
Understanding the Role of Estrogen Receptor Status in PRODH/POX-Dependent Apoptosis/Survival in Breast Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
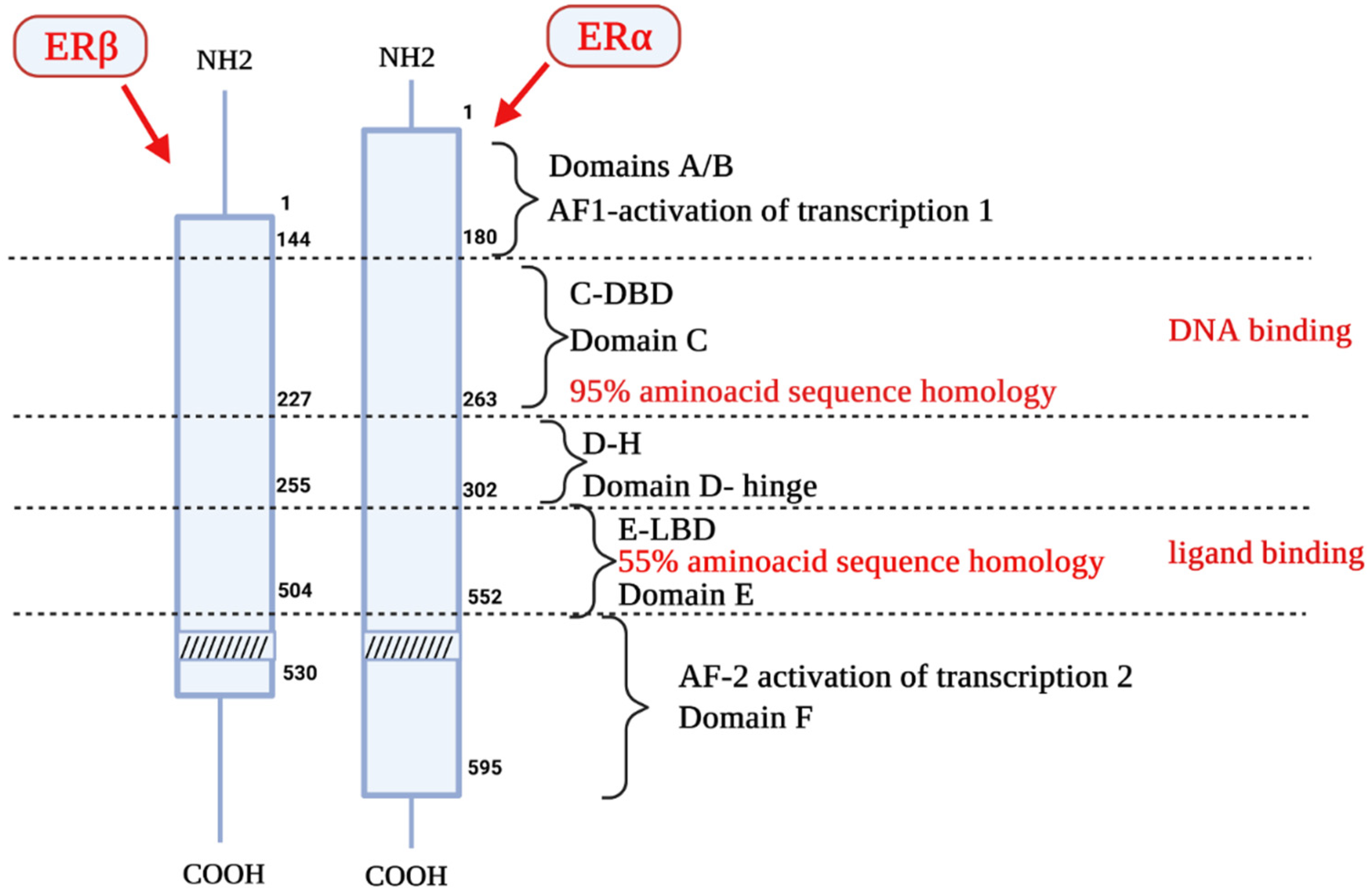
