
Targeted RNA-Seq Reveals the M. tuberculosis Transcriptome from an In Vivo Infection Model

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacteria Cultures
2.2. Mice Infection
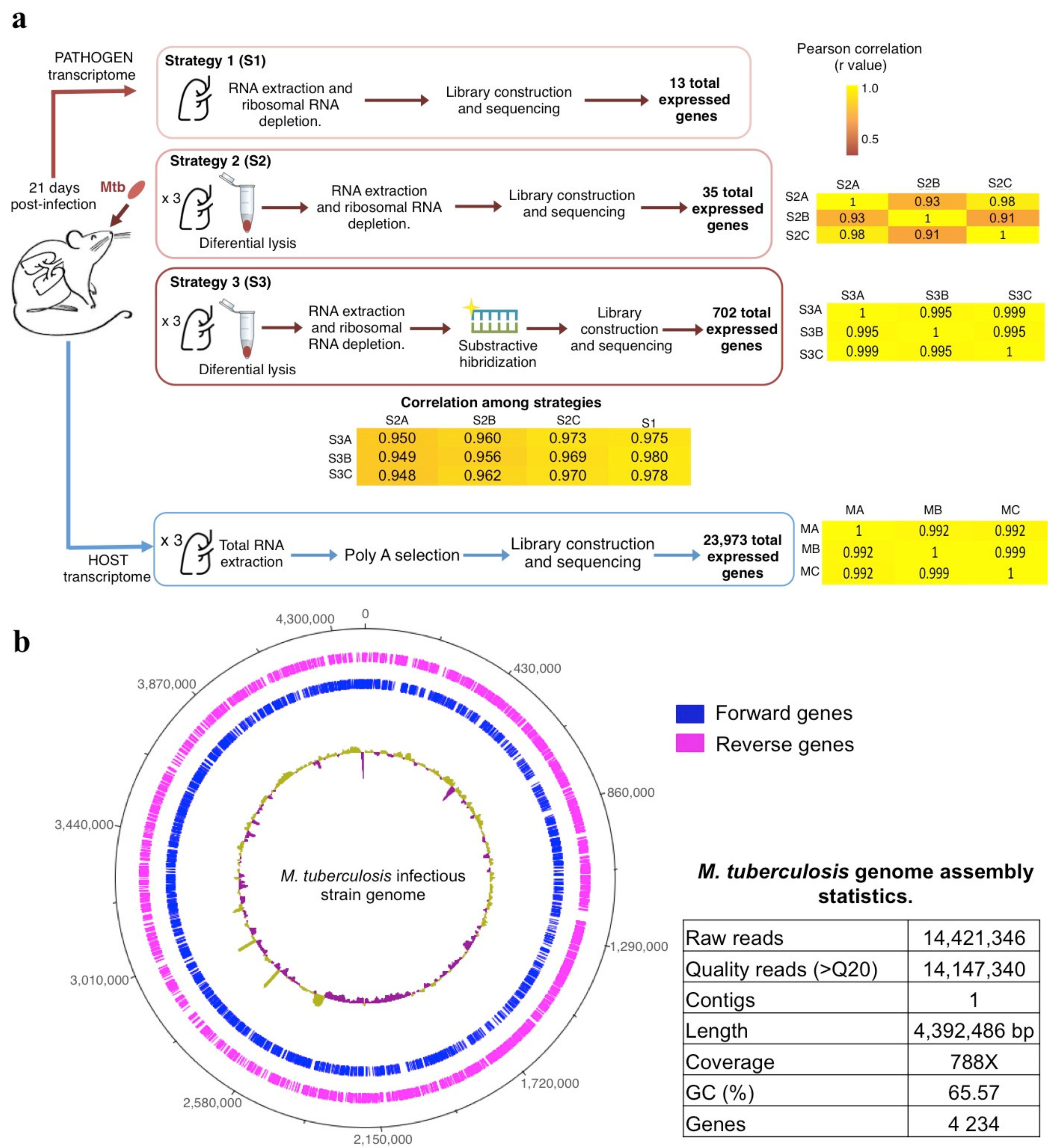
2.3. M. tuberculosis Transcriptome
2.4. I. M. tuberculosis Transcriptome
- (i)
- RNA extracted without differential lysis and centrifugation (Strategy 1): We pulverized one infected left lung using sterile mortars and pestles frozen with liquid nitrogen and placed the homogenate in a microfuge tube. Then, total RNA was extracted directly using the Quick RNA miniprep Kit (Zymo Research, Irvine, CA, USA; Cat.R1055) following the manufacturer’s recommendations, including the digestion with DNAseI to remove the contaminating DNA. Finally, we quantified and assessed the quality of the resulting RNA by Qubit Fluorometer (Invitrogen, Waltham, MA, USA, Cat. Q32851) and Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA, Cat. 5067-4626), respectively.
