
Gene Co-Expression Analysis of Human RNASEH2A Reveals Functional Networks Associated with DNA Replication, DNA Damage Response, and Cell Cycle Regulation



Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Co-Expression Correlation Analysis
2.2. Co-Expression Correlation Analysis Verification with STRING Database
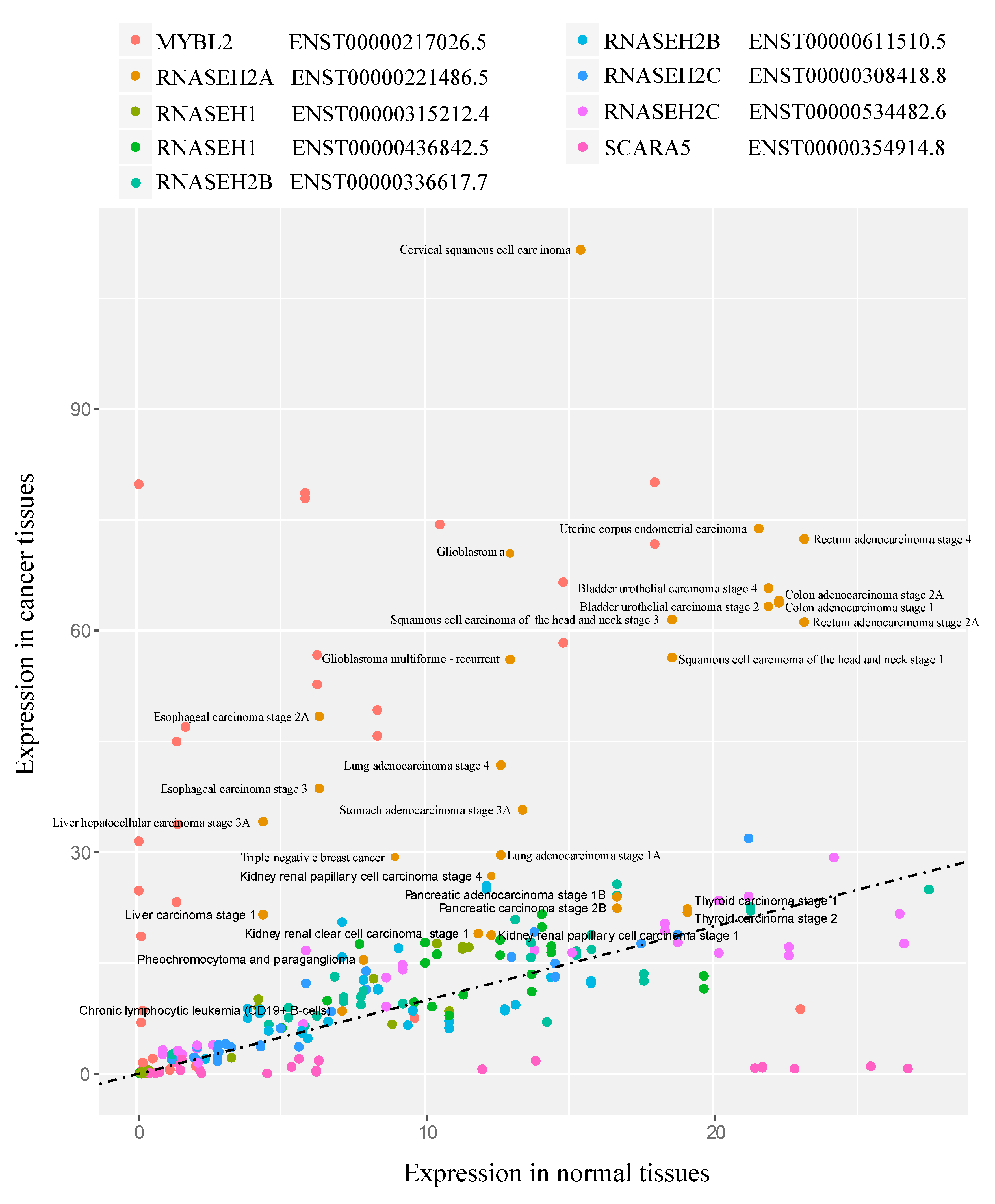
2.3. Gene Expression Analysis in Cancer
2.4. RNASEH2A Copy Number Alterations Analysis in TCGA Pan Cancer Dataset
2.5. Cell and Protein Extracts
2.6. Plasmids and Transfection
2.7. Co-Immunoprecipitation
2.8. Protein Analysis (Western Blot)
2.9. Mass Spectrometry Analysis
2.10. RNASEH2A Correlation with Cancer Progression and Cell Cycle Related Genes in CCLE and TCGA Pan Cancer Dataset
3. Results
3.1. RNASEH2A Is Co-Expressed with Genes that Function in Cell Cycle Regulation
3.2. RNASEH2A Expression Is Increased in Actively Cycling Cells and Tissues
3.3. RNASEH2A Gene Amplifications Has Higher Prevalence in Multiple Cancer Types
3.4. Mass Spectrometry Analysis Identified RNASEH2A Binding Partners Involved in Mitosis Regulation
3.5. RNASEH2A Expression Positively Correlates with Cancer Proliferation Markers and Cell Cycle Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burgess, D.J. Gene expression: Principles of gene regulation across tissues. Nat. Rev. Genet. 2017, 18, 701. [Google Scholar] [CrossRef]
- Sonawane, A.R.; Platig, J.; Fagny, M.; Chen, C.-Y.; Paulson, J.N.; Lopes-Ramos, C.M.; DeMeo, D.L.; Quackenbush, J.; Glass, K.; Kuijjer, M.L. Understanding Tissue-Specific Gene Regulation. Cell Rep. 2017, 21, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium. The genotype-tissue expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Cerritelli, S.M.; Crouch, R.J. Ribonuclease H: The enzymes in Eukaryotes. FEBS J. 2009, 276, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Sparks, J.L.; Chon, H.; Cerritelli, S.M.; Kunkel, T.A.; Johansson, E.; Crouch, R.J.; Burgers, P.M. RNase H2-initiated ribonucleotide excision repair. Mol. Cell 2012, 47, 980–986. [Google Scholar] [CrossRef] [PubMed]
- Chon, H.; Vassilev, A.; Depamphilis, M.L.; Zhao, Y.; Zhang, J.; Burgers, P.M.; Crouch, R.J.; Cerritelli, S.M. Contributions of the two accessory subunits, RNASEH2B and RNASEH2C, to the activity and properties of the human RNase H2 complex. Nucleic Acids Res. 2009, 37, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Sollier, J.; Stork, C.T.; García-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef]
