
ILeukin10Pred: A Computational Approach for Predicting IL-10-Inducing Immunosuppressive Peptides Using Combinations of Amino Acid Global Features




Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Roles of Interleukin (IL)-10 in Immune Responses
1.2. Literature Review
1.3. Challenges in Predicting IL-10-Inducing Peptides
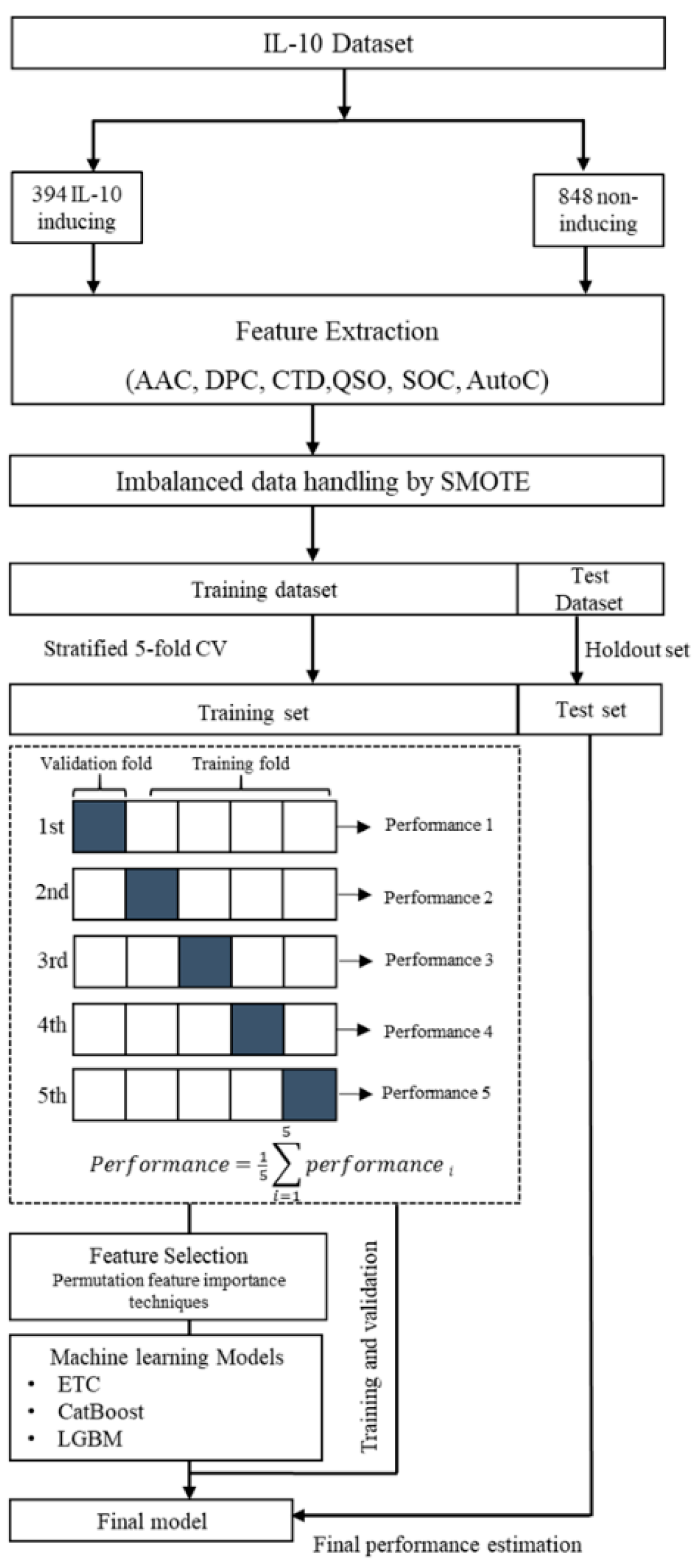
2. Materials and Methods
2.1. Dataset Acquisition and Preprocessing
2.2. Feature Encoding
2.2.1. Amino Acid Composition (AAC)
2.2.2. Dipeptide Composition (DPC)
2.2.3. Composition, Transition, and Distribution (CTD)
2.2.4. Quasi-Sequence Order (QSO)
2.2.5. Sequence Order Coupling (SOC) Number
2.2.6. Autocorrelation (AutoC)
2.3. Imbalanced Dataset Handling by Oversampling
2.4. Machine Learning Algorithms
2.5. Computational Framework
2.6. Performance Evaluation
3. Results
3.1. Hyper-Parameters Tuning
3.1.1. ET Classifier
3.1.2. LGBM Classifier
3.1.3. CatBoost Classifier
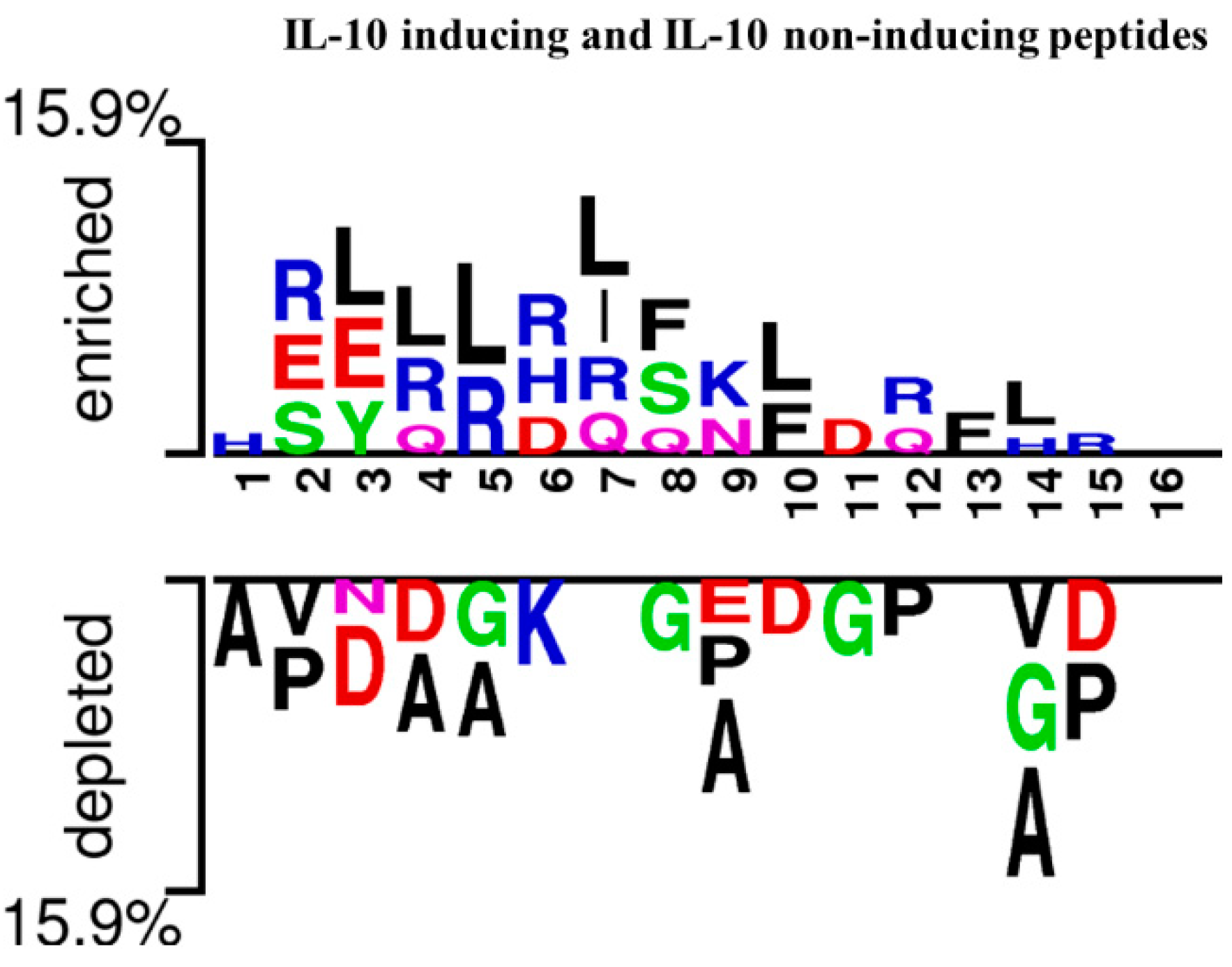
3.2. Analysis of Amino Acid Position Preferences
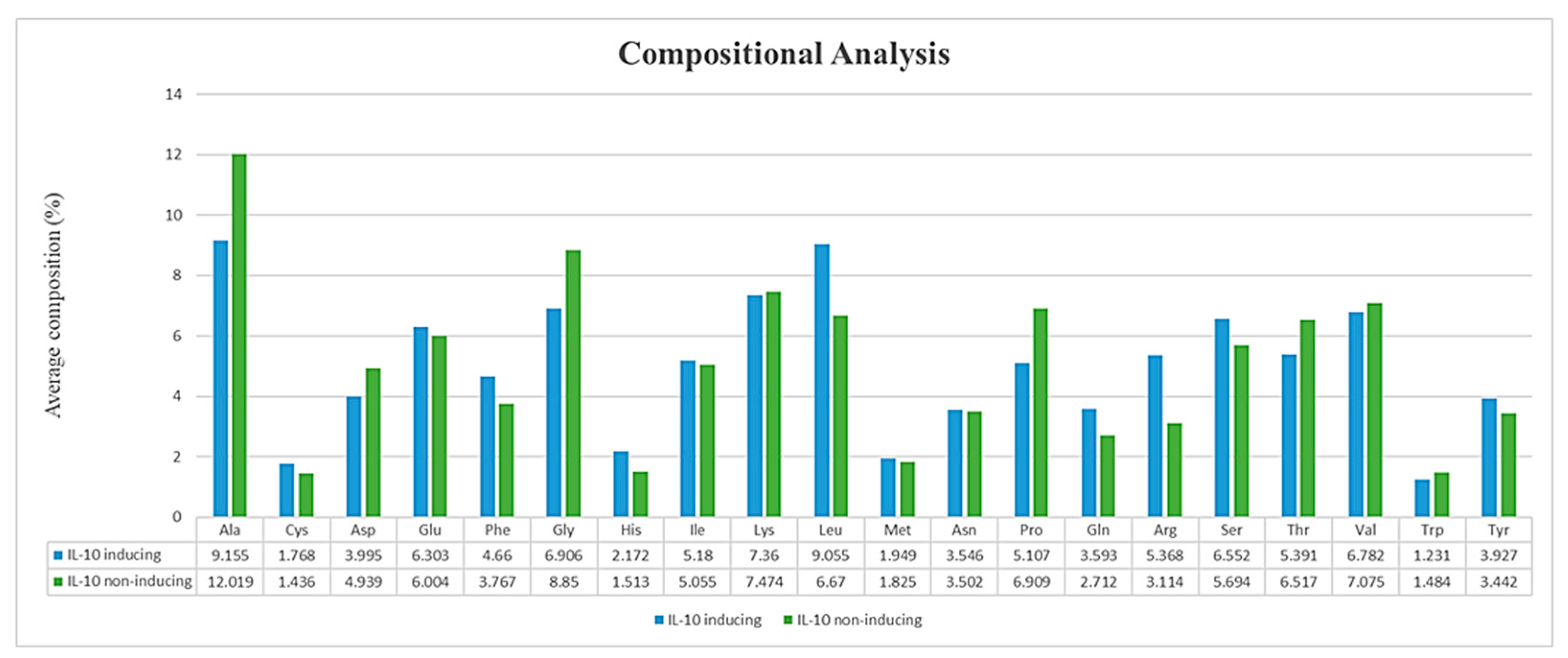
3.3. Compositional Analysis of IL-10 Datasets
3.4. Machine Learning Model Prediction of Single Feature Types
3.5. Machine Learning Predictions of Hybrid Feature Types
4. Discussion
4.1. Position Preference and Composition Analysis of Amino Acids
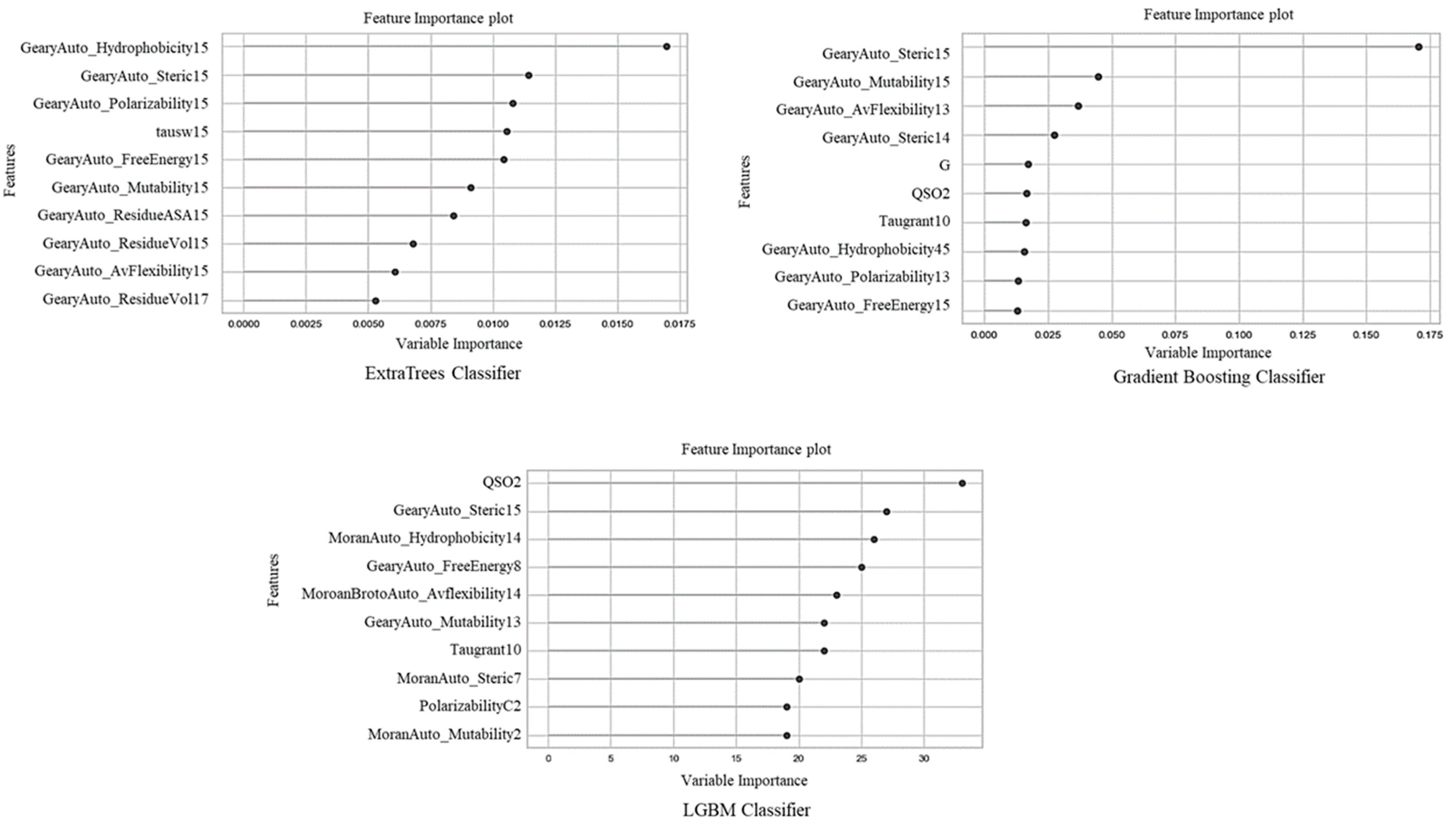
4.2. Biological Assessment of the Top 10 Selected Features
4.3. Evaluation of Single-Feature Types versus Hybrid-Feature Types
4.4. Performance Comparison with the State-of-the-Art Method
4.5. Advantages and Limitations of ILeukin10Pred
4.6. Future Work
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rodriguez-Cortez, V.C.; Hernando, H.; de la Rica, L.; Vento, R.; Ballestar, E. Epigenomic deregulation in the immune system. Epigenomics 2011, 3, 697–713. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.-Y.; El-Gebali, S.; Fraser, M.I.; et al. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef] [Green Version]
- Conti, P.; Kempuraj, D.; Kandere, K.; Di Gioacchino, M.; Barbacane, R.C.; Castellani, M.L.; Felaco, M.; Boucher, W.; Letourneau, R.; Theoharides, T.C. IL-10, an inflammatory/inhibitory cytokine, but not always. Immunol. Lett. 2003, 86, 123–129. [Google Scholar] [CrossRef]