- (ii)
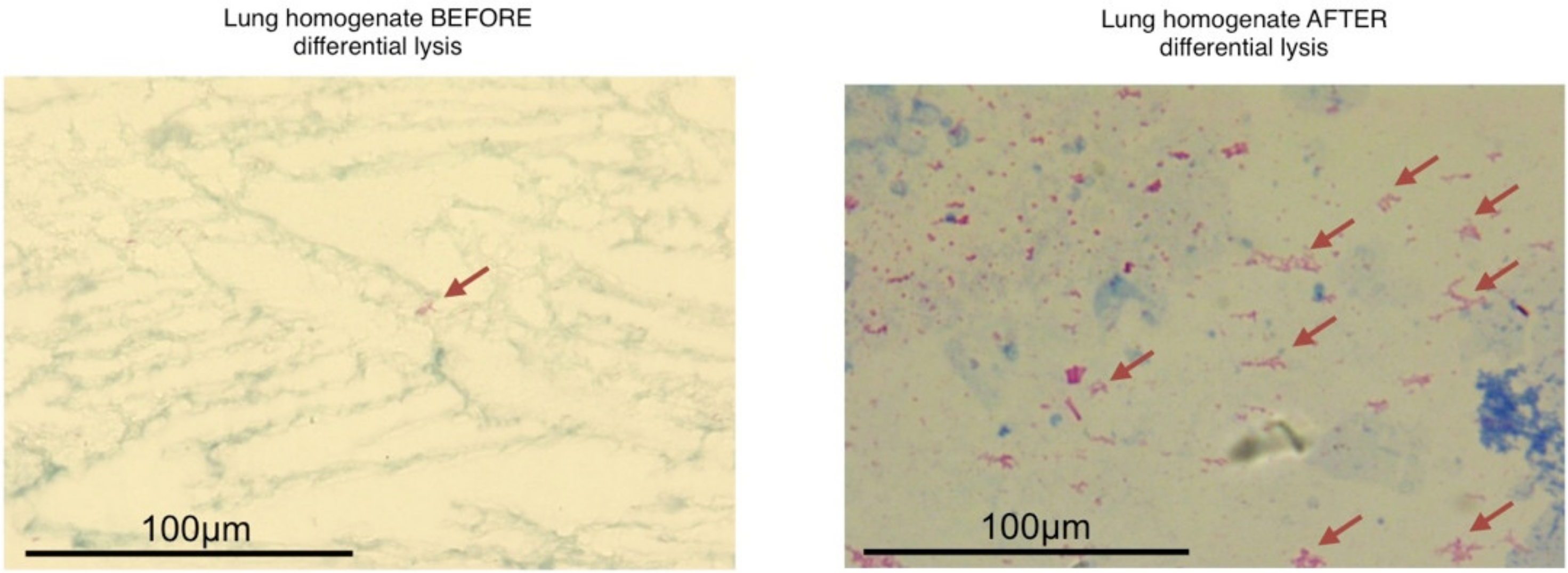
- RNA extracted with differential lysis and centrifugation (Strategy 2): We pulverized six infected left lungs independently using sterile mortars and pestles frozen with liquid nitrogen and placed each homogenate in microfuge tubes. Then, we added RLT buffer (Qiagen, Hilden, Germany, Cat.79216) with β mercaptoethanol as the initial lysis buffer to each tube and centrifuged at 14,000 rpm/4 °C for 5 min. We discarded the supernatant and kept the cream-colored pellet on ice. This procedure allowed the enrichment of bacterial cells in the pellet due to the mild-lysis produced by the RLT, which breaks most mouse cells keeping the bacterial cells intact due to its thicker wall membrane. Immediately after centrifugation, we continued the RNA extraction from the pellet using the Quick RNA miniprep Kit (Zymo Research, Irvine, CA, USA; Cat.R1055) following the manufacturer’s recommendations, including the digestion with DNAseI to remove the contaminating DNA. Finally, we quantified and assessed the quality of the resulting RNA by Qubit Fluorometer and Agilent 2100 Bioanalyzer, respectively.
2.5. Mouse Transcriptome
2.6. Sequencing and Assembly of the M. tuberculosis Infectious Strain
2.7. Analysis of Host and Pathogen RNA-Seq Data
2.7.1. Analysis of M. tuberculosis Transcriptome
2.7.2. Analysis of Mouse Transcriptome
3. Results and Discussion
3.1. The Differential Cell Lysis Protocol Concentrated the Number of Mycobacterial Cells and Increased the Extracted Bacterial RNA
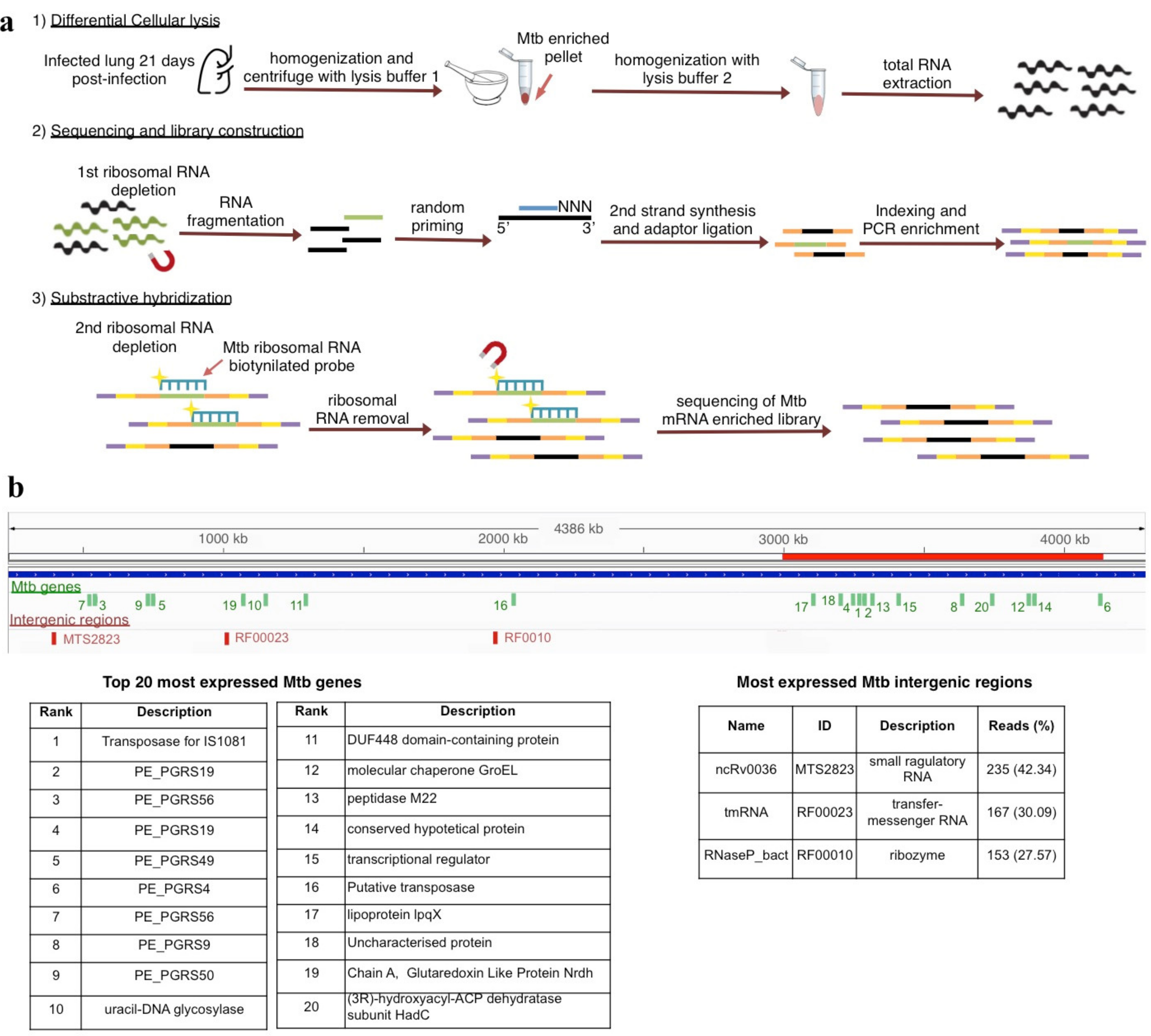
3.2. The MTb Gene Expression Profile Showed a High Abundance of Genes Codifiyng PE-PGRS Members and the Insertion Sequence IS1081
3.3. An Increased Proportion of Expressed Genes Belong to the MTb Secretome
3.4. A High Expression of Three ncRNAs during Mtb In Vivo Infection
3.5. The Host Gene Expression Showed an Inflammatory Profile Consistent with the Early Stage of Infection
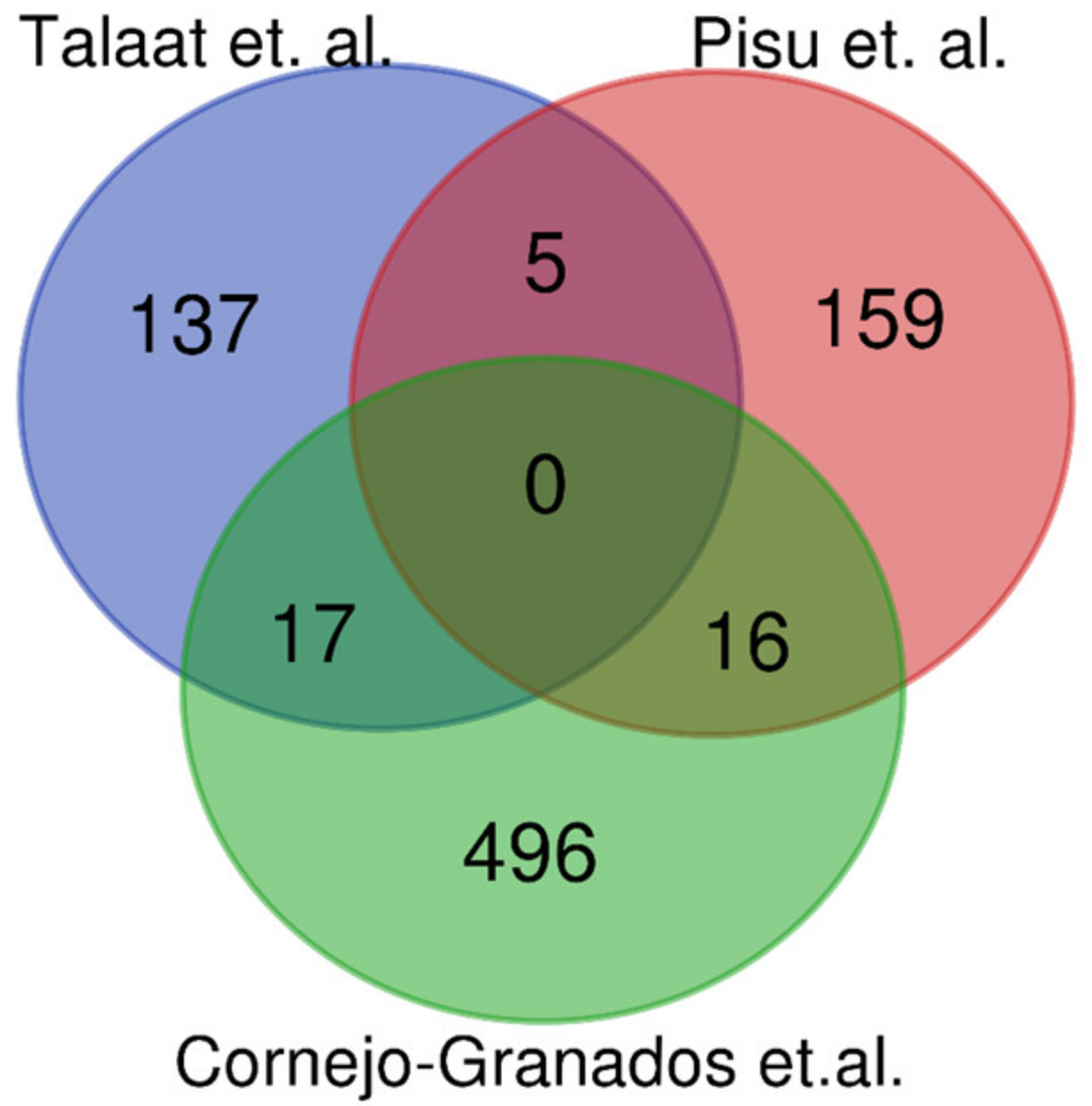
3.6. Comparisson of Mtb Gene Expression Using Other RNA-Seq Methods during In Vivo Infections
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Avraham, R.; Haseley, N.; Fan, A.; Bloom-Ackermann, Z.; Livny, J.; Hung, D.T. A highly multiplexed and sensitive RNA-seq protocol for simultaneous analysis of host and pathogen transcriptomes. Nat. Protoc. 2016, 11, 1477–1491. [Google Scholar] [CrossRef] [Green Version]
- Westermann, A.J.; Gorski, S.A.; Vogel, J. Dual RNA-seq of pathogen and host. Nat. Rev. Microbiol. 2012, 10, 618–630. [Google Scholar] [CrossRef]
- Schnappinger, D.; Ehrt, S.; Voskuil, M.I.; Liu, Y.; Mangan, J.A.; Monahan, I.M.; Dolganov, G.; Efron, B.; Butcher, P.D.; Nathan, C.; et al. Transcriptional Adaptation of Mycobacterium tuberculosis within Macrophages: Insights into the Phagosomal Environment. J. Exp. Med. 2003, 198, 693–704. [Google Scholar] [CrossRef] [Green Version]