- Hamperl, S.; Bocek, M.J.; Saldivar, J.C.; Swigut, T.; Cimprich, K.A. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell 2017, 170, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Meers, C.; Keskin, H.; Storici, F. DNA repair by RNA: Templated, or not templated, that is the question. DNA Repair 2016, 44, 17–21. [Google Scholar] [CrossRef]
- Wahba, L.; Amon, J.D.; Koshland, D.; Vuica-Ross, M. RNase H and multiple RNA biogenesis factors cooperate to prevent RNA: DNA hybrids from generating genome instability. Mol. Cell 2011, 44, 978–988. [Google Scholar] [CrossRef]
- Cornelio, D.A.; Sedam, H.N.; Ferrarezi, J.A.; Sampaio, N.M.; Argueso, J.L. Both R-loop removal and ribonucleotide excision repair activities of RNase H2 contribute substantially to chromosome stability. DNA Repair 2017, 52, 110–114. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, K.; Jinks-Robertson, S.; Petes, T.D. Elevated Genome-Wide Instability in Yeast Mutants Lacking RNase H Activity. Genetics 2015, 201, 963–975. [Google Scholar] [CrossRef]
- Zimmer, A.D.; Koshland, D. Differential roles of the RNases H in preventing chromosome instability. Proc. Natl. Acad. Sci. USA 2016, 113, 12220–12225. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, K.; Knittler, K.; Borowski, C.; Rudnik, S.; Damme, M.; Aden, K.; Spehlmann, M.E.; Frey, N.; Saftig, P.; Chalaris, A.; et al. Absence of RNase H2 triggers generation of immunogenic micronuclei removed by autophagy. Hum. Mol. Genet. 2017, 26, 3960–3972. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.; Patrick, T.; Parmar, R.; Taylor, C.F.; Aeby, A.; Aicardi, J.; Artuch, R.; Montalto, S.A.; Bacino, C.A.; Barroso, B.; et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am. J. Hum. Genet. 2007, 81, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Goutières, F. Aicardi-Goutières syndrome. Brain Dev. 2005, 27, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Pulliero, A.; Fazzi, E.; Cartiglia, C.; Orcesi, S.; Balottin, U.; Uggetti, C.; La Piana, R.; Olivieri, I.; Galli, J.; Izzotti, A. The Aicardi-Goutières syndrome. Molecular and clinical features of RNAse deficiency and microRNA overload. Mutat. Res. 2011, 717, 99–108. [Google Scholar] [CrossRef]
- Flanagan, J.M.; Funes, J.M.; Henderson, S.; Wild, L.; Carey, N.; Boshoff, C. Genomics screen in transformed stem cells reveals RNASEH2A, PPAP2C, and ADARB1 as putative anticancer drug targets. Mol. Cancer Ther. 2009, 8, 249–260. [Google Scholar] [CrossRef]
- Xu, J.; Liu, H.; Yang, Y.; Wang, X.; Liu, P.; Li, Y.; Meyers, C.; Banerjee, N.S.; Wang, H.-K.; Cam, M.; et al. Genome-Wide Profiling of Cervical RNA-Binding Proteins Identifies Human Papillomavirus Regulation of RNASEH2A Expression by Viral E7 and E2F1. mBio 2019, 10, e02687-18. [Google Scholar] [CrossRef]