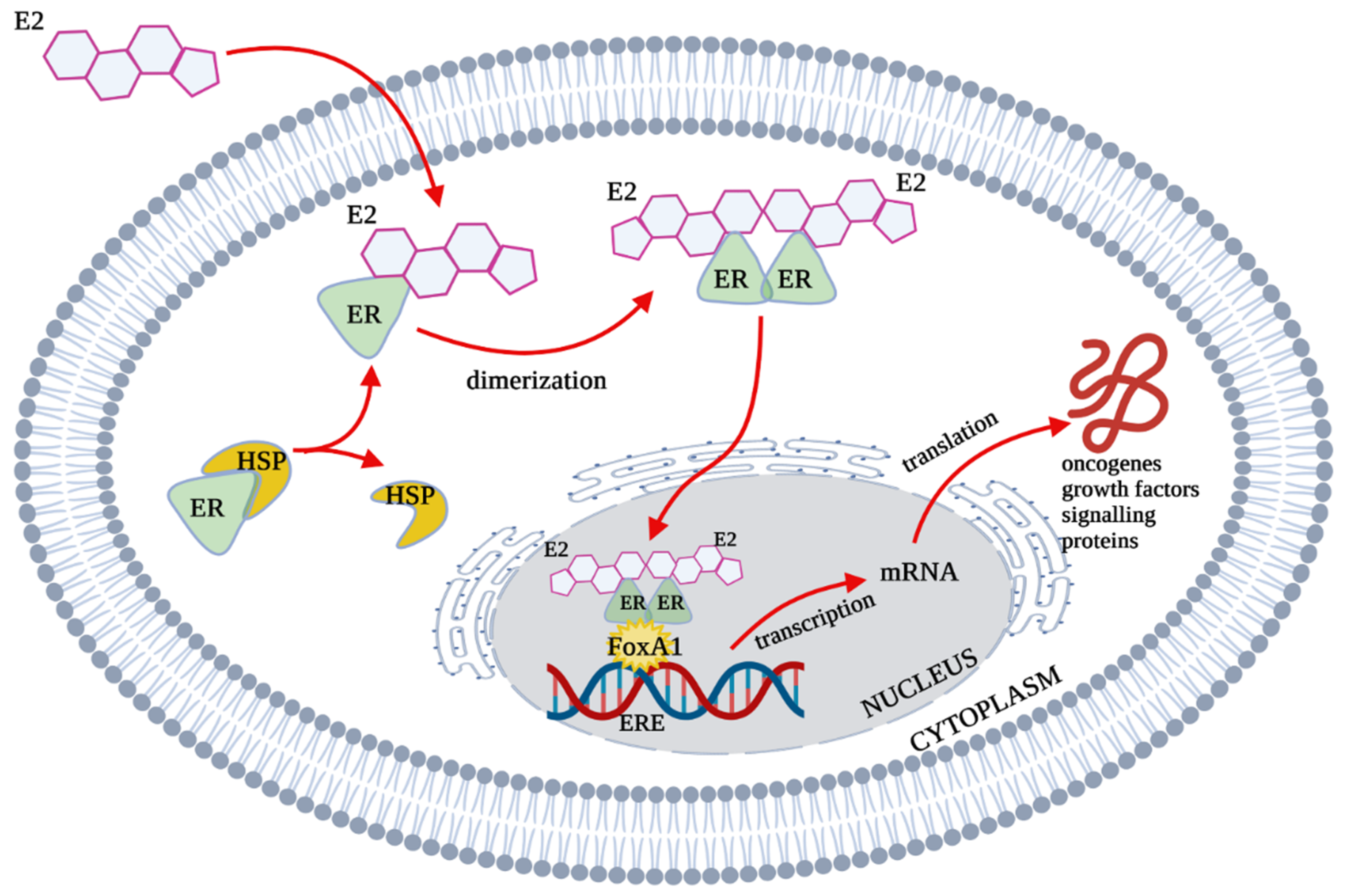
2. Estrogen Receptors Structure, Location and Function
3. Apoptosis
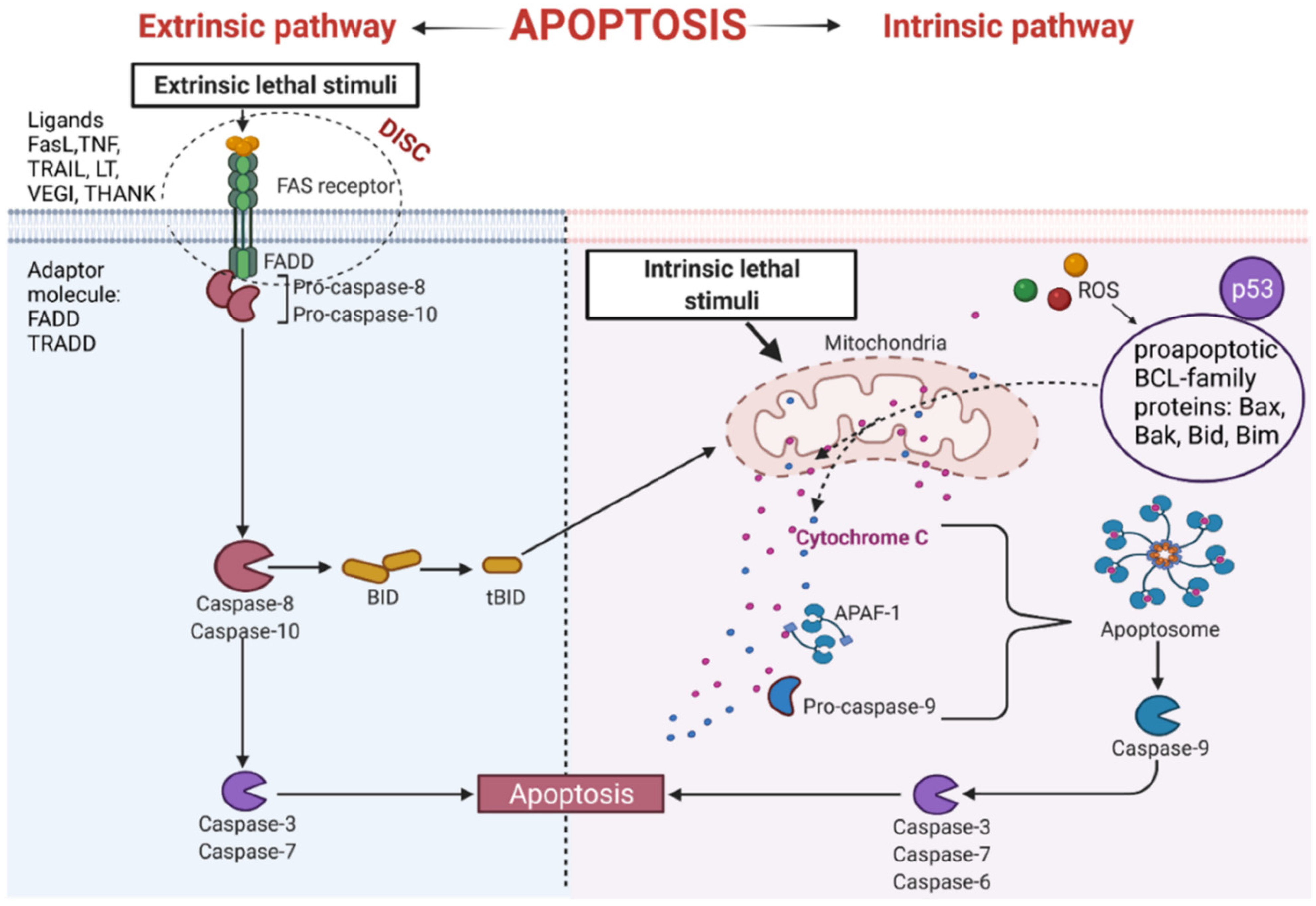
3.1. The Extrinsic Apoptosis Pathway
3.2. The Intrinsic Apoptosis Pathway