- Rohde, K.H.; Abramovitch, R.B.; Russell, D.G. Mycobacterium tuberculosis invasion of macrophages: Linking bacterial gene expression to environmental cues. Cell Host Microbe 2007, 2, 352–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisu, D.; Huang, L.; Grenier, J.K.; Russell, D.G. Dual RNA-Seq of Mtb-Infected Macrophages In Vivo Reveals Ontologically Distinct Host-Pathogen Interactions. Cell Rep. 2020, 30, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Talaat, A.M.; Lyons, R.; Howard, S.T.; Johnston, S.A. The temporal expression profile of Mycobacterium tuberculosis infection in mice. Proc. Natl. Acad. Sci. USA 2004, 101, 4602–4607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waddell, S.J.; Laing, K.; Senner, C.; Butcher, P.D. Microarray analysis of defined Mycobacterium tuberculosis populations using RNA amplification strategies. BMC Genom. 2008, 9, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Pando, R.; Orozco, H.; Arriaga, K.; Sampieri, A.; Larriva-Sahd, J.; Madrid-Marina, V. Analysis of the local kinetics and localization of interleukin-1 alpha, tumour necrosis factor-alpha and transforming growth factor-beta, during the course of experimental pulmonary tuberculosis. Immunology 1997, 90, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Pando, R.; Orozcoe, H.; Sampieri, A.; Pavon, L.; Velasquillo, C.; Larriva-Sahd, J.; Alcocer, J.M.; Madrid, M.V. Correlation between the kinetics of Th1, Th2 cells and pathology in a murine model of experimental pulmonary tuberculosis. Immunology 1996, 89, 26–33. [Google Scholar] [PubMed]
- Bini, E.I.; Mata Espinosa, D.; Marquina Castillo, B.; Barrios Payan, J.; Colucci, D.; Cruz, A.F.; Zatarain, Z.L.; Alfonseca, E.; Pardo, M.R.; Bottasso, O.; et al. The influence of sex steroid hormones in the immunopathology of experimental pulmonary tuberculosis. PLoS ONE 2014, 9, e93831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, B.; Aguilar, D.; Orozco, H.; Burger, M.; Espitia, C.; Ritacco, V.; Barrera, L.; Kremer, K.; Hernandez-Pando, R.; Huygen, K.; et al. A marked difference in pathogenesis and immune response induced by different Mycobacterium tuberculosis genotypes. Clin. Exp. Immunol. 2003, 133, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Delcher, A.L.; Harmon, D.; Kasif, S.; White, O.; Salzberg, S.L. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999, 27, 4636–4641. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef] [PubMed]
- Kapopoulou, A.; Lew, J.M.; Cole, S.T. The MycoBrowser portal: A comprehensive and manually annotated resource for mycobacterial genomes. Tuberculosis 2011, 91, 8–13. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [Green Version]
- Cornejo-Granados, F.; Zatarain-Barron, Z.L.; Cantu-Robles, V.A.; Mendoza-Vargas, A.; Molina-Romero, C.; Sanchez, F.; Del Pozo-Yauner, L.; Hernandez-Pando, R.; Ochoa-Leyva, A. Secretome Prediction of Two M. tuberculosis Clinical Isolates Reveals Their High Antigenic Density and Potential Drug Targets. Front. Microbiol. 2017, 8, 128. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Dong, X.; Zhu, Y.; Wang, C.; Sun, G.; Luo, T.; Tian, W.; Zheng, H.; Gao, Q. Revealing of Mycobacterium marinum transcriptome by RNA-seq. PLoS ONE 2013, 8, e75828. [Google Scholar] [CrossRef] [Green Version]
- Russell, D.G.; VanderVen, B.C.; Lee, W.; Abramovitch, R.B.; Kim, M.J.; Homolka, S.; Niemann, S.; Rohde, K.H. Mycobacterium tuberculosis wears what it eats. Cell Host Microbe 2010, 8, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Roychowdhury, T.; Mandal, S.; Bhattacharya, A. Analysis of IS6110 insertion sites provide a glimpse into genome evolution of Mycobacterium tuberculosis. Sci. Rep. 2015, 5, 12567. [Google Scholar] [CrossRef]