- Williams, K.A.; Lee, M.; Hu, Y.; Andreas, J.; Patel, S.J.; Zhang, S.; Chines, P.; Elkahloun, A.; Chandrasekharappa, S.; Gutkind, J.S.; et al. A systems genetics approach identifies CXCL14, ITGAX, and LPCAT2 as novel aggressive prostate cancer susceptibility genes. PLoS Genet. 2014, 10, e1004809. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, Y.; Cheng, L.; Cheng, Y.; Zhou, H.H.; Tan, Z.R. Identification of Common Genes Refers to Colorectal Carcinogenesis with Paired Cancer and Noncancer Samples. Dis. Markers 2018, 3452739. [Google Scholar] [CrossRef] [PubMed]
- Yuan, N.; Zhang, G.; Bie, F.; Ma, M.; Ma, Y.; Jiang, X.; Wang, Y.; Hao, X. Integrative analysis of lncRNAs and miRNAs with coding RNAs associated with ceRNA crosstalk network in triple negative breast cancer. Onco. Targets Ther. 2017, 10, 5883–5897. [Google Scholar] [CrossRef]
- Lockhart, A.; Pires, V.B.; Bento, F.; Kellner, V.; Luke-Glaser, S.; Yakoub, G.; Ulrich, H.D.; Luke, B. RNase H1 and H2 Are Differentially Regulated to Process RNA-DNA Hybrids. Cell Rep. 2019, 29, 2890–2900.e5. [Google Scholar] [CrossRef]
- Pearson’s Correlation Coefficient. In Encyclopedia of Public Health; Kirch, W., Ed.; Springer: Dordrecht, The Netherlands, 2008. [Google Scholar] [CrossRef]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-R.; Sun, C.-H.; Li, W.; Chao, R.-F.; Huang, C.-C.; Zhou, X.J.; Liu, C.-C. Cancer RNA-Seq Nexus: A database of phenotype-specific transcriptome profiling in cancer cells. Nucleic Acids Res. 2016, 44, D944–D951. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404, Erratum in: Cancer Discov. 2012, 2, 960. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, l1. [Google Scholar] [CrossRef] [PubMed]
- Garcia, B.A.; Smalley, D.M.; Cho, H.; Shabanowitz, J.; Ley, K.; Hunt, D.F. The platelet microparticle proteome. J. Proteome Res. 2005, 4, 1516–1521. [Google Scholar] [CrossRef]
- Mukherjee, A.; Stathos, M.E.; Varner, C.; Arsiwala, A.; Frey, S.; Hu, Y.; Smalley, D.M.; Schaffer, D.V.; Kane, R.S. One-pot synthesis of heterodimeric agonists that activate the canonical Wnt signaling pathway. Chem. Commun. 2020, 56, 3685–3688. [Google Scholar] [CrossRef] [PubMed]
- Van Ooijen, M.P.; Jong, V.L.; Eijkemans, M.J.C.; Heck, A.J.R.; Andeweg, A.C.; Binai, N.A.; Van den Ham, H.J. Identification of differentially expressed peptides in high-throughput proteomics data. Brief Bioinform. 2018, 19, 971–981. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 25 June 2020).