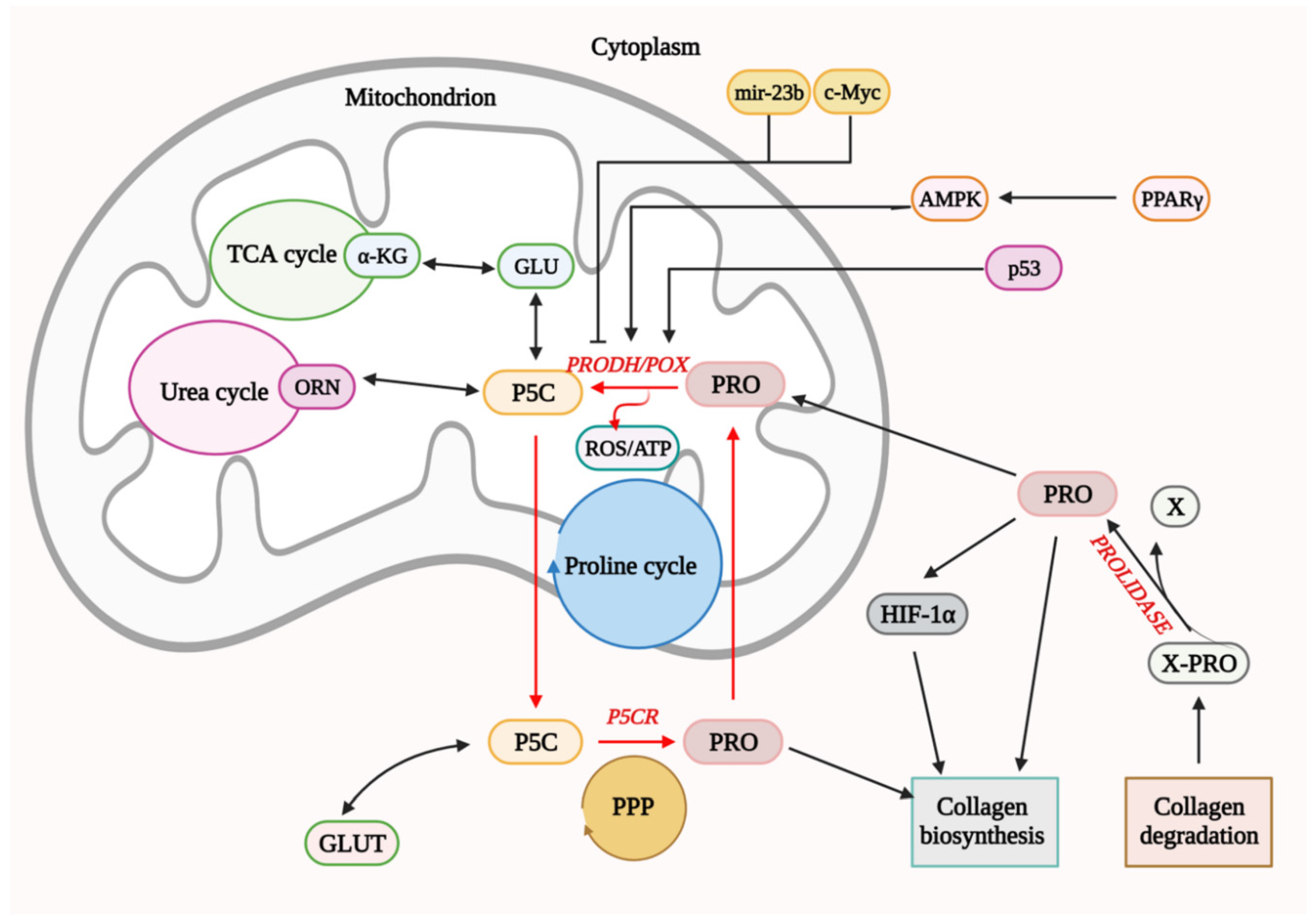
4. Functional Significance of PRODH/POX in Cell Metabolism
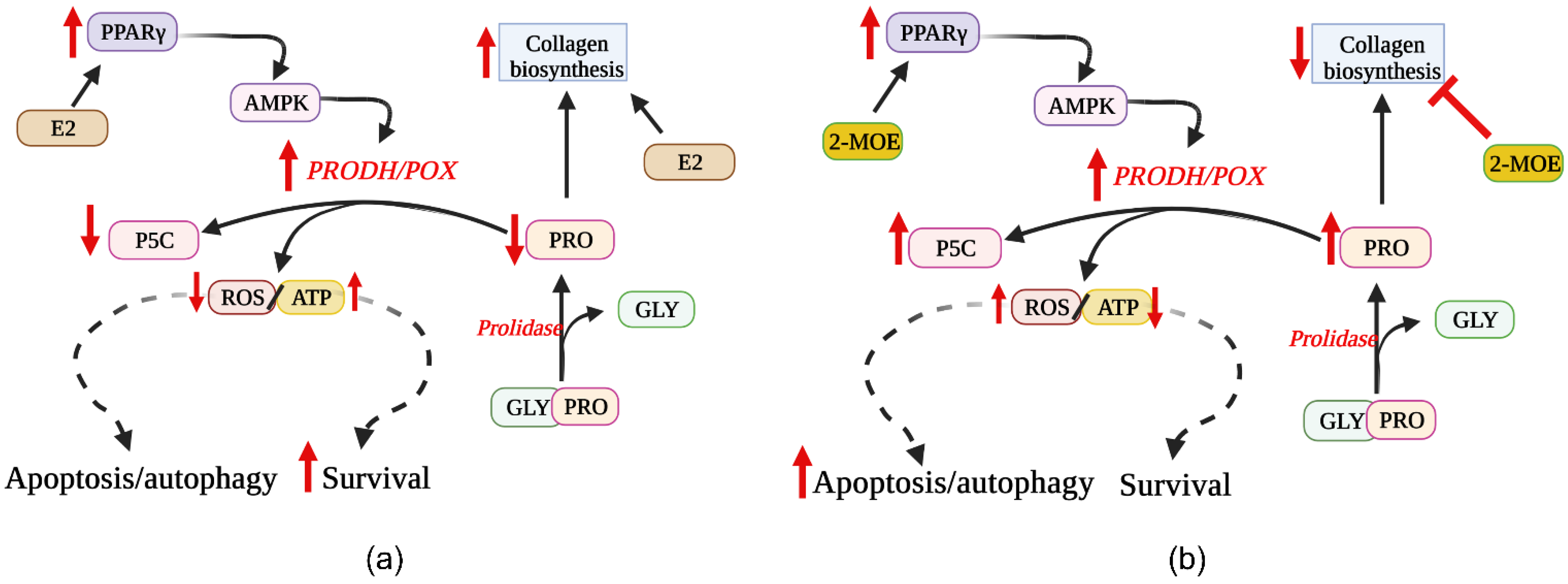
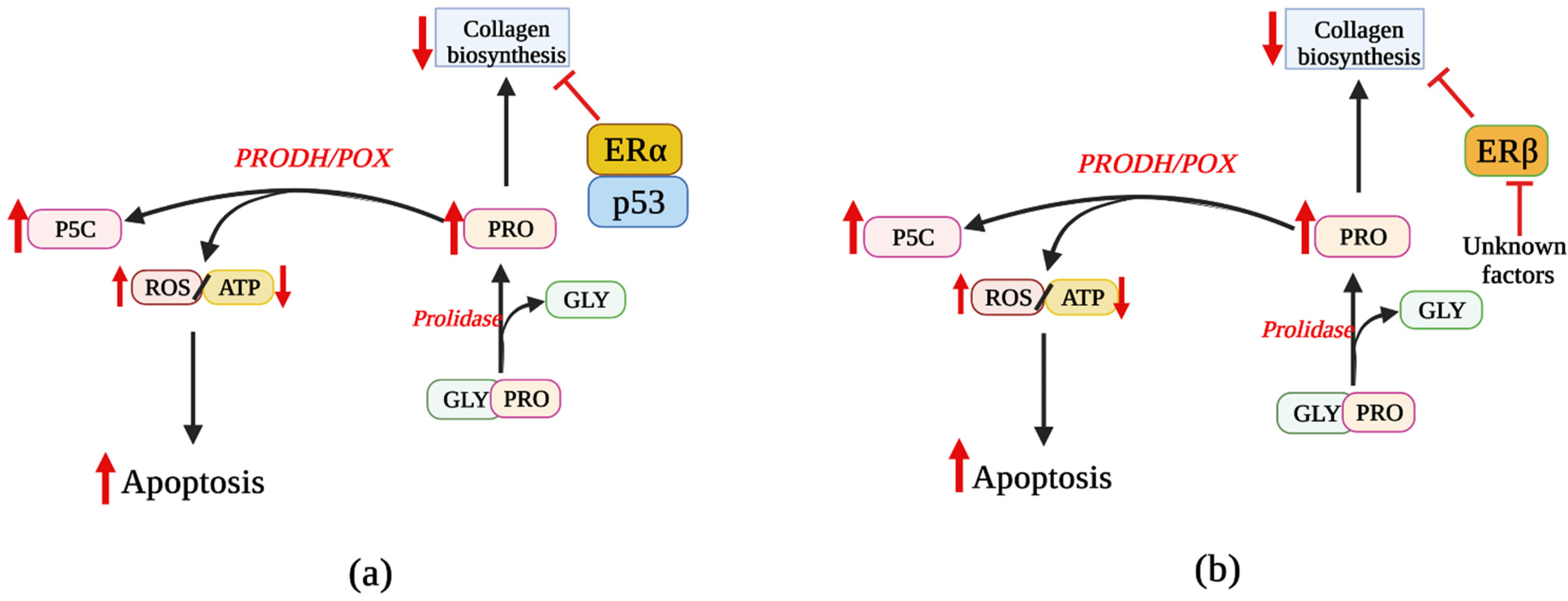
5. Involvement of ER Agonists in PRODH/POX-Dependent Apoptosis
6. Effects of ER Modulators on PRODH/POX-Dependent Apoptosis
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Cancer Day: Breast Cancer Overtakes Lung Cancer in Terms of Number of New Cancer Cases Worldwide. IARC Showcases Key Research Projects to Address Breast Cancer; WHO (World Health Organization). Available online: https://www.iarc.who.int/news-events/world-cancer-day-2021 (accessed on 24 November 2021).