- Gonzalo-Asensio, J.; Perez, I.; Aguilo, N.; Uranga, S.; Pico, A.; Lampreave, C.; Cebollada, A.; Otal, I.; Samper, S.; Martin, C. New insights into the transposition mechanisms of IS6110 and its dynamic distribution between Mycobacterium tuberculosis Complex lineages. PLoS Genet. 2018, 14, e1007282. [Google Scholar] [CrossRef] [Green Version]
- Delogu, G.; Sanguinetti, M.; Pusceddu, C.; Bua, A.; Brennan, M.J.; Zanetti, S.; Fadda, G. PE_PGRS proteins are differentially expressed by Mycobacterium tuberculosis in host tissues. Microbes Infect. 2006, 8, 2061–2067. [Google Scholar] [CrossRef]
- Bettencourt, P.; Muller, J.; Nicastri, A.; Cantillon, D.; Madhavan, M.; Charles, P.D.; Fotso, C.B.; Wittenberg, R.; Bull, N.; Pinpathomrat, N.; et al. Identification of antigens presented by MHC for vaccines against tuberculosis. NPJ Vaccines 2020, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Kruh, N.A.; Troudt, J.; Izzo, A.; Prenni, J.; Dobos, K.M. Portrait of a pathogen: The Mycobacterium tuberculosis proteome in vivo. PLoS ONE 2010, 5, e13938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitter, W.; Houben, E.N.; Luirink, J.; Appelmelk, B.J. Type VII secretion in mycobacteria: Classification in line with cell envelope structure. Trends Microbiol. 2009, 17, 337–338. [Google Scholar] [CrossRef] [PubMed]
- Ocampo, M.; Curtidor, H.; Vanegas, M.; Patarroyo, M.A.; Patarroyo, M.E. Specific interaction between Mycobacterium tuberculosis lipoprotein-derived peptides and target cells inhibits mycobacterial entry in vitro. Chem. Biol. Drug Des. 2014, 84, 626–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, G.R.; Wilkinson, K.A.; Newton, S.M.; Sullivan, S.M.; Neyrolles, O.; Wain, J.R.; Patel, J.; Pool, K.L.; Young, D.B.; Wilkinson, R.J. Effect of deletion or overexpression of the 19-kilodalton lipoprotein Rv3763 on the innate response to Mycobacterium tuberculosis. Infect. Immun. 2005, 73, 6831–6837. [Google Scholar] [CrossRef] [Green Version]
- Noss, E.H.; Pai, R.K.; Sellati, T.J.; Radolf, J.D.; Belisle, J.; Golenbock, D.T.; Boom, W.H.; Harding, C.V. Toll-like receptor 2-dependent inhibition of macrophage class II MHC expression and antigen processing by 19-kDa lipoprotein of Mycobacterium tuberculosis. J. Immunol. 2001, 167, 910–918. [Google Scholar] [CrossRef] [Green Version]
- Gehring, A.J.; Rojas, R.E.; Canaday, D.H.; Lakey, D.L.; Harding, C.V.; Boom, W.H. The Mycobacterium tuberculosis 19-kilodalton lipoprotein inhibits gamma interferon-regulated HLA-DR and Fc gamma R1 on human macrophages through Toll-like receptor 2. Infect. Immun. 2003, 71, 4487–4497. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Wei, J.; Liu, H.; Li, G.; Guo, Q.; Qiu, Y.; Zhao, L.; Li, M.; Zhao, X.; Dou, X.; et al. Polymorphisms in the PE35 and PPE68 antigens in Mycobacterium tuberculosis strains may affect strain virulence and reflect ongoing immune evasion. Mol. Med. Rep. 2016, 13, 947–954. [Google Scholar] [CrossRef]
- Arnvig, K.B.; Comas, I.; Thomson, N.; Houghton, J.; Boshoff, H.I.; Croucher, N.; Rose, G.; Perkins, T.T.; Parkhill, J.; Dougan, G.; et al. Sequence-Based Analysis Uncovers an Abundance of Non-Coding RNA in the Total Transcriptome of Mycobacterium tuberculosis. PLoS Pathog. 2011, 7, e1002342. [Google Scholar] [CrossRef] [Green Version]