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., 3rd; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Harrell, F.E., Jr.; With contributions from Charles Dupont and Many Others. 2020 Hmisc: Harrell Miscellaneous. R Package Version 4.4-1. Available online: https://CRAN.R-project.org/package=Hmisc (accessed on 18 December 2020).
- Arora, R.; Lee, Y.; Wischnewski, H.; Brun, C.M.; Schwarz, T.; Azzalin, C.M. RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat. Commun. 2014, 5, 5220. [Google Scholar] [CrossRef] [PubMed]
- Cusanelli, E.; Chartrand, P. Telomeric repeat-containing RNA TERRA: A noncoding RNA connecting telomere biology to genome integrity. Front. Genet. 2015, 6, 143. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Sun, Q.; Wang, X. Transcriptional landscape of human cancers. Oncotarget 2017, 8, 34534–34551. [Google Scholar] [CrossRef]
- Shao, X.; Lv, N.; Liao, J.; Long, J.; Xue, R.; Ai, N.; Xu, D.; Fan, X. Copy number variation is highly correlated with differential gene expression: A pan-cancer study. BMC Med. Genet. 2019, 20, 175. [Google Scholar] [CrossRef]
- Freund, A.; Zhong, F.L.; Venteicher, A.S.; Meng, Z.; Veenstra, T.D.; Frydman, J.; Artandi, S.E. Proteostatic control of telomerase function through TRiC-mediated folding of TCAB1. Cell 2014, 159, 1389–1403. [Google Scholar] [CrossRef]
- Kaisari, S.; Sitry-Shevah, D.; Miniowitz-Shemtov, S.; Teichner, A.; Hershko, A. Role of CCT chaperonin in the disassembly of mitotic checkpoint complexes. Proc. Natl. Acad. Sci. USA 2017, 114, 956–961. [Google Scholar] [CrossRef]
- Yang, S.; Ren, X.; Liang, Y.; Yan, Y.; Zhou, Y.; Hu, J.; Wang, Z.; Song, F.; Wang, F.; Liao, W.; et al. KNK437 restricts the growth and metastasis of colorectal cancer via targeting DNAJA1/CDC45 axis. Oncogene 2020, 39, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Madungwe, N.B.; Feng, Y.; Lie, M.; Tombo, N.; Liu, L.; Kaya, F.; Bopassa, J.C. Mitochondrial inner membrane protein (mitofilin) knockdown induces cell death by apoptosis via an AIF-PARP-dependent mechanism and cell cycle arrest. Am. J. Physiol. Cell Physiol. 2018, 315, C28–C43. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Whitfield, M.L.; George, L.K.; Grant, G.D.; Perou, C.M. Common markers of proliferation. Nat. Rev. Cancer. 2006, 2, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, G.; Feng, X.; Shepherd, P.; Zhang, J.; Tang, M.; Chen, Z.; Srivastava, M.; McLaughlin, M.E.; Navone, N.M.; et al. Genome-wide CRISPR screens reveal synthetic lethality of RNASEH2 deficiency and ATR inhibition. Oncogene 2019, 38, 2451–2463. [Google Scholar] [CrossRef]
- Arudchandran, A.; Cerritelli, S.M.; Narimatsu, S.K.; Itayaa, M.; Shinb, D.-Y.; Shimadac, Y.; Crouch, R.J. The absence of ribonuclease H1 or H2 alters the sensitivity of Saccharomyces cerevisiae to hydroxyurea, caffeine and ethyl methanesulphonate: Implications for roles of RNases H in DNA replication and repair. Genes Cells 2000, 5, 789–802. [Google Scholar] [CrossRef]