- Breast Cancer Now Most Common Form of Cancer: WHO Taking Action. WHO (World Health Organization). Available online: https://www.who.int/news/item/03-02-2021-breast-cancer-now-most-common-form-of-cancer-who-taking-action (accessed on 3 August 2021).
- Shi, J.; Kobayashi, L.C.; Grundy, A.; Richardson, H.; SenGupta, S.K.; Lohrisch, C.A.; Spinelli, J.J.; Aronson, K.J. Lifetime moderate-to-vigorous physical activity and ER/PR/HER-defined post-menopausal breast cancer risk. Breast Cancer Res. Treat. 2017, 165, 201–213. [Google Scholar] [CrossRef]
- Fuller, P.J. The steroid receptor superfamily: Mechanisms of diversity. FASEB J. 1991, 5, 3092–3099. [Google Scholar] [CrossRef]
- Pratt, S.E.; Pollak, M.N. Estrogen and antiestrogen modulation of MCF7 human breast cancer cell proliferation is associated with specific alterations in accumulation of insulin-like growth factor-binding proteins in conditioned media. Cancer Res. 1993, 53, 5193–5198. [Google Scholar] [PubMed]
- Kumar, U.; Ardasheva, A.; Mahmud, Z.; Coombes, R.C.; Yague, E. FOXA1 is a determinant of drug resistance in breast cancer cells. Breast Cancer Res. Treat. 2021, 186, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.J.; Qiu, J. Estrogen signaling in hypothalamic circuits controlling reproduction. Brain Res. 2010, 1364, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, G.; El-Alfy, M. Immunocytochemical localization of estrogen receptors alpha and beta in the human reproductive organs. J. Clin. Endocrinol. Metab. 2000, 85, 4835–4840. [Google Scholar] [CrossRef][Green Version]
- Friend, K.E.; Chiou, Y.K.; Lopes, M.B.; Laws, E.R., Jr.; Hughes, K.M.; Shupnik, M.A. Estrogen receptor expression in human pituitary: Correlation with immunohistochemistry in normal tissue, and immunohistochemistry and morphology in macroadenomas. J. Clin. Endocrinol. Metab. 1994, 78, 1497–1504. [Google Scholar] [CrossRef] [PubMed]
- Ahlbory-Dieker, D.L.; Stride, B.D.; Leder, G.; Schkoldow, J.; Trolenberg, S.; Seidel, H.; Otto, C.; Sommer, A.; Parker, M.G.; Schutz, G.; et al. DNA binding by estrogen receptor-alpha is essential for the transcriptional response to estrogen in the liver and the uterus. Mol. Endocrinol. 2009, 23, 1544–1555. [Google Scholar] [CrossRef]
- Hiroi, H.; Tsutsumi, O.; Momoeda, M.; Takai, Y.; Osuga, Y.; Taketani, Y. Differential interactions of bisphenol A and 17beta-estradiol with estrogen receptor alpha (ERalpha) and ERbeta. Endocr. J. 1999, 46, 773–778. [Google Scholar] [CrossRef]
- Jensen, E.V.; Jordan, V.C. The estrogen receptor: A model for molecular medicine. Clin. Cancer Res. 2003, 9, 1980–1989. [Google Scholar]
- Leygue, E.; Murphy, L.C. A bi-faceted role of estrogen receptor β in breast cancer. Endocr.-Relat. Cancer 2013, 20, R127–R139. [Google Scholar] [CrossRef]
- Marino, M.; Ascenzi, P. Membrane association of estrogen receptor alpha and beta influences 17beta-estradiol-mediated cancer cell proliferation. Steroids 2008, 73, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Taheri, M.; Shoorei, H.; Dinger, M.E.; Ghafouri-Fard, S. Perspectives on the role of non-coding RNAs in the regulation of expression and function of the estrogen receptor. Cancers 2020, 12, 2162. [Google Scholar] [CrossRef] [PubMed]
- Curtis, S.W.; Washburn, T.; Sewall, C.; DiAugustine, R.; Lindzey, J.; Couse, J.F.; Korach, K.S. Physiological coupling of growth factor and steroid receptor signaling pathways: Estrogen receptor knockout mice lack estrogen-like response to epidermal growth factor. Proc. Natl. Acad. Sci. USA 1996, 93, 12626–12630. [Google Scholar] [CrossRef]