- Ignatov, D.V.; Salina, E.G.; Fursov, M.V.; Skvortsov, T.A.; Azhikina, T.L.; Kaprelyants, A.S. Dormant non-culturable Mycobacterium tuberculosis retains stable low-abundant mRNA. BMC Genom. 2015, 16, 954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasula, R.; Downing, J.F.; Wright, J.R.; Kachel, D.L.; Davis, T.E., Jr.; Martin, W.J. Surfactant protein A (SP-A) mediates attachment of Mycobacterium tuberculosis to murine alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 1997, 17, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Gaynor, C.D.; McCormack, F.; Voelker, D.R.; McGowan, S.; Schlesinger, L.S. Pulmonary surfactant protein A mediates enhanced phagocytosis of Mycobacterium tuberculosis by a direct interaction with human macrophages. J. Immunol. 1995, 155, 5343–5351. [Google Scholar]
- Shepherd, V.L.; Lopez, J.P. The Role of Surfactant-Associated Protein A in Pulmonary Host Defense. Immunol. Res. 2001, 23, 111–120. [Google Scholar] [CrossRef]
- Pasula, R.; Wright, J.R.; Kachel, D.L.; Martin, W.J. Surfactant protein A suppresses reactive nitrogen intermediates by alveolar macrophages in response to Mycobacterium tuberculosis. J. Clin. Investig. 1999, 103, 483–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepelkova, G.; Pommerenke, C.; Alberts, R.; Geffers, R.; Evstifeev, V.; Apt, A.; Schughart, K.; Wilk, E. Analysis of the lung transcriptome in Mycobacterium tuberculosis-infected mice reveals major differences in immune response pathways between TB-susceptible and resistant hosts. Tuberculosis 2013, 93, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Shafiani, S.; Tucker-Heard, G.; Kariyone, A.; Takatsu, K.; Urdahl, K.B. Pathogen-specific regulatory T cells delay the arrival of effector T cells in the lung during early tuberculosis. J. Exp. Med. 2010, 207, 1409–1420. [Google Scholar] [CrossRef] [Green Version]
- Ulrichs, T.; Kaufmann, S.H. New insights into the function of granulomas in human tuberculosis. J. Pathol. 2005, 208, 261–269. [Google Scholar] [CrossRef]
- Malardo, T.; Gardinassi, L.G.; Moreira, B.P.; Padilha, É.; Lorenzi, J.C.C.; Soares, L.S.; Gembre, A.F.; Fontoura, I.C.; De Almeida, L.P.; Santos, I.K.F.d.M.; et al. MicroRNA expression signatures in lungs of mice infected with Mycobacterium tuberculosis. Tuberculosis 2016, 101, 151–159. [Google Scholar] [CrossRef]
- Ghorpade, D.S.; Leyland, R.; Kurowska-Stolarska, M.; Patil, S.A.; Balaji, K.N. MicroRNA-155 Is Required for Mycobacterium bovis BCG-Mediated Apoptosis of Macrophages. Mol. Cell. Biol. 2012, 32, 2239–2253. [Google Scholar] [CrossRef] [Green Version]
- Rajaram, M.; Ni, B.; Morris, J.D.; Brooks, M.N.; Carlson, T.K.; Bakthavachalu, B.; Schoenberg, D.; Torrelles, J.B.; Schlesinger, L.S. Mycobacterium tuberculosis lipomannan blocks TNF biosynthesis by regulating macrophage MAPK-activated protein kinase 2 (MK2) and microRNA miR-125b. Proc. Natl. Acad. Sci. USA 2011, 108, 17408–17413. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Halder, P.; Sahu, S.K.; Kumar, M.; Kumari, M.; Jana, K.; Ghosh, Z.; Sharma, P.; Kundu, M.; Basu, J. Identification of a novel role of ESAT-6-dependent miR-155 induction during infection of macrophages withMycobacterium tuberculosis. Cell. Microbiol. 2012, 14, 1620–1631. [Google Scholar] [CrossRef]