- Spits, H.; Malefyt, R.D.W. Functional Characterization of Human IL. Int. Arch. Allergy Immunol. 1992, 99, 8–15. [Google Scholar] [CrossRef]
- Shao, Y.; Cheng, Z.; Li, X.; Chernaya, V.; Wang, H.; Yang, X.-F. Immunosuppressive/anti-inflammatory cytokines directly and indirectly inhibit endothelial dysfunction- a novel mechanism for maintaining vascular function. J. Hematol. Oncol. 2014, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti, P.; Kempuraj, D.; Frydas, S.; Kandere, K.; Boucher, W.; Letourneau, R.; Madhappan, B.; Sagimoto, K.; Christodoulou, S.; Theoharides, T. IL-10 subfamily members: IL-19, IL-20, IL-22, IL-24 and IL. Immunol. Lett. 2003, 88, 171–174. [Google Scholar] [CrossRef]
- Ouyang, W.; O’Garra, A. IL-10 Family Cytokines IL-10 and IL-22: From Basic Science to Clinical Translation. Immunity 2019, 50, 871–891. [Google Scholar] [CrossRef]
- Tan, J.; Indelicato, S.; Narula, S.; Zavodny, P.; Chou, C. Characterization of interleukin-10 receptors on human and mouse cells. J. Biol. Chem. 1993, 268, 21053–21059. [Google Scholar] [CrossRef]
- Yamaoka, K.; Saharinen, P.; Pesu, M.; Holt, V.E.T.; Silvennoinen, O.; O’Shea, J.J. The Janus kinases (Jaks). Genome Biol. 2004, 5, 253. [Google Scholar] [CrossRef] [Green Version]
- Clarke, C.J.; Hales, A.; Hunt, B.; Foxwell, M. IL-10-mediated suppression of TNF-α production is independent of its ability to inhibit NFκB activity. Eur. J. Immunol. 1998, 28, 1719–1726. [Google Scholar] [CrossRef]
- Kühn, R.; Löhler, J.; Rennick, D.M.; Rajewsky, K.; Muller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993, 75, 263–274. [Google Scholar] [CrossRef]
- Bai, X.-F.; Zhu, J.; Zhang, G.-X.; Kaponides, G.; Höjeberg, B.; Van Der Meide, P.H.; Link, H. IL-10 Suppresses Experimental Autoimmune Neuritis and Down-regulates TH1-Type Immune Responses. Clin. Immunol. Immunopathol. 1997, 83, 117–126. [Google Scholar] [CrossRef]
- Beebe, A.M.; Cua, D.J.; Malefyt, R.D.W. The role of interleukin-10 in autoimmune disease: Systemic lupus erythematosus (SLE) and multiple sclerosis (MS). Cytokine Growth Factor Rev. 2002, 13, 403–412. [Google Scholar] [CrossRef]
- Bettelli, E.; Das, M.P.; Howard, E.D.; Weiner, H.L.; Sobel, R.A.; Kuchroo, V.K. IL-10 is critical in the regulation of autoimmune encephalomyelitis as demonstrated by studies of IL-10- and IL-4-deficient and transgenic mice. J. Immunol. 1998, 161, 3299–3306. [Google Scholar]