- Reijns, M.A.; Rabe, B.; Rigby, R.E.; Mill, P.; Astell, K.R.; Lettice, L.A.; Boyle, S.; Leitch, A.; Keighren, M.; Kilanowski, F.; et al. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 2012, 149, 1008–1022. [Google Scholar] [CrossRef]
- McElhinny, S.A.N.; Watts, B.E.; Kumar, D.; Watt, D.L.; Lundström, E.-B.; Burgers, P.M.J.; Johansson, E.; Chabes, A.; Kunkel, T.A. Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proc. Natl. Acad. Sci. USA 2010, 107, 4949–4954. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.S.; Lujan, S.A.; Kunkel, T.A. Processing ribonucleotides incorporated during eukaryotic DNA replication. Nat. Rev. Mol. Cell Biol. 2016, 17, 350–363. [Google Scholar] [CrossRef]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell. 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Vijayraghavan, S.; Tsai, F.L.; Schwacha, A. A Checkpoint-Related Function of the MCM Replicative Helicase Is Required to Avert Accumulation of RNA:DNA Hybrids during S-phase and Ensuing DSBs during G2/M. PLoS Genet. 2016, 12, e1006277. [Google Scholar] [CrossRef] [PubMed]
- Ciferri, C.; Pasqualato, S.; Screpanti, E.; Varetti, G.; Santaguida, S.; Dos Reis, G.; Maiolica, A.; Polka, J.; De Luca, J.G.; De Wulf, P.; et al. Implications for kinetochore-microtubule attachment from the structure of an engineered Ndc80 complex. Cell 2008, 133, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Foltz, D.R.; Jansen, L.E.; Black, B.E.; Bailey, A.O.; Yates, J.R., 3rd; Cleveland, D.W. The human CENP-A centromeric nucleosome-associated complex. Nat. Cell Biol. 2006, 8, 458–469. [Google Scholar] [CrossRef]
- Miki, H.; Setou, M.; Kaneshiro, K.; Hirokawa, N. All kinesin superfamily protein, KIF, genes in mouse and human. Proc. Natl. Acad. Sci. USA 2001, 98, 7004–7011. [Google Scholar] [CrossRef]
- Dai, B.; Zhang, P.; Zhang, Y.; Pan, C.; Meng, G.; Xiao, X.; Wu, Z.; Jia, W.; Zhang, J.; Zhang, L. RNaseH2A is involved in human gliomagenesis through the regulation of cell proliferation and apoptosis. Oncol. Rep. 2016, 36, 173–180. [Google Scholar] [CrossRef]
- Shen, J.; Lin, S.; Liu, L.; Wang, C. Ribonuclease H2 Subunit A impacts invasiveness and chemoresistance resulting in poor survivability of breast cancer in ER dependent manner. Am. J. Transl Res. 2020, 12, 2281–2294. [Google Scholar] [PubMed]
- Kessler, M.; Zietlow, R.; Meyer, T.F. Stem cells in the reproductive system. In Adults Stem Cell Niches; Wislet, S., Ed.; IntechOpen: London, UK, 2014; Chapter 6; pp. 139–169. [Google Scholar]
- Stepanenko, A.A.; Dmitrenko, V.V. HEK293 in cell biology and cancer research: Phenotype, karyotype, tumorigenicity, and stress-induced genome-phenotype evolution. Gene 2015, 569, 182–190. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Process | |||
| GO Term | Description | p-Value | FDR q-Value |
| GO 1903047 | mitotic cell cycle process | 2.80 × 10−14 | 5.93 × 10−11 |
| GO 0070507 | regulation of microtubule cytoskeleton organ | 1.39 × 10−7 | 8.40 × 10−5 |
| GO 0006974 | cellular response to DNA damage stimulus | 2.77 × 10−5 | 5.86 × 10−3 |
| GO 0007059 | chromosome segregation | 9.00 × 10−5 | 1.36 × 10−2 |