- Wang, J.; Yang, K.; Zhou, L.; Wu, M.; Wu, Y.; Zhu, M.; Lai, X.; Chen, T.; Feng, L.; Li, M.; et al. MicroRNA-155 Promotes Autophagy to Eliminate Intracellular Mycobacteria by Targeting Rheb. PLoS Pathog. 2013, 9, e1003697. [Google Scholar] [CrossRef] [Green Version]
- Iwai, H.; Funatogawa, K.; Matsumura, K.; Kato-Miyazawa, M.; Kirikae, F.; Kiga, K.; Sasakawa, C.; Miyoshi-Akiyama, T.; Kirikae, T. MicroRNA-155 knockout mice are susceptible to Mycobacterium tuberculosis infection. Tuberculosis 2015, 95, 246–250. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| InterPro ID | Category | Description | p-Value |
|---|---|---|---|
| IPR014043 | domain | Acyl transferase | 6.267 × 10−7 |
| IPR009081 | domain | Phosphopantetheine binding ACP domain | 1.438 × 10−6 |
| IPR014030 | domain | Beta-ketoacyl synthase, N-terminal | 2.747 × 10−5 |
| IPR014031 | domain | Beta-ketoacyl synthase, C-terminal | 4.885 × 10−5 |
| IPR013968 | domain | Polyketide synthase, ketoreductase domain | 1.624 × 10−4 |
| IPR023836 | family | EccCa-like, Actinobacteria | 1.087 × 10−3 |
| IPR023837 | family | EccCb-like, Actinobacteria | 1.087 × 10−3 |
| IPR032821 | domain | Ketoacyl-synthetase, C-terminal extension | 1.190 × 10−3 |
| IPR006091 | domain | Acyl-CoA oxidase/dehydrogenase, central domain | 1.363 × 10−3 |
| IPR020807 | domain | Polyketide synthase, dehydratase domain | 1.757 × 10−3 |
| IPR009075 | domain | Acyl-CoA dehydrogenase/oxidase C-terminal | 1.995 × 10−3 |
| IPR002543 | domain | FtsK domain | 6.180 × 10−3 |
| IPR003029 | domain | S1 domain | 7.039 × 10−3 |
| IPR003495 | domain | CobW/HypB/UreG, nucleotide-binding domain | 7.039 × 10−3 |
| IPR023753 | domain | FAD/NAD(P) | 1.360 × 10-2 |
| IPR000788 | domain | Ribonucleotide reductase large subunit, C-terminal | 1.558 × 10−2 |
| IPR001030 | domain | Aconitase/3-isopropylmalate dehydratase large subunit, alpha/beta/alpha domain | 1.558 × 10−2 |
| IPR002300 | domain | Aminoacyl-tRNA synthetase, class Ia | 1.558 × 10−2 |
| IPR003714 | domain | PhoH-like protein | 1.558 × 10−2 |
| IPR004100 | domain | ATPase, F1/V1/A1 complex, alpha/beta subunit, N-terminal domain | 1.558 × 10−2 |
| GO ID | GO Name | p−Value |
|---|---|---|
| A. Biological Process | ||
| GO:0031295 | T-cell co-stimulation | 1.765 × 10−5 |
| GO:0031294 | Lymphocyte co-stimulation | 1.765 × 10−5 |
| GO:0009081 | Branched-chain amino acid metabolic process | 4.007 × 10−5 |
| GO:0009083 | Branched-chain amino acid catabolic process | 1.102 × 10−4 |
| GO:0006549 | Isoleucine metabolic process | 1.197 × 10−4 |
| GO:0000288 | Nuclear-transcribed mRNA catabolic process, Deadenylation-dependent decay | 2.663 × 10−4 |
| GO:0006573 | Valine metabolic process | 3.513 × 10−4 |
| GO:0060184 | Cell cycle switching | 4.878 × 10−4 |
| GO:0051728 | Cell cycle switching, mitotic to meiotic cell cycle | 4.878 × 10−4 |
| GO:0006574 | Valine catabolic process | 5.108 × 10−4 |
| B. Cellular Component | ||
| GO:0005643 | Nuclear pore | 4.20 × 10−3 |
| GO:0000932 | P-body | 5.39 × 10−3 |
| GO:0009986 | Cell surface | 6.00 × 10−3 |
| GO:0098978 | Glutamatergic synapse | 6.18 × 10−3 |
| GO:1990527 | Tec1p-Ste12p-Dig1p complex | 7.04 × 10−3 |
| GO:0110165 | Cellular anatomical entity | 1.23 × 10−2 |
| GO:1990526 | Ste12p-Dig1p-Dig2p complex | 1.60 × 10−2 |
| GO:0030496 | Midbody | 1.81 × 10−2 |
| GO:0009325 | Nitrate reductase complex | 1.83 × 10−2 |
| GO:0016020 | Membrane | 2.56 × 10−2 |
| C. Molecular Function | ||