- Ignar-Trowbridge, D.M.; Pimentel, M.; Parker, M.G.; McLachlan, J.A.; Korach, K.S. Peptide growth factor cross-talk with the estrogen receptor requires the A/B domain and occurs independently of protein kinase C or estradiol. Endocrinology 1996, 137, 1735–1744. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Collins, J.; Hudlicky, T.; Pandey, S. Enhancement of apoptotic and autophagic induction by a novel synthetic C-1 analogue of 7-deoxypancratistatin in human breast adenocarcinoma and neuroblastoma cells with tamoxifen. J. Vis. Exp. 2012, 30, 3586. [Google Scholar] [CrossRef]
- Sen, S. Programmed cell death: Concept, mechanism and control. Biol. Rev. Camb. Philos. Soc. 1992, 67, 287–319. [Google Scholar] [CrossRef]
- Parton, M.; Dowsett, M.; Smith, I. Studies of apoptosis in breast cancer. BMJ Open 2001, 322, 1528–1532. [Google Scholar] [CrossRef]
- Reed, J.C. Mechanisms of apoptosis. Am. J. Pathol. 2000, 157, 1415–1430. [Google Scholar] [CrossRef]
- Shi, Y. Apoptosome: The cellular engine for the activation of caspase-9. Structure 2002, 10, 285–288. [Google Scholar] [CrossRef]
- Reed, J.C. Apoptosis mechanisms: Implications for cancer drug discovery. Oncology 2004, 18, 11–20. [Google Scholar]
- Wang, Z.; Zhang, X.; Shen, P.; Loggie, B.W.; Chang, Y.; Deuel, T.F. Identification, cloning, and expression of human estrogen receptor-alpha36, a novel variant of human estrogen receptor-alpha66. Biochem. Biophys. Res. Commun. 2005, 336, 1023–1027. [Google Scholar] [CrossRef]
- Eissing, T.; Conzelmann, H.; Gilles, E.D.; Allgower, F.; Bullinger, E.; Scheurich, P. Bistability analyses of a caspase activation model for receptor-induced apoptosis. J. Biol. Chem. 2004, 279, 36892–36897. [Google Scholar] [CrossRef] [PubMed]
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies within. Science 1998, 281, 1312–1316. [Google Scholar] [CrossRef]
- Beurel, E.; Jope, R.S. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog. Neurobiol. 2006, 79, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Dixit, V.M. Apoptosis control by death and decoy receptors. Curr. Opin. Cell Biol. 1999, 11, 255–260. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.K.; Chun, H.J.; Zheng, L.; Siegel, R.M.; Bui, K.L.; Lenardo, M.J. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science 2000, 288, 2351–2354. [Google Scholar] [CrossRef]
- Piotrowska, A.; Izykowska, I.; Podhorska-Okolow, M.; Zabel, M.; Dziegiel, P. The structure of NF- kappaB family proteins and their role in apoptosis. Postep. Hig. Med. Dosw. 2008, 62, 64–74. [Google Scholar]
- Fang, J.; Seki, T.; Maeda, H. Therapeutic strategies by modulating oxygen stress in cancer and inflammation. Adv. Drug Deliv. Rev. 2009, 61, 290–302. [Google Scholar] [CrossRef]
- Haupt, S.; Berger, M.; Goldberg, Z.; Haupt, Y. Apoptosis-the p53 network. J. Cell Sci. 2003, 116, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B. Signalling pathways of the TNF superfamily: A double-edged sword. Nat. Rev. Immunol. 2003, 3, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Peter, M.E.; Krammer, P.H. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 2003, 10, 26–35. [Google Scholar] [CrossRef]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Wu, G.S.; Kim, K.; el-Deiry, W.S. KILLER/DR5, a novel DNA-damage inducible death receptor gene, links the p53-tumor suppressor to caspase activation and apoptotic death. Adv. Exp. Med. Biol. 2000, 465, 143–151. [Google Scholar] [CrossRef]
- Daniel, P.T. Dissecting the pathways to death. Leukemia 2000, 14, 2035–2044. [Google Scholar] [CrossRef]
- Jiang, P.; Gan, M.; Lin, W.L.; Yen, S.H. Nutrient deprivation induces alpha-synuclein aggregation through endoplasmic reticulum stress response and SREBP2 pathway. Front. Aging Neurosci. 2014, 6, 268. [Google Scholar] [CrossRef]
- Storz, P. Reactive oxygen species in tumor progression. Front. Biosci. 2005, 10, 1881–1896. [Google Scholar] [CrossRef] [PubMed]