| GO 0006260 | DNA replication | 2.66 × 10−4 | 3.12 × 10−2 |
| Function | |||
| GO Term | Description | p-Value | FDR q-Value |
| GO 0140097 | catalytic activity, acting on DNA | 9.59 × 10−6 | 9.06 × 10−3 |
| GO 0008017 | microtubule binding | 4.52 × 10−5 | 1.42 × 10−2 |
| GO 0019900 | kinase binding | 3.28 × 10−4 | 7.74 × 10−2 |
| Component | |||
| GO Term | Description | p-Value | FDR q-Value |
| GO 0044427 | chromosomal part | 4.06 × 10−8 | 1.36 × 10−5 |
| GO 0000922 | spindle pole | 1.13 × 10−6 | 2.52 × 10−4 |
| GO 0005654 | nucleoplasm | 3.52 × 10−6 | 5.88 × 10−4 |
| GO 0030496 | midbody | 5.01 × 10−6 | 6.70 × 10−4 |
| GO 0005815 | microtubule organizing center | 5.13 × 10−6 | 5.72 × 10−4 |
| GO 0000940 | condensed chromosome outer kinetochore | 8.72 × 10−6 | 7.28 × 10−4 |
| GO 0000796 | condensin complex | 8.71 × 10−5 | 4.16 × 10−3 |
| RNASEH1 | RNASEH2A | RNASEH2B | RNASEH2C | RNASEH1 | RNASEH2A | RNASEH2B | RNASEH2C | ||
|---|---|---|---|---|---|---|---|---|---|
| Liver | 5.193 | 3.947 | 3.878 | 11.57 | Adipose-Subcutaneous | 13.845 | 15.96 | 16.585 | 30.145 |
| Muscle-Skeletal | 11.72 | 5.9535 | 5.51 | 9.215 | Lung | 12.92 | 14.51 | 19.34 | 30.99 |
| Heart-Left Ventricle | 7.251 | 4.262 | 8.466 | 12.71 | Breast-Mammary Tissue | 12.93 | 16.39 | 16.65 | 33.895 |
| Pancreas | 5.3175 | 6.44 | 10.175 | 11.48 | Pituitary | 8.271 | 11.02 | 14.5 | 52.95 |
| Whole Blood | 2.726 | 7.958 | 6.471 | 16.33 | Vagina | 12.08 | 20.33 | 16.85 | 39.11 |
| Brain-Putamen (basal ganglia) | 4.6985 | 5.5965 | 7.875 | 15.635 | Esophagus-Muscularis | 14.91 | 8.687 | 22.84 | 46.885 |
| Brain-Caudate (basal ganglia) | 5.1025 | 6.1935 | 9.2325 | 17.175 | Artery-Tibial | 16.91 | 8.638 | 17.8 | 50.75 |
| Brain-Substantia nigra | 6.0725 | 7.4295 | 8.123 | 16.355 | Thyroid | 12.615 | 16.935 | 24.905 | 43.205 |
| Brain-Amygdala | 5.0275 | 7.4755 | 8.2985 | 17.285 | Esophagus-Gastroesophageal Junction | 13.75 | 8.3635 | 22.05 | 53.92 |
| Brain-Hippocampus | 5.907 | 6.655 | 7.567 | 18.76 | Bladder | 15.4 | 15.95 | 23.68 | 43.32 |
| Brain-Anterior cingulate cortex (BA24) | 5.921 | 6.326 | 9.554 | 19.13 | Artery-Coronary | 13.65 | 10.5 | 18.58 | 56.4 |
| Brain-Hypothalamus | 6.903 | 7.451 | 8.639 | 18.47 | Artery-Aorta | 14.63 | 9.235 | 17.31 | 58.05 |
| Kidney-Cortex | 6.303 | 6.632 | 6.614 | 22.18 | Prostate | 11.11 | 11.475 | 20.27 | 57.625 |
| Brain-Nucleus accumbens (basal ganglia) | 6.557 | 7.162 | 12.03 | 20.17 | Nerve-Tibial | 15.18 | 19.24 | 19.55 | 48.22 |
| Heart-Atrial Appendage | 8.609 | 5.969 | 9.644 | 22.33 | Fallopian Tube | 14.57 | 13.31 | 25.31 | 49.21 |
| Brain-Cortex | 5.5355 | 6.8145 | 11.095 | 25.555 | Colon-Sigmoid | 13.24 | 10 | 24.46 | 55.42 |
| Brain-Frontal Cortex (BA9) | 7.597 | 6.766 | 13.7 | 23.46 | Cells-Transformed fibroblasts | 29.98 | 34.04 | 17.95 | 25.17 |
| Stomach | 8.19 | 9.396 | 12.55 | 24.365 | Brain-Cerebellum | 9.384 | 16.21 | 12.41 | 70.97 |
| Brain-Spinal cord (cervical c-1) | 9.183 | 13.54 | 10.54 | 22.75 | Spleen | 10.935 | 25.695 | 24.555 | 49.11 |