| GO:0004085 | Butyryl-CoA dehydrogenase activity | 1.62 × 10−4 |
| GO:0005488 | Binding | 6.96 × 10−4 |
| GO:0052890 | Oxidoreductase activity, acting on the CH-CH group of donors, with a flavin as acceptor | 7.83 × 10−4 |
| GO:0043168 | Anion binding | 1.16 × 10−3 |
| GO:0003955 | NAD(P)H dehydrogenase (quinone) activity | 1.94 × 10−3 |
| GO:0017056 | Structural constituent of nuclear pore | 2.87 × 10−3 |
| GO:0004962 | Endothelin receptor activity | 2.94 × 10−3 |
| GO:0036094 | Small molecule binding | 3.15 × 10−3 |
| GO:1901363 | Heterocyclic compound binding | 4.09 × 10−3 |
| GO:0097159 | Organic cyclic compound binding | 4.10 × 10−3 |
| KEGG Id | Pathway | % of the Pathway Represented by the Expressed Genes |
|---|---|---|
| 03020 | RNA polymerase | 75.00 |
| 00630 | Glyoxylate and dicarboxylate metabolism | 34.15 |
| 03018 | RNA degradation | 33.33 |
| 00562 | Inositol phosphate metabolism | 33.33 |
| 00910 | Nitrogen metabolism | 31.82 |
| 05152 | Tuberculosis | 30.77 |
| 00680 | Methane metabolism | 28.57 |
| 00290 | Valine, leucine and isoleucine biosynthesis | 28.57 |
| 00430 | Taurine and hypotaurine metabolism | 28.57 |
| 00020 | Citrate cycle (TCA cycle) | 28.13 |
| 00730 | Thiamine metabolism | 27.27 |
| 00983 | Drug metabolism—other enzymes | 27.27 |
| 00061 | Fatty acid biosynthesis | 26.67 |
| 02024 | Quorum sensing | 25.00 |
| 00010 | Glycolysis/Gluconeogenesis | 24.24 |
| 00270 | Cysteine and methionine metabolism | 24.24 |
| 00260 | Glycine, serine, and threonine metabolism | 24.00 |
| 03060 | Protein export | 23.53 |
| 01053 | Biosynthesis of siderophore group nonribosomal peptides | 22.22 |
| 03070 | Bacterial secretion system | 21.43 |
| Expression Ranking | Gen ID | Gene Description | Mean RPKM |
|---|---|---|---|
| 1 | Rv2512c | Transposase for insertion sequence element IS1081 | 6.84 × 105 |
| 2 | Rv1067c | PE_PGRS19 PE-PGR | 3.45 × 105 |
| 3 | Rv3512 | PE_PGRS56 PE-PGRS | 1.91 × 105 |
| 4 | Rv1067c | PE_PGRS19 PE-PGR | 1.65 × 105 |
| 5 | Rv3344c | PE_PGRS49 PE-PGR | 1.63E × 105 |
| 6 | Rv0279c | PE_PGRS4 PE-PGRS | 1.10E × 105 |
| 7 | Rv3512 | PE_PGRS56 PE-PGRS | 5.01 × 104 |
| 8 | Rv0746 | PE_PGRS9 PE-PGRS | 4.35 × 104 |
| 9 | Rv3345c | PE_PGRS50 PE-PGR | 1.94 × 104 |
| 10 | Rv0105c | uracil-DNA glycosylase | 7.61 × 103 |
| 11 | Rv2840c | DUF448 domain-containing protein | 6.94 × 103 |
| 12 | Rv0440 | molecular chaperone GroEL | 5.71 × 103 |
| 13 | Rv3620c | peptidase M22 | 4.88 × 103 |
| 14 | Rv0454 | conserved hypotetical protein | 4.58 × 103 |
| 15 | Rv0967 | transcriptional regulator | 4.12 × 103 |
| 16 | Rv2424c | Putative transposase | 4.05 × 103 |
| 17 | Rv1228 | lipoprotein lpqX | 3.66 × 103 |
| 18 | Rv2424c | Uncharacterized protein | 3.11 × 103 |
| 19 | Rv3053c | Chain A, Glutaredoxin Like Protein Nrdh | 3.09 × 103 |
| 20 | Rv0637 | (3R)-hydroxyacyl-ACP dehydratase subunit HadC | 2.96 × 103 |
| Talaat et al., 2004 [6] | Pisu et al., 2020 [5] | Cornejo-Granados et al., 2021 | |
|---|---|---|---|
| Mtb strain | H37Rv | Erdman ATCC 35801 mCherry | H37Rv |
| Day post-infection analyzed | 7, 14, 21 and 28 | 14 | 21 |
| Murine model used | BALB/c and BALB/c SCID/SCID | C57BL/6J WT | BALB/c |
| Route of infection | Intranasal | Intranasal | Intratracheal |
| Infected tissue analyzed | Complete infected lungs | Alveolar and interstitial macrophages isolated from infected lungs | Complete infected lungs |