- Hata, H.; Sakaguchi, N.; Yoshitomi, H.; Iwakura, Y.; Sekikawa, K.; Azuma, Y.; Kanai, C.; Moriizumi, E.; Nomura, T.; Nakamura, T.; et al. Distinct contribution of IL-6, TNF-α, IL-1, and IL-10 to T cell–mediated spontaneous autoimmune arthritis in mice. J. Clin. Investig. 2004, 114, 582–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, A.; Khosraviani, S.; Noel, S.; Mohan, D.; Donner, T.; Hamad, A.R.A. Interleukin-10 paradox: A potent immunoregulatory cytokine that has been difficult to harness for immunotherapy. Cytokine 2015, 74, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Soria-Guerra, R.E.; Nieto-Gomez, R.; Govea-Alonso, D.; Rosales-Mendoza, S. An overview of bioinformatics tools for epitope prediction: Implications on vaccine development. J. Biomed. Inform. 2015, 53, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinkernagel, R.M.; Doherty, P.C. The discovery of MHC restriction. Immunol. Today 1997, 18, 14–17. [Google Scholar] [CrossRef]
- Falk, K.; Rötzschke, O.; Stevanovié, S.; Jung, G.; Rammensee, H.-G. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nat. Cell Biol. 1991, 351, 290–296. [Google Scholar] [CrossRef]
- De Groot, A.S.; Moise, L. New tools, new approaches and new ideas for vaccine development. Expert Rev. Vaccines 2007, 6, 125–127. [Google Scholar] [CrossRef]
- Nagpal, G.; Usmani, S.S.; Dhanda, S.; Kaur, H.; Singh, S.; Sharma, M.; Raghava, G.P.S. Computer-aided designing of immunosuppressive peptides based on IL-10 inducing potential. Sci. Rep. 2017, 7, 42851. [Google Scholar] [CrossRef] [Green Version]
- Bhasin, M.; Raghava, G. Prediction of CTL epitopes using QM, SVM and ANN techniques. Vaccine 2004, 22, 3195–3204. [Google Scholar] [CrossRef] [PubMed]
- Stranzl, T.; Larsen, M.V.; Lundegaard, C.; Nielsen, M. NetCTLpan: Pan-specific MHC class I pathway epitope predictions. Immunogenetics 2010, 62, 357–368. [Google Scholar] [CrossRef] [Green Version]
- Bhasin, M.; Raghava, G.P.S. A hybrid approach for predicting promiscuous MHC class I restricted T cell epitopes. J. Biosci. 2007, 32, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Raghava, G. ProPred1: Prediction of promiscuous MHC Class-I binding sites. Bioinformatics 2003, 19, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Bhasin, M.; Lata, S.; Raghava, G. TAPPred Prediction of TAP-Binding Peptides in Antigens. In Immunoinformatics; Springer: Singapore, 2007; Volume 409, pp. 381–386. [Google Scholar]