| Skin-Sun Exposed (Lower leg) | 11.62 | 18.75 | 10.87 | 19.18 | Ovary | 12.4 | 19.46 | 20.07 | 59.51 |
| Skin-Not Sun Exposed (Suprapubic) | 10.72 | 18.3 | 11.7 | 19.98 | Cervix-Ectocervix | 14.765 | 24.625 | 23.54 | 51.435 |
| Minor Salivary Gland | 10.33 | 13.21 | 13.03 | 24.15 | Brain-Cerebellar Hemisphere | 13.325 | 17.52 | 14.98 | 68.75 |
| Esophagus-Mucosa | 10.58 | 24.62 | 11.92 | 18.2 | Cervix-Endocervix | 15.15 | 19 | 26.62 | 55 |
| Colon-Transverse | 9.484 | 12.52 | 14.885 | 28.96 | Testis | 14.12 | 49.84 | 23.42 | 31.66 |
| Adrenal Gland | 10.045 | 9.7865 | 12.39 | 35.965 | Uterus | 16.6 | 21.33 | 30.43 | 66.03 |
| Adipose-Visceral (Omentum) | 12.25 | 12.14 | 14.71 | 32.23 | Cells-Epstein-Barr virus (EBV)-transformed lymphocytes | 27.94 | 79.945 | 41.36 | 32.195 |
| Small Intestine-Terminal Ileum | 9.633 | 14.12 | 18.32 | 30.46 |
| Cancer Type | Percentage of Patients with Copy Number Alterations | ||||
|---|---|---|---|---|---|
| Deep Deletion | Shallow Deletion | Diploid | Gain | Amplification | |
| Ovarian Epithelial Tumor | 0.34% | 29.15% | 26.10% | 36.27% | 8.14% |
| Endometrial Carcinoma | - | 12.13% | 69.67% | 14.90% | 3.29% |
| Adrenocortical Carcinoma | - | 1.32% | 34.21% | 61.84% | 2.63% |
| Pleural Mesothelioma | - | 8.05% | 73.56% | 16.09% | 2.30% |
| Esophageal Squamous Cell Carcinoma | - | 34.04% | 44.68% | 19.15% | 2.13% |
| Cervical Squamous Cell Carcinoma | 0.41% | 26.23% | 57.79% | 13.52% | 2.05% |
| Diffuse Glioma | 0.20% | 3.33% | 73.92% | 20.78% | 1.76% |
| Sarcoma | - | 10.36% | 50.20% | 37.85% | 1.59% |
| Invasive Breast Carcinoma | 0.09% | 21.25% | 60.11% | 17.23% | 1.31% |
| Ocular Melanoma | - | 3.75% | 92.50% | 2.50% | 1.25% |
| Thymic Epithelial Tumor | - | 2.52% | 94.12% | 2.52% | 0.84% |
| Esophagogastric Adenocarcinoma | 0.62% | 33.40% | 57.73% | 7.42% | 0.82% |
| Head and Neck Squamous Cell Carcinoma | 0.20% | 19.45% | 66.99% | 12.57% | 0.79% |
| Glioblastoma | - | 8.11% | 58.11% | 33.11% | 0.68% |
| Non-Small Cell Lung Cancer | 0.30% | 44.10% | 43.39% | 11.71% | 0.50% |
| Bladder Urothelial Carcinoma | - | 29.21% | 53.71% | 16.58% | 0.50% |
| Prostate Adenocarcinoma | 0.20% | 5.74% | 92.21% | 1.64% | 0.20% |
| Cervical Adenocarcinoma | 2.17% | 26.09% | 65.22% | 6.52% | - |
| Cholangiocarcinoma | - | 13.89% | 72.22% | 13.89% | - |
| Colorectal Adenocarcinoma | 0.51% | 11.02% | 71.86% | 16.61% | - |
| Encapsulated Glioma | - | - | 100.00% | - | - |
| Fibrolamellar Carcinoma | - | - | 100.00% | - | - |
| Hepatocellular Carcinoma | 0.28% | 21.51% | 64.25% | 13.97% | - |
| Leukemia | - | 1.81% | 95.18% | 3.01% | - |
| Mature B-Cell Neoplasms | - | 2.08% | 91.67% | 6.25% | - |
| Melanoma | - | 22.62% | 61.31% | 16.08% | - |
| Miscellaneous Neuroepithelial Tumor | - | - | 96.30% | 3.70% | - |
| Non-Seminomatous Germ Cell Tumor | - | 52.33% | 33.72% | 13.95% | - |
| Pancreatic Adenocarcinoma | - | 14.77% | 77.27% | 7.95% | - |
| Pheochromocytoma | - | 0.75% | 84.33% | 14.93% | - |
| Renal Clear Cell Carcinoma | - | 2.17% | 88.56% | 9.27% | - |