| RNA extraction method | Total RNA extraction with Tri Reagent (Trizol) from groups of 50 mice | Isolated cells were treated with Trizol and centrifuged to pellet mycobacterial cells. ~80% of the Trizol (containing host RNA) was removed. Finally, total RNA was extracted with fresh Trizol, and a proportion of the host RNA was added back. | Used a differential centrifugation of individual lungs with a mild-lysis buffer to pellet mycobacterial cells. Plus, removing the supernatant containing host RNA followed by total RNA extraction from the mycobacterial cells with a commercial kit. |
| Ribosomal RNA elimination | - | Ribo-Zero Gold rRNA Removal Kit (Epidemiology) | Ribo-Zero Gold rRNA Removal Kit (Epidemiology) and In-house Mtb ribosomal probes |
| Methodology for gene expression analysis | DNA Microarrays with oligonucleotides representing Mtb coding sequences. | RNA-seq | RNA-seq |
| Key insights | -The expression profile of Mtb in SCID mice is most similar to the profile when grown in broth. -Around 49 genes were only expressed in vivo, and 20 of these are contiguous in a delimited area of the Mtb genome. | -In vivo signature of 180 genes upregulated in macrophages. -Transcriptional signatures varied with macrophage ontology. -Alveolar macrophages showed a distinct up regulation of genes for the acquisition and use of fatty acids. -Interstitial macrophages showed an up regulation of genes for iron sequestration. | -About 62.59% of non-ribosomal sequences represented 702 genes, while 38.41% represented intergenic regions. -The transposase for IS1081 was the most expressed gene. -Eight genes for PE-PGRS members are among the most expressed genes.-Three highly expressed ncRNAs (MTS2823, RF00023, and RF00010) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cornejo-Granados, F.; López-Leal, G.; Mata-Espinosa, D.A.; Barrios-Payán, J.; Marquina-Castillo, B.; Equihua-Medina, E.; Zatarain-Barrón, Z.L.; Molina-Romero, C.; Hernández-Pando, R.; Ochoa-Leyva, A. Targeted RNA-Seq Reveals the M. tuberculosis Transcriptome from an In Vivo Infection Model. Biology 2021, 10, 848. https://doi.org/10.3390/biology10090848
Cornejo-Granados F, López-Leal G, Mata-Espinosa DA, Barrios-Payán J, Marquina-Castillo B, Equihua-Medina E, Zatarain-Barrón ZL, Molina-Romero C, Hernández-Pando R, Ochoa-Leyva A. Targeted RNA-Seq Reveals the M. tuberculosis Transcriptome from an In Vivo Infection Model. Biology. 2021; 10(9):848. https://doi.org/10.3390/biology10090848
Chicago/Turabian StyleCornejo-Granados, Fernanda, Gamaliel López-Leal, Dulce A. Mata-Espinosa, Jorge Barrios-Payán, Brenda Marquina-Castillo, Edgar Equihua-Medina, Zyanya L. Zatarain-Barrón, Camilo Molina-Romero, Rogelio Hernández-Pando, and Adrian Ochoa-Leyva. 2021. "Targeted RNA-Seq Reveals the M. tuberculosis Transcriptome from an In Vivo Infection Model" Biology 10, no. 9: 848. https://doi.org/10.3390/biology10090848
APA StyleCornejo-Granados, F., López-Leal, G., Mata-Espinosa, D. A., Barrios-Payán, J., Marquina-Castillo, B., Equihua-Medina, E., Zatarain-Barrón, Z. L., Molina-Romero, C., Hernández-Pando, R., & Ochoa-Leyva, A. (2021). Targeted RNA-Seq Reveals the M. tuberculosis Transcriptome from an In Vivo Infection Model. Biology, 10(9), 848. https://doi.org/10.3390/biology10090848