- Dhanda, S.K.; Gupta, S.; Vir, P.; Raghava, G.P.S. Prediction of IL4 Inducing Peptides. Clin. Dev. Immunol. 2013, 2013, 263952. [Google Scholar] [CrossRef] [PubMed]
- Dhall, A.; Patiyal, S.; Sharma, N.; Usmani, S.S.; Raghava, G.P.S. Computer-aided prediction and design of IL-6 inducing peptides: IL-6 plays a crucial role in COVID-19. Brief. Bioinform. 2021, 22, 936–945. [Google Scholar] [CrossRef]
- Minshawi, F.; Lanvermann, S.; McKenzie, E.; Jeffery, R.; Couper, K.; Papoutsopoulou, S.; Roers, A.; Muller, W. The Generation of an Engineered Interleukin-10 Protein with Improved Stability and Biological Function. Front. Immunol. 2020, 11, 1794. [Google Scholar] [CrossRef] [PubMed]
- Fioranelli, M.; Roccia, M.G. Twenty-five years of studies and trials for the therapeutic application of IL-10 immunomodulating properties. From high doses administration to low dose medicine new paradigm. J. Integr. Cardiol. 2014, 1, 2–6. [Google Scholar]
- Chukwudozie, O.S.; Gray, C.M.; Fagbayi, T.A.; Chukwuanukwu, R.C.; Oyebanji, V.O.; Bankole, T.T.; Adewole, R.A.; Daniel, E.M. Immuno-informatics design of a multimeric epitope peptide based vaccine targeting SARS-CoV-2 spike glycoprotein. PLoS ONE 2021, 16, e0248061. [Google Scholar] [CrossRef] [PubMed]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef] [Green Version]
- Barnes, J. Azure Machine Learning. In Microsoft Azure Essentials, 1st ed; Microsoft, One Microsoft Way: Redmond, WA, USA, 2015. [Google Scholar]
- Dong, J.; Yao, Z.-J.; Wen, M.; Zhu, M.-F.; Wang, N.-N.; Miao, H.-Y.; Lu, A.-P.; Zeng, W.-B.; Cao, D.-S. BioTriangle: A web-accessible platform for generating various molecular representations for chemicals, proteins, DNAs/RNAs and their interactions. J. Chemin. 2016, 8, 34. [Google Scholar] [CrossRef] [Green Version]
- Bhasin, M.; Raghava, G.P.S. Classification of Nuclear Receptors Based on Amino Acid Composition and Dipeptide Composition. J. Biol. Chem. 2004, 279, 23262–23266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saravanan, V.; Gautham, N. Harnessing Computational Biology for Exact Linear B-Cell Epitope Prediction: A Novel Amino Acid Composition-Based Feature Descriptor. OMICS J. Integr. Biol. 2015, 19, 648–658. [Google Scholar] [CrossRef]