| Renal Non-Clear Cell Carcinoma | - | 5.17% | 87.36% | 7.47% | - |
| Seminoma | - | 1.59% | 55.56% | 42.86% | - |
| Undifferentiated Stomach Adenocarcinoma | - | 25.00% | 58.33% | 16.67% | - |
| Well-Differentiated Thyroid Cancer | - | 1.21% | 97.37% | 1.41% | - |
| All Cancer types | 0.17% | 17.69% | 66.83% | 14.30% | 1.01% |
| Protein | Gene | Description |
|---|---|---|
| Q5TBB1 | RNASEH2B | Ribonuclease H2 subunit B |
| E9PN81 | RNASEH2C | Ribonuclease H2 subunit C |
| P31689 | DNAJA1 | DnaJ homolog subfamily A member 1 |
| O95831 | AIFM1 | Apoptosis-inducing factor 1, mitochondrial |
| P50990 | CCT8 | T-complex protein 1 subunit theta |
| Gene Name (s) | Function/Role | References |
|---|---|---|
| CDKN2A, CDKN2B, CCND1 | G1 Cell Cycle Phase | A. Subramanian et al. Gene Set Enrichment Analysis (GSEA Database) [46] |
| DHFR, CCNE1 | G1/S Cell Cycle Phase | A. Subramanian et al. (GSEA Database) [46] |
| AKT1-3, E2F4-5 | S Cell Cycle Phase | A. Subramanian et al. (GSEA Database) [46] |
| CDKN2D, MDM2 | G2 Cell Cycle Phase | A. Subramanian et al. (GSEA Database) [46] |
| CCNB2, TOPBP1 | G2/M Cell Cycle Phase | A. Subramanian et al. (GSEA Database) [46] |
| APC, BUB1 | M Cell Cycle Phase | A. Subramanian et al. (GSEA Database) [46] |
| E2F2, E2F3, CCNB1 | M/G1 Cell Cycle Phase | A. Subramanian et al. (GSEA Database) [46] |
| MYBL2, FOXM1, BUB1, AURKA, AURKB | Upregulated in cancer | M. Li et al. [40] |
| SCARA5, MYOM1 | Downregulated in cancer | M. Li et al. [40] |
| PCNA, MKI67(Ki67), MCM2–MCM6, E2F1 | Proliferative markers in cancer | M.L. Whitfield et al. [47] |
| CCNE1, CCND1, CCNB1 | Cell cycle markers associated with cancer (G1/S, G2, and M) | M.L. Whitfield et al. [47] |
| RNASEH2A, RNASEH2B, RNASEH2C, RNASEH1 | Target genes in this study | This study |
| CCT8, DNAJA1, AIFM1 | Predicted Binding partners of RNASEH2A | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marsili, S.; Tichon, A.; Kundnani, D.; Storici, F. Gene Co-Expression Analysis of Human RNASEH2A Reveals Functional Networks Associated with DNA Replication, DNA Damage Response, and Cell Cycle Regulation. Biology 2021, 10, 221. https://doi.org/10.3390/biology10030221
Marsili S, Tichon A, Kundnani D, Storici F. Gene Co-Expression Analysis of Human RNASEH2A Reveals Functional Networks Associated with DNA Replication, DNA Damage Response, and Cell Cycle Regulation. Biology. 2021; 10(3):221. https://doi.org/10.3390/biology10030221
Chicago/Turabian StyleMarsili, Stefania, Ailone Tichon, Deepali Kundnani, and Francesca Storici. 2021. "Gene Co-Expression Analysis of Human RNASEH2A Reveals Functional Networks Associated with DNA Replication, DNA Damage Response, and Cell Cycle Regulation" Biology 10, no. 3: 221. https://doi.org/10.3390/biology10030221
APA StyleMarsili, S., Tichon, A., Kundnani, D., & Storici, F. (2021). Gene Co-Expression Analysis of Human RNASEH2A Reveals Functional Networks Associated with DNA Replication, DNA Damage Response, and Cell Cycle Regulation. Biology, 10(3), 221. https://doi.org/10.3390/biology10030221