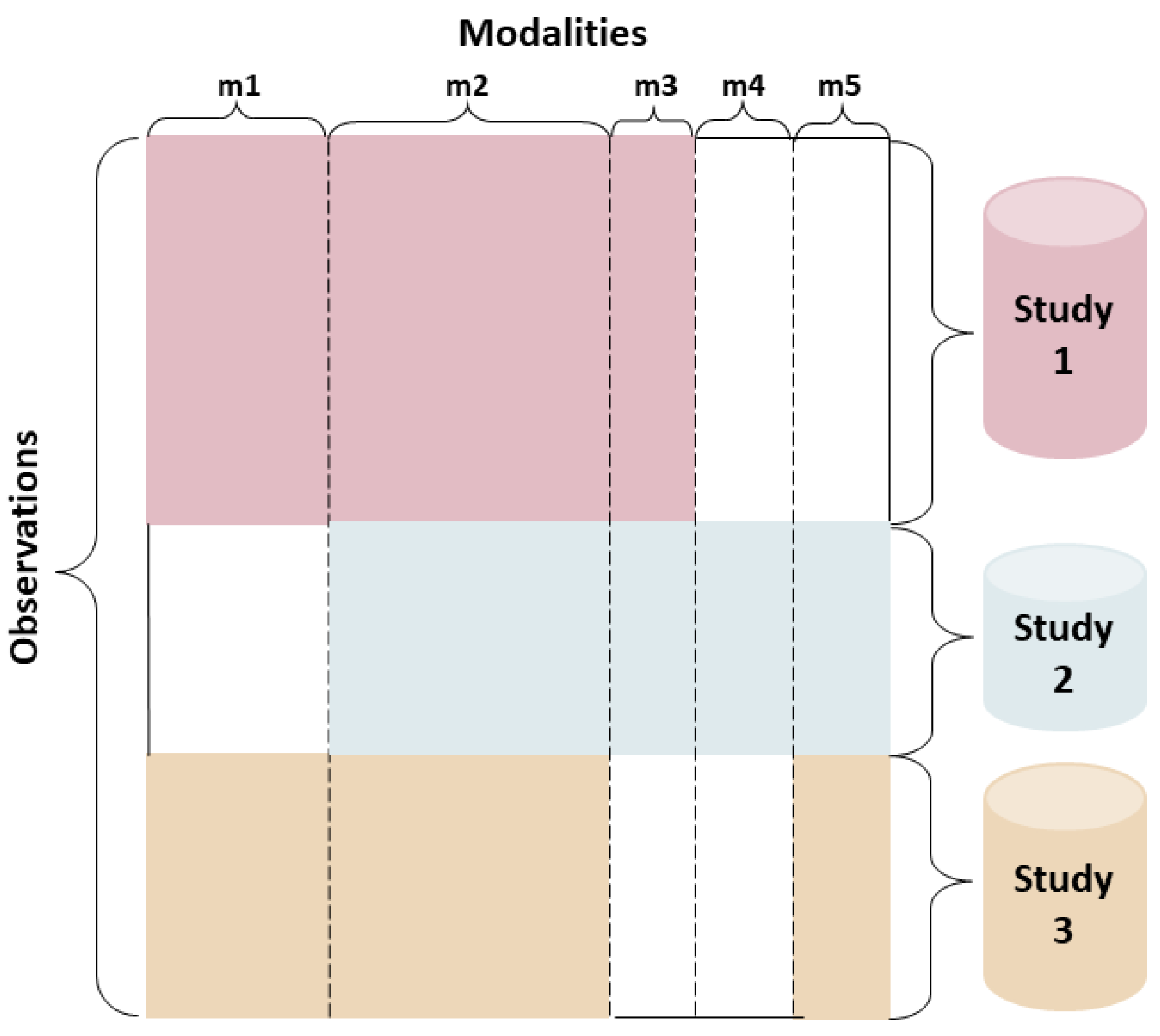
3.1. Data Preparation
3.2. Implementation Details
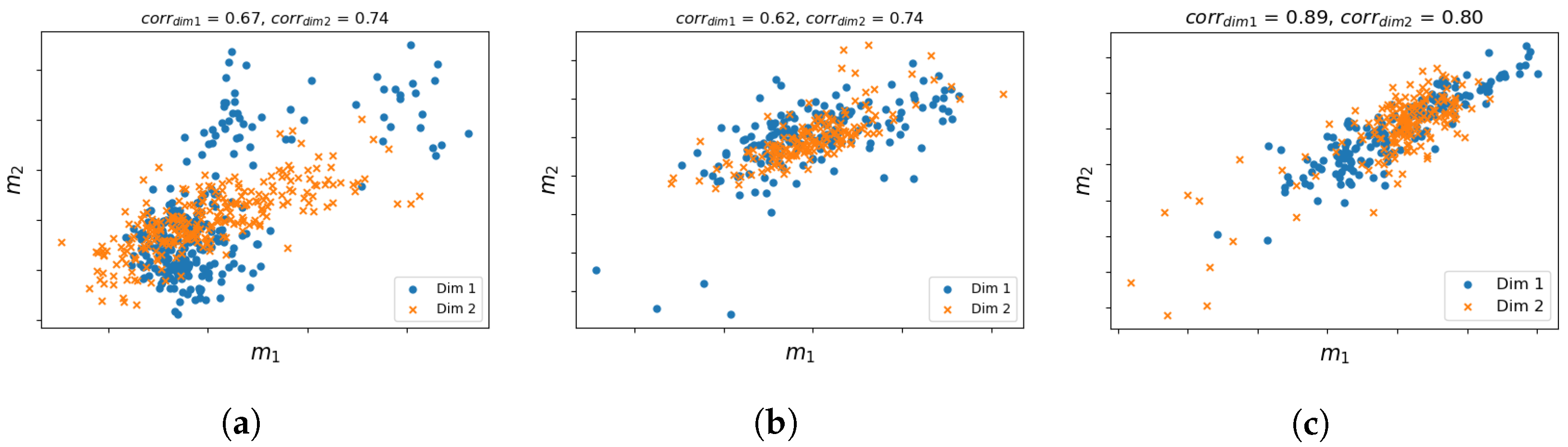
3.3. Correlation between Shared and Unshared Modalities
3.4. Evaluation Metrics
3.5. Prediction Performances
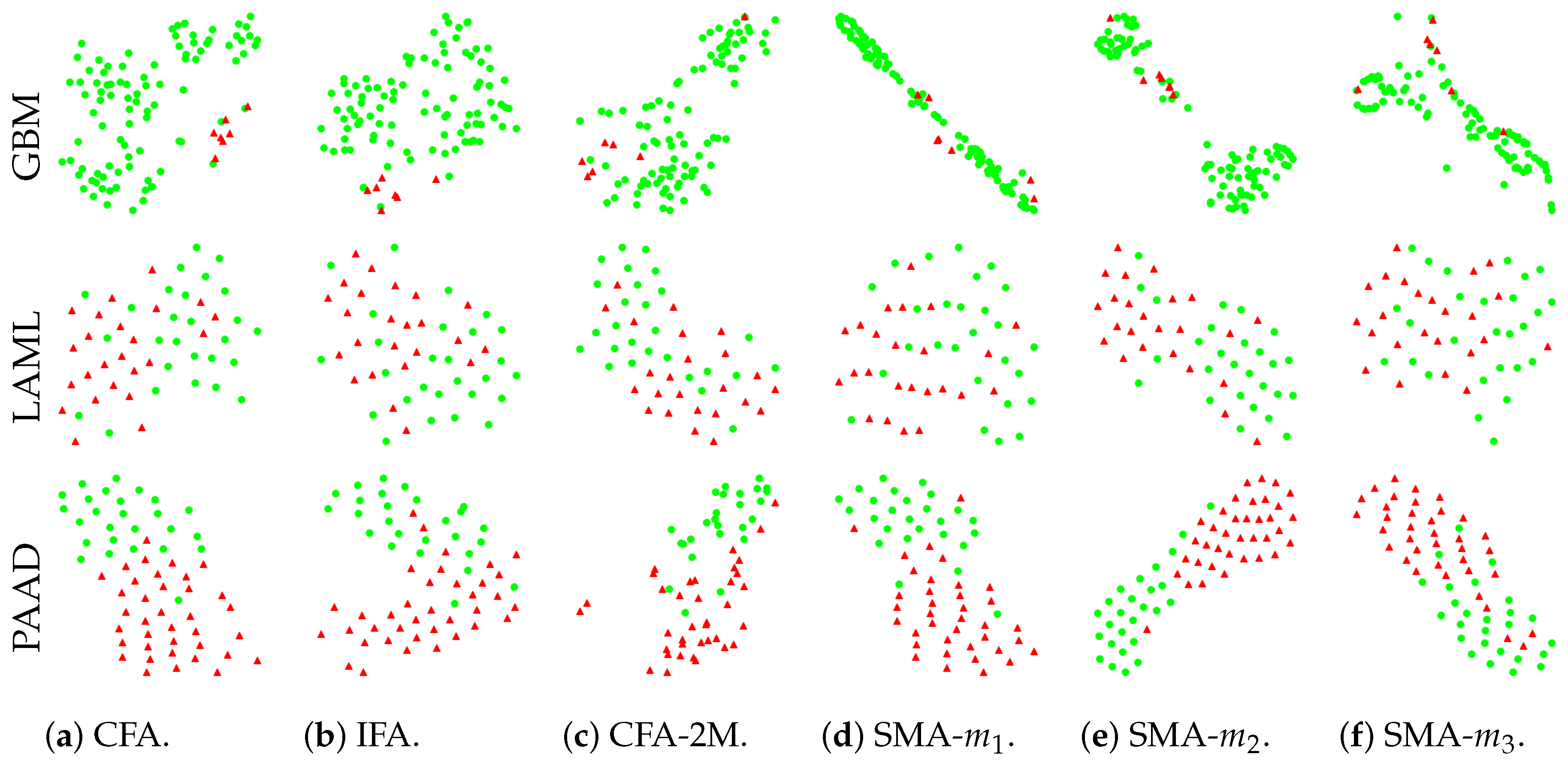
3.6. Effective Compression
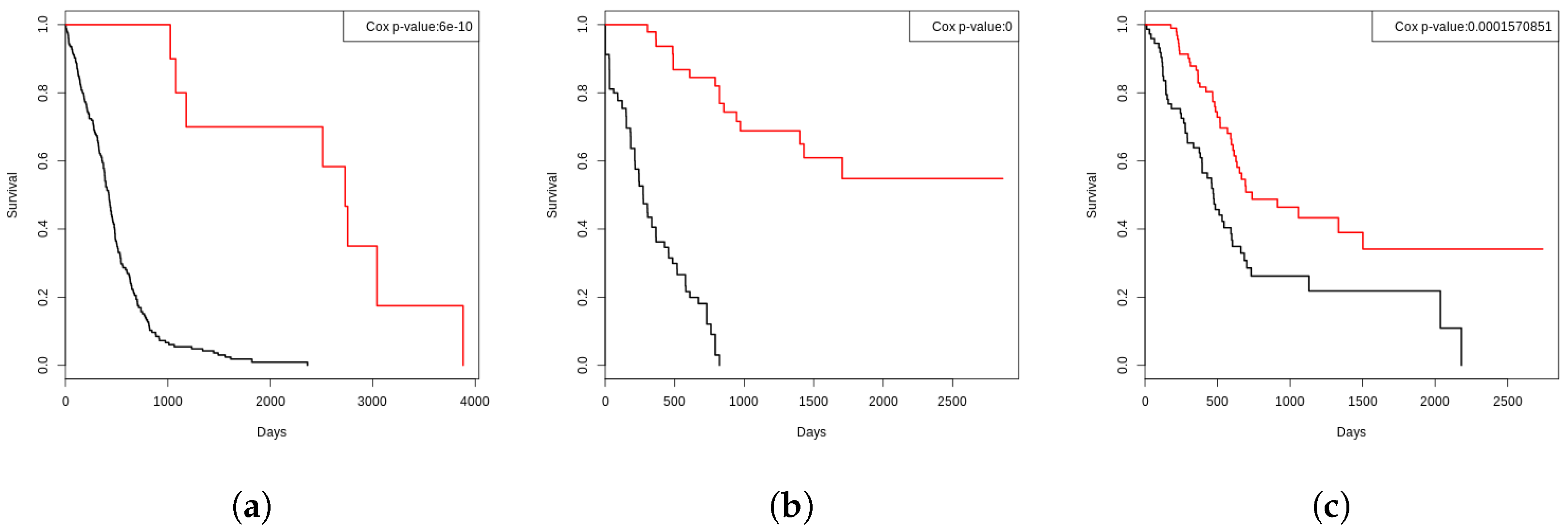
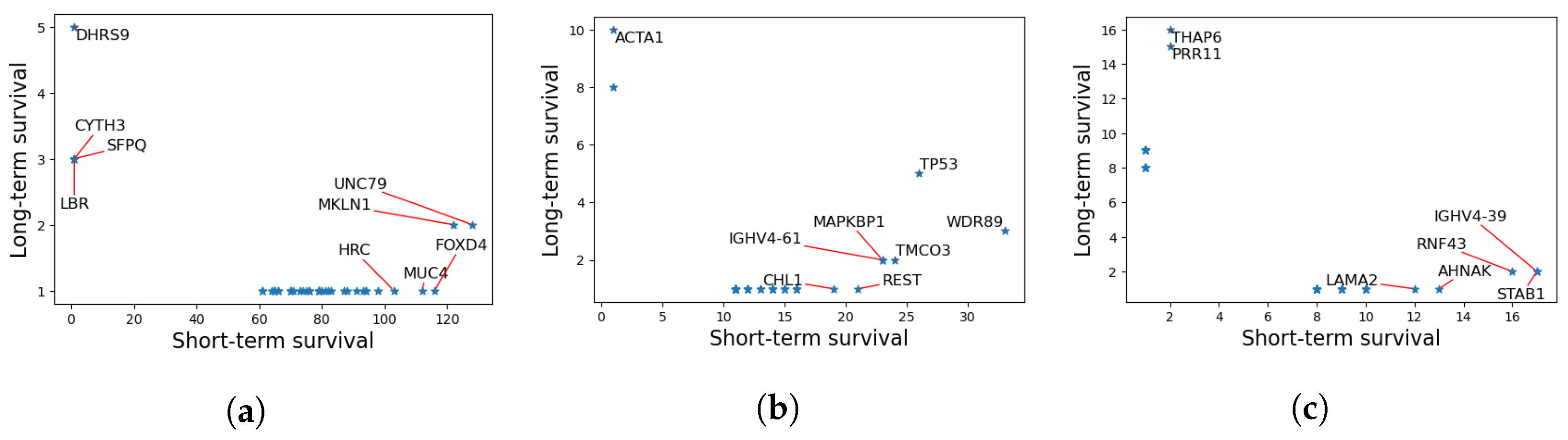
3.7. Functional Analysis
3.8. Ablation Study
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dieterich, M.; Stubert, J.; Reimer, T.; Erickson, N.; Berling, A. Influence of lifestyle factors on breast cancer risk. Breast Care 2014, 9, 407–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leitzmann, M.F.; Rohrmann, S. Risk factors for the onset of prostatic cancer: Age, location, and behavioral correlates. Clin. Epidemiol. 2012, 4, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van IJzendoorn, D.G.; Szuhai, K.; Briaire-de Bruijn, I.H.; Kostine, M.; Kuijjer, M.L.; Bovée, J.V. Machine learning analysis of gene expression data reveals novel diagnostic and prognostic biomarkers and identifies therapeutic targets for soft tissue sarcomas. PLoS Comput. Biol. 2019, 15, e1006826. [Google Scholar] [CrossRef] [PubMed]
- López-García, G.; Jerez, J.M.; Franco, L.; Veredas, F.J. Transfer learning with convolutional neural networks for cancer survival prediction using gene-expression data. PLoS ONE 2020, 15, e0230536. [Google Scholar] [CrossRef]
- Zhou, L.; Guo, Z.; Wang, B.; Wu, Y.; Li, Z.; Yao, H.; Fang, R.; Yang, H.; Cao, H.; Cui, Y. Risk Prediction in Patients with Heart Failure with Preserved Ejection Fraction Using Gene Expression Data and Machine Learning. Front. Genet. 2021, 12, 412. [Google Scholar] [CrossRef]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef]