- Cai, C.; Han, L.; Ji, Z.; Chen, Y. Enzyme family classification by support vector machines. Proteins Struct. Funct. Bioinform. 2004, 55, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Dubchak, I.; Muchnik, I.; Holbrook, S.R.; Kim, S.H. Prediction of protein folding class using global description of amino acid sequence. Proc. Natl. Acad. Sci. USA 1995, 92, 8700–8704. [Google Scholar] [CrossRef] [Green Version]
- Dubchak, I.; Muchnik, I.; Mayor, C.; Dralyuk, I.; Hou-Kim, S. Recognition of a protein fold in the context of the SCOP classification. Proteins: Structure, Function, and Bioinformatics 1999, 35, 401–407. [Google Scholar] [CrossRef]
- Han, L.Y.; Cai, C.Z.; Lo, S.L.; Chung, M.C.; Chen, Y.Z. Prediction of RNA-binding proteins from primary sequence by a support vector machine approach. RNA 2004, 10, 355–368. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.R.; Lin, H.H.; Han, L.Y.; Jiang, L.; Chen, X.; Chen, Y.Z. PROFEAT: A web server for computing structural and physicochemical features of proteins and peptides from amino acid sequence. Nucleic Acids Res. 2006, 34, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Chou, K.-C. Prediction of Protein Subcellular Locations by Incorporating Quasi-Sequence-Order Effect. Biochem. Biophys. Res. Commun. 2000, 278, 477–483. [Google Scholar] [CrossRef]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. SMOTE: Synthetic Minority Over-sampling Technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Rastogi, R.; Shim, K. Public: A Decision Tree Classifier that Integrates Building and Pruning. Data Min. Knowl. Discov. 2000, 4, 315–344. [Google Scholar] [CrossRef]
- Geurts, P.; Ernst, D.; Wehenkel, L. Extremely randomized trees. Mach. Learn. 2006, 63, 3–42. [Google Scholar] [CrossRef] [Green Version]
- Prokhorenkova, L.; Gusev, G.; Vorobev, A.; Dorogush, A.V.; Gulin, A. CatBoost: Unbiased boosting with categorical features. In Advances in Neural Information Processing Systems; Curran Associates Inc.: Red Hook, NY, USA, 2018. [Google Scholar]
- Ke, G.; Meng, Q.; Finley, T.; Wang, T.; Chen, W.; Ma, W.; Ye, Q.; Liu, T.-Y. Lightgbm: A highly efficient gradient boosting decision tree. Adv. Neural Inf. Process. Syst. 2017, 30, 3146–3154. [Google Scholar]