- Schattenberg, J.M.; Galle, P.R.; Schuchmann, M. Apoptosis in liver disease. Liver Int. 2006, 26, 904–911. [Google Scholar] [CrossRef]
- Faubert, B.; Vincent, E.E.; Poffenberger, M.C.; Jones, R.G. The AMP-activated protein kinase (AMPK) and cancer: Many faces of a metabolic regulator. Cancer Lett. 2015, 356, 165–170. [Google Scholar] [CrossRef]
- Phang, J.M.; Donald, S.P.; Pandhare, J.; Liu, Y. The metabolism of proline, a stress substrate, modulates carcinogenic pathways. Amino Acids 2008, 35, 681–690. [Google Scholar] [CrossRef]
- Arentson, B.W.; Sanyal, N.; Becker, D.F. Substrate channeling in proline metabolism. Front. Biosci. 2012, 17, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Pandhare, J.; Donald, S.P.; Cooper, S.K.; Phang, J.M. Regulation and function of proline oxidase under nutrient stress. J. Cell. Biochem. 2009, 107, 759–768. [Google Scholar] [CrossRef]
- Vogt, S.; Rhiel, A.; Weber, P.; Ramzan, R. Revisiting Kadenbach: Electron flux rate through cytochrome c-oxidase determines the ATP-inhibitory effect and subsequent production of ROS. Bioessays 2016, 38, 556–567. [Google Scholar] [CrossRef]
- Hancock, C.N.; Liu, W.; Alvord, W.G.; Phang, J.M. Co-regulation of mitochondrial respiration by proline dehydrogenase/oxidase and succinate. Amino Acids 2016, 48, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Phang, J.M.; Liu, W.; Hancock, C.; Christian, K.J. The proline regulatory axis and cancer. Front. Oncol. 2012, 2, 60. [Google Scholar] [CrossRef]
- Maxwell, S.A.; Rivera, A. Proline oxidase induces apoptosis in tumor cells, and its expression is frequently absent or reduced in renal carcinomas. J. Biol. Chem. 2003, 278, 9784–9789. [Google Scholar] [CrossRef]
- Liu, W.; Glunde, K.; Bhujwalla, Z.M.; Raman, V.; Sharma, A.; Phang, J.M. Proline oxidase promotes tumor cell survival in hypoxic tumor microenvironments. Cancer Res. 2012, 72, 3677–3686. [Google Scholar] [CrossRef]
- Pandhare, J.; Cooper, S.K.; Phang, J.M. Proline oxidase, a proapoptotic gene, is induced by troglitazone: Evidence for both peroxisome proliferator-activated receptor gamma-dependent and -independent mechanisms. J. Biol. Chem. 2006, 281, 2044–2052. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Liu, Y.; Borchert, G.L.; Surazynski, A.; Hu, C.A.; Phang, J.M. Proline oxidase activates both intrinsic and extrinsic pathways for apoptosis: The role of ROS/superoxides, NFAT and MEK/ERK signaling. Oncogene 2006, 25, 5640–5647. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.A.; Donald, S.P.; Yu, J.; Lin, W.W.; Liu, Z.; Steel, G.; Obie, C.; Valle, D.; Phang, J.M. Overexpression of proline oxidase induces proline-dependent and mitochondria-mediated apoptosis. Mol. Cell. Biochem. 2007, 295, 85–92. [Google Scholar] [CrossRef]
- Liu, Y.; Borchert, G.L.; Donald, S.P.; Diwan, B.A.; Anver, M.; Phang, J.M. Proline oxidase functions as a mitochondrial tumor suppressor in human cancers. Cancer Res. 2009, 69, 6414–6422. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Lupi, A.; Rossi, A.; Vaghi, P.; Gallanti, A.; Cetta, G.; Forlino, A. N-benzyloxycarbonyl-L-proline: An in vitro and in vivo inhibitor of prolidase. Biochim. Biophys. Acta 2005, 1744, 157–163. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Besio, R.; Maruelli, S.; Gioia, R.; Villa, I.; Grabowski, P.; Gallagher, O.; Bishop, N.J.; Foster, S.; De Lorenzi, E.; Colombo, R.; et al. Lack of prolidase causes a bone phenotype both in human and in mouse. Bone 2015, 72, 53–64. [Google Scholar] [CrossRef]
- Karna, E.; Szoka, L.; Huynh, T.Y.L.; Palka, J.A. Proline-dependent regulation of collagen metabolism. Cell. Mol. Life Sci. 2020, 77, 1911–1918. [Google Scholar] [CrossRef]
- Liu, Y.; Borchert, G.L.; Donald, S.P.; Surazynski, A.; Hu, C.A.; Weydert, C.J.; Oberley, L.W.