- Lauber, C.; Correia, N.; Trumpp, A.; Rieger, M.A.; Dolnik, A.; Bullinger, L.; Roeder, I.; Seifert, M. Survival differences and associated molecular signatures of DNMT3A-mutant acute myeloid leukemia patients. Sci. Rep. 2020, 10, 12761. [Google Scholar] [CrossRef]
- Jonckheere, N.; Auwercx, J.; Hadj Bachir, E.; Coppin, L.; Boukrout, N.; Vincent, A.; Neve, B.; Gautier, M.; Treviño, V.; Van Seuningen, I. Unsupervised hierarchical clustering of pancreatic adenocarcinoma dataset from TCGA defines a mucin expression profile that impacts overall survival. Cancers 2020, 12, 3309. [Google Scholar] [CrossRef]
- Plotnikova, O.; Baranova, A.; Skoblov, M. Comprehensive analysis of human microRNA–mRNA interactome. Front. Genet. 2019, 10, 933. [Google Scholar] [CrossRef]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef]
- Aure, M.R.; Fleischer, T.; Bjørklund, S.; Ankill, J.; Castro-Mondragon, J.A.; Børresen-Dale, A.L.; Tost, J.; Sahlberg, K.K.; Mathelier, A.; Tekpli, X.; et al. Crosstalk between microRNA expression and DNA methylation drives the hormone-dependent phenotype of breast cancer. Genome Med. 2021, 13, 72. [Google Scholar] [CrossRef] [PubMed]
- Bengio, Y.; Courville, A.; Vincent, P. Representation learning: A review and new perspectives. IEEE Trans. Pattern Anal. Mach. Intell. 2013, 35, 1798–1828. [Google Scholar] [CrossRef] [PubMed]
- LeCun, Y.; Ranzato, M. Deep learning tutorial. In Tutorials in International Conference on Machine Learning (ICML 2013); Citeseer: Atlanta, GA, USA, 2013; pp. 1–29. [Google Scholar]
- Xu, C.; Tao, D.; Xu, C. A survey on multi-view learning. arXiv 2013, arXiv:1304.5634. [Google Scholar]
- Zheng, V.W.; Zheng, Y.; Xie, X.; Yang, Q. Collaborative location and activity recommendations with gps history data. In Proceedings of the 19th International Conference on World Wide Web, Raleigh, NC, USA, 26–30 April 2010; pp. 1029–1038. [Google Scholar]
- Pan, S.J.; Yang, Q. A survey on transfer learning. IEEE Trans. Knowl. Data Eng. 2009, 22, 1345–1359. [Google Scholar] [CrossRef]
- Arslanturk, S.; Draghici, S.; Nguyen, T. Integrated Cancer Subtyping using Heterogeneous Genome-Scale Molecular Datasets. Pac. Symp. Biocomput. 2020, 25, 551–562. [Google Scholar]
- Nguyen, T.; Tagett, R.; Diaz, D.; Draghici, S. A novel approach for data integration and disease subtyping. Genome Res. 2017, 27, 2025–2039. [Google Scholar] [CrossRef]
- Zhou, R.; Shen, Y.D. End-to-end adversarial-attention network for multi-modal clustering. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, Seattle, WA, USA, 14–19 June 2020; pp. 14619–14628. [Google Scholar]
- Wang, Y.; Huang, W.; Sun, F.; Xu, T.; Rong, Y.; Huang, J. Deep multimodal fusion by channel exchanging. Adv. Neural Inf. Process. Syst. 2020, 33, 4835–4845. [Google Scholar]
- Zheng, Y. Methodologies for cross-domain data fusion: An overview. IEEE Trans. Big Data 2015, 1, 16–34. [Google Scholar] [CrossRef]
- Lahat, D.; Adali, T.; Jutten, C. Multimodal data fusion: An overview of methods, challenges, and prospects. Proc. IEEE 2015, 103, 1449–1477. [Google Scholar] [CrossRef] [Green Version]
- Mariappan, R.; Rajan, V. Deep collective matrix factorization for augmented multi-view learning. Mach. Learn. 2019, 108, 1395–1420. [Google Scholar] [CrossRef] [Green Version]
- Eisen, M.B.; Spellman, P.T.; Brown, P.O.; Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 14863–14868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acar, E.; Kolda, T.G.; Dunlavy, D.M. All-at-once optimization for coupled matrix and tensor factorizations. arXiv 2011, arXiv:1105.3422. [Google Scholar]
- Beutel, A.; Talukdar, P.P.; Kumar, A.; Faloutsos, C.; Papalexakis, E.E.; Xing, E.P. Flexifact: Scalable flexible factorization of coupled tensors on hadoop. In Proceedings of the 2014 SIAM International Conference on Data Mining. SIAM, Philadelphia, PA, USA, 24–26 April 2014; pp. 109–117. [Google Scholar]
- Papalexakis, E.E.; Faloutsos, C.; Sidiropoulos, N.D. Tensors for data mining and data fusion: Models, applications, and scalable algorithms. ACM Trans. Intell. Syst. Technol. (TIST) 2016, 8, 1–44. [Google Scholar] [CrossRef] [Green Version]
- Ray, P.; Zheng, L.; Lucas, J.; Carin, L. Bayesian joint analysis of heterogeneous genomics data. Bioinformatics 2014, 30, 1370–1376. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhan, D.C.; Sheng, X.R.; Jiang, Y. Semi-Supervised Multi-Modal Learning with Incomplete Modalities. In Proceedings of the Twenty-Seventh International Joint Conference on Artificial Intelligence (IJCAI-18), Stockholm, Sweden, 13–19 July 2018; pp. 2998–3004. [Google Scholar]
- Angermueller, C.; Lee, H.J.; Reik, W.; Stegle, O. DeepCpG: Accurate prediction of single-cell DNA methylation states using deep learning. Genome Biol. 2017, 18, 67. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Xu, C.; Deng, H.W.; Shen, H. A novel computational strategy for DNA methylation imputation using mixture regression model (MRM). BMC Bioinform. 2020, 21, 552. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chai, H.; Zhao, H.; Luo, C.H.; Yang, Y. Imputing missing RNA-sequencing data from DNA methylation by using a transfer learning–based neural network. GigaScience 2020, 9, giaa076. [Google Scholar] [CrossRef] [PubMed]
- Bischke, B.; Helber, P.; Koenig, F.; Borth, D.; Dengel, A. Overcoming missing and incomplete modalities with generative adversarial networks for building footprint segmentation. In Proceedings of the 2018 IEEE International Conference on Content-Based Multimedia Indexing (CBMI), La Rochelle, France, 4–6 September 2018; pp. 1–6. [Google Scholar]
- Ma, M.; Ren, J.; Zhao, L.; Tulyakov, S.; Wu, C.; Peng, X. SMIL: Multimodal learning with severely missing modality. In Proceedings of the AAAI Conference on Artificial Intelligence, Virtual, 2–9 February 2021; Volume 35, pp. 2302–2310. [Google Scholar]
- Tran, L.; Liu, X.; Zhou, J.; Jin, R. Missing modalities imputation via cascaded residual autoencoder. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Honolulu, HI, USA, 21–26 July 2017; pp. 1405–1414. [Google Scholar]
- Wang, C.; Niepert, M.; Li, H. LRMM: Learning to recommend with missing modalities. arXiv 2018, arXiv:1808.06791. [Google Scholar]
- Azarkhalili, B.; Saberi, A.; Chitsaz, H.; Sharifi-Zarchi, A. DeePathology: Deep multi-task learning for inferring molecular pathology from cancer transcriptome. Sci. Rep. 2019, 9, 16526. [Google Scholar] [CrossRef]
- Zhou, K.; Arslanturk, S.; Craig, D.B.; Heath, E.; Draghici, S. Discovery of primary prostate cancer biomarkers using cross cancer learning. Sci. Rep. 2021, 11, 10433. [Google Scholar] [CrossRef]
- Cadena, C.; Dick, A.R.; Reid, I.D. Multi-modal Auto-Encoders as Joint Estimators for Robotics Scene Understanding. Robot. Sci. Syst. 2016, 5, 1. [Google Scholar]
- Ioffe, S.; Szegedy, C. Batch normalization: Accelerating deep network training by reducing internal covariate shift. In Proceedings of the International Conference on Machine Learning, Lille, France, 6–11 July 2015; Volume 37, pp. 448–456. [Google Scholar]
- Wold, S.; Sjöström, M.; Eriksson, L. PLS-regression: A basic tool of chemometrics. Chemom. Intell. Lab. Syst. 2001, 58, 109–130. [Google Scholar] [CrossRef]
- Xu, W.; Xu, M.; Wang, L.; Zhou, W.; Xiang, R.; Shi, Y.; Zhang, Y.; Piao, Y. Integrative analysis of DNA methylation and gene expression identified cervical cancer-specific diagnostic biomarkers. Signal Transduct. Target. Ther. 2019, 4, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastasiadi, D.; Esteve-Codina, A.; Piferrer, F. Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenetics Chromatin 2018, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.K.; Guda, C. Genome-wide DNA methylation analysis reveals molecular subtypes of pancreatic cancer. Oncotarget 2017, 8, 28990. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.C.; Jimeno, A.; Lin, S.H.; Wheelhouse, J.; Chan, F.; Solomon, A.; Rajeshkumar, N.; Rubio-Viqueira, B.; Hidalgo, M. Characterizing DNA methylation patterns in pancreatic cancer genome. Mol. Oncol. 2009, 3, 425–438. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, S.M.; Lee, S.I. A unified approach to interpreting model predictions. Adv. Neural Inf. Process. Syst. 2017, 4765–4774. [Google Scholar]
- Sundararajan, M.; Taly, A.; Yan, Q. Axiomatic attribution for deep networks. In Proceedings of the International Conference on Machine Learning, Sydney, Australia, 6–11 August 2017. [Google Scholar]
- Liu, K.; Fu, Y.; Wang, P.; Wu, L.; Bo, R.; Li, X. Automating Feature Subspace Exploration via Multi-Agent Reinforcement Learning. In Proceedings of the ACM International Conference on Knowledge Discovery & Data Mining, Anchorage, AK, USA, 4–8 August 2019; pp. 207–215. [Google Scholar]
- Chinnaiyan, P.; Kensicki, E.; Bloom, G.; Prabhu, A.; Sarcar, B.; Kahali, S.; Eschrich, S.; Qu, X.; Forsyth, P.; Gillies, R. The metabolomic signature of malignant glioma reflects accelerated anabolic metabolism. Cancer Res. 2012, 72, 5878–5888. [Google Scholar] [CrossRef] [Green Version]
- Cammarata, F.P.; Torrisi, F.; Forte, G.I.; Minafra, L.; Bravatà, V.; Pisciotta, P.; Savoca, G.; Calvaruso, M.; Petringa, G.; Cirrone, G.A.; et al. Proton therapy and src family kinase inhibitor combined treatments on U87 human glioblastoma multiforme cell line. Int. J. Mol. Sci. 2019, 20, 4745. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.P.; Dong, J.J.; Xie, T.; Guan, X. Integrative Analysis of MUC4 to Prognosis and Immune Infiltration in Pan-Cancer: Friend or Foe? Front. Cell Dev. Biol. 2021, 9, 695544. [Google Scholar] [CrossRef]
- Li, W.; Wu, C.; Yao, Y.; Dong, B.; Wei, Z.; Lv, X.; Zhang, J.; Xu, Y. MUC4 modulates human glioblastoma cell proliferation and invasion by upregulating EGFR expression. Neurosci. Lett. 2014, 566, 82–87. [Google Scholar] [CrossRef]
- King, R.J.; Yu, F.; Singh, P.K. Genomic alterations in mucins across cancers. Oncotarget 2017, 8, 67152. [Google Scholar] [CrossRef] [Green Version]
- Seifert, M.; Schackert, G.; Temme, A.; Schröck, E.; Deutsch, A.; Klink, B. Molecular characterization of astrocytoma progression towards secondary glioblastomas utilizing patient-matched tumor pairs. Cancers 2020, 12, 1696. [Google Scholar] [CrossRef]
- Barbosa, K.; Li, S.; Adams, P.D.; Deshpande, A.J. The role of TP53 in acute myeloid leukemia: Challenges and opportunities. Genes Chromosomes Cancer 2019, 58, 875–888. [Google Scholar] [CrossRef] [Green Version]
- Tu, J.; Park, S.; Yu, W.; Zhang, S.; Wu, L.; Carmon, K.; Liu, Q.J. The most common RNF43 mutant G659Vfs* 41 is fully functional in inhibiting Wnt signaling and unlikely to play a role in tumorigenesis. Sci. Rep. 2019, 9, 18557. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Module | Neurons in Layer 1 | Neurons in Layer 2 |
|---|---|---|
| Encoder 1 | 1024 | 256 |
| Encoder 2 | 1024 | 256 |
| Encoder 3 | 64 | - |
| Fusion Encoder | 576 | 36 |
| Fusion Decoder | 512 | - |
| Cancer Type | Gene Expression () | DNA Methylation () | miRNA () | Short-Term Survival | Long-Term Survival |
|---|---|---|---|---|---|
| GBM | 12,042 | 22,833 | 534 | 253 | 20 |
| LAML | 16,818 | 22,288 | 552 | 91 | 52 |
| PAAD | 14,105 | 20,006 | 257 | 75 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, K.; Kottoori, B.S.; Munj, S.A.; Zhang, Z.; Draghici, S.; Arslanturk, S. Integration of Multimodal Data from Disparate Sources for Identifying Disease Subtypes. Biology 2022, 11, 360. https://doi.org/10.3390/biology11030360
Zhou K, Kottoori BS, Munj SA, Zhang Z, Draghici S, Arslanturk S. Integration of Multimodal Data from Disparate Sources for Identifying Disease Subtypes. Biology. 2022; 11(3):360. https://doi.org/10.3390/biology11030360
Chicago/Turabian StyleZhou, Kaiyue, Bhagya Shree Kottoori, Seeya Awadhut Munj, Zhewei Zhang, Sorin Draghici, and Suzan Arslanturk. 2022. "Integration of Multimodal Data from Disparate Sources for Identifying Disease Subtypes" Biology 11, no. 3: 360. https://doi.org/10.3390/biology11030360
APA StyleZhou, K., Kottoori, B. S., Munj, S. A., Zhang, Z., Draghici, S., & Arslanturk, S. (2022). Integration of Multimodal Data from Disparate Sources for Identifying Disease Subtypes. Biology, 11(3), 360. https://doi.org/10.3390/biology11030360