- Ialenti, A.; Santagada, V.; Caliendo, G.; Severino, B.; Fiorino, F.; Maffia, P.; Ianaro, A.; Morelli, F.; Di Micco, B.; Cartenì, M.; et al. Synthesis of novel anti-inflammatory peptides derived from the amino-acid sequence of the bioactive protein SV-IV. JBIC J. Biol. Inorg. Chem. 2001, 268, 3399–3406. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, Y.; Yao, Y.; Xu, M.; Du, H.; Zhang, M.; Tu, Y. Anti-inflammatory activity of di-peptides derived from ovotransferrin by simulated peptide-cut in TNF-α-induced Caco-2 cells. J. Funct. Foods 2017, 37, 424–432. [Google Scholar] [CrossRef]
- Govindan, G.; Nair, A.S. Composition, Transition and Distribution (CTD)—A Dynamic Feature for Predictions Based on Hier-archical Structure of Cellular Sorting. In Proceedings of the 2011 Annual IEEE India Conference, Hyderabad, India, 16–18 December 2011. [Google Scholar]
- Schneider, G.; Wrede, P. The rational design of amino acid sequences by artificial neural networks and simulated molecular evolution: De novo design of an idealized leader peptidase cleavage site. Biophys. J. 1994, 66, 335–344. [Google Scholar] [CrossRef] [Green Version]
- Gesser, B.; Leffers, H.; Jinquan, T.; Vestergaard, C.; Kirstein, N.; Sindet-Pedersen, S.; Jensen, S.L.; Thestrup-Pedersen, K.; Larsen, C.G. Identification of functional domains on human interleukin. Proc. Natl. Acad. Sci. USA 1997, 94, 14620–14625. [Google Scholar] [CrossRef] [Green Version]
- Hibbs, J.B.; Taintor, R.R.; Vavrin, Z. Macrophage Cytotoxicity: Role for L-Arginine Deiminase and Imino Nitrogen Oxidation to Nitrite. Science 1987, 235, 473–476. [Google Scholar] [CrossRef]
- Dowling, J.K.; Afzal, R.; Gearing, L.J.; Cervantes-Silva, M.P.; Annett, S.; Davis, G.M.; De Santi, C.; Assmann, N.; Dettmer, K.; Gough, D.J.; et al. Mitochondrial arginase-2 is essential for IL-10 metabolic reprogramming of inflammatory macrophages. Nat. Commun. 2021, 12, 1460. [Google Scholar] [CrossRef]
- Zdanov, A.; Schalk-Hihi, C.; Gustchina, A.; Tsang, M.; Weatherbee, J.; Wlodawer, A. Crystal structure of interleukin-10 reveals the functional dimer with an unexpected topological similarity to interferon γ. Structure 1995, 3, 591–601. [Google Scholar] [CrossRef]
- Andou, A.; Hisamatsu, T.; Okamoto, S.; Chinen, H.; Kamada, N.; Kobayashi, T.; Hashimoto, M.; Okutsu, T.; Shimbo, K.; Takeda, T.; et al. Dietary histidine ameliorates murine colitis by inhibition of proinflammatory cytokine production from macro-phages. Gastroenterology 2009, 136, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Glocker, E.-O.; Kotlarz, D.; Boztug, K.; Gertz, E.M.; Schäffer, A.A.; Noyan, F.; Perro, M.; Diestelhorst, J.; Allroth, A.; Murugan, D.; et al. Inflammatory Bowel Disease and Mutations Affecting the Interleukin-10 Receptor. N. Engl. J. Med. 2009, 361, 2033–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tien, M.; Meyer, A.G.; Sydykova, D.; Spielman, S.; Wilke, C.O. Maximum Allowed Solvent Accessibilites of Residues in Proteins. PLoS ONE 2013, 8, e80635. [Google Scholar] [CrossRef] [Green Version]
- Rosenblum, M.D.; Gratz, I.K.; Paw, J.S.; Abbas, A.K. Treating Human Autoimmunity: Current Practice and Future Prospects. Sci. Transl. Med. 2012, 4, 125sr1. [Google Scholar] [CrossRef] [Green Version]
- Metzler, B.; Wraith, D.C. Inhibition of experimental autoimmune encephalomyelitis by inhalation but not oral administration of the encephalitogenic peptide: Influence of MHC binding affinity. Int. Immunol. 1993, 5, 1159–1165. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Training Set | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Feature | ETC | CatBoost | LGBM | ||||||
| Acc. % | AUC | MCC | Acc. % | AUC | MCC | Acc. % | AUC | MCC | |
| AAC | 82.3 ± 0.022 | 0.906 ± 0.013 | 0.647 ± 0.046 | 86.1 ± 0.022 | 0.920 ± 0.018 | 0.722 ± 0.045 | 85.3 ± 0.017 | 0.919 ± 0.014 | 0.707 ± 0.035 |
| DPC | 86.5 ± 0.008 | 0.942 ± 0.004 | 0.730 ± 0.017 | 84.2 ± 0.010 | 0.922 ± 0.011 | 0.685 ± 0.021 | 85.4 ± 0.015 | 0.920 ± 0.012 | 0.709 ± 0.029 |
| CTD | 84.6 ± 0.014 | 0.915 ± 0.005 | 0.693 ± 0.027 | 85.4 ± 0.013 | 0.912 ± 0.010 | 0.708 ± 0.025 | 85.6 ± 0.023 | 0.913 ± 0.012 | 0.704 ± 0.046 |
| AutoC | 82.9 ± 0.021 | 0.905 ± 0.012 | 0.664 ± 0.042 | 84.9 ± 0.014 | 0.903 ± 0.009 | 0.699 ± 0.029 | 84.8 ± 0.015 | 0.907 ± 0.014 | 0.696 ± 0.029 |
| QSO | 86.8 ± 0.013 | 0.925 ± 0.009 | 0.7369 ± 0.025 | 84.8 ± 0.029 | 0.912 ± 0.016 | 0.695 ± 0.055 | 84.3 ± 0.021 | 0.911 ± 0.019 | 0.687 ± 0.041 |
| SOC | 82.3 ± 0.004 | 0.887 ± 0.005 | 0.649 ± 0.008 | 80.7 ± 0.014 | 0.876 ± 0.008 | 0.619 ± 0.027 | 80.4 ± 0.016 | 0.869 ± 0.014 | 0.608 ± 0.033 |
| Test Set | |||||||||
| Feature | ETC | CatBoost | LGBM | ||||||
| Acc. % | AUC | MCC | Acc. % | AUC | MCC | Acc. % | AUC | MCC | |
| AAC | 83.5 | 0.912 | 0.674 | 85.1 | 0.919 | 0.705 | 85.4 | 0.903 | 0.712 |
| DPC | 86.6 | 0.943 | 0.733 | 84.5 | 0.925 | 0.689 | 84.8 | 0.919 | 0.695 |
| CTD | 83.8 | 0.913 | 0.678 | 83.8 | 0.887 | 0.677 | 83.8 | 0.891 | 0.677 |
| AutoC | 84.8 | 0.922 | 0.698 | 85.9 | 0.909 | 0.719 | 84.8 | 0.916 | 0.695 |
| QSO | 86.3 | 0.924 | 0.726 | 82.9 | 0.906 | 0.658 | 86.3 | 0.910 | 0.725 |
| SOC | 87.8 | 0.952 | 0.757 | 89.9 | 0.936 | 0.801 | 86.9 | 0.932 | 0.737 |
| Training Set | ||||||
|---|---|---|---|---|---|---|
| Model | Acc. (%) | AUC | Recall/Sen. (%) | Specificity (%) | Precision (%) | MCC |
| ETC | 86.5 ± 0.013 | 0.929 ± 0.015 | 82.2 ± 0.004 | 89.8 ± 0.025 | 88.3 ± 0.025 | 0.724 ± 0.027 |
| LGBM | 86.3 ± 0.015 | 0.918 ± 0.013 | 83.8 ± 0.016 | 88.6 ± 0.029 | 87.3 ± 0.025 | 0.726 ± 0.030 |
| CatBoost | 86.2 ± 0.019 | 0.916 ± 0.019 | 83.1 ± 0.009 | 88.9 ± 0.034 | 87.6 ± 0.033 | 0.724 ± 0.039 |
| Test Set | ||||||
| Model | Acc. (%) | AUC | Recall/Sen. (%) | Specificity (%) | Precision (%) | MCC |
| IL-10Pred | 81.2 | 0.880 | 79.7 | 81.9 | N/A * | 0.590 |
| ETC | 87.5 | 0.931 | 80.4 | 94.7 | 92.7 | 0.755 |
| LGBM | 87.2 | 0.929 | 81.0 | 91.7 | 91.4 | 0.747 |
| CatBoost | 86.6 | 0.923 | 79.1 | 92.9 | 91.9 | 0.737 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, O.; Hsu, W.-L.; Su, E.C.-Y. ILeukin10Pred: A Computational Approach for Predicting IL-10-Inducing Immunosuppressive Peptides Using Combinations of Amino Acid Global Features. Biology 2022, 11, 5. https://doi.org/10.3390/biology11010005
Singh O, Hsu W-L, Su EC-Y. ILeukin10Pred: A Computational Approach for Predicting IL-10-Inducing Immunosuppressive Peptides Using Combinations of Amino Acid Global Features. Biology. 2022; 11(1):5. https://doi.org/10.3390/biology11010005
Chicago/Turabian StyleSingh, Onkar, Wen-Lian Hsu, and Emily Chia-Yu Su. 2022. "ILeukin10Pred: A Computational Approach for Predicting IL-10-Inducing Immunosuppressive Peptides Using Combinations of Amino Acid Global Features" Biology 11, no. 1: 5. https://doi.org/10.3390/biology11010005
APA StyleSingh, O., Hsu, W.-L., & Su, E. C.-Y. (2022). ILeukin10Pred: A Computational Approach for Predicting IL-10-Inducing Immunosuppressive Peptides Using Combinations of Amino Acid Global Features. Biology, 11(1), 5. https://doi.org/10.3390/biology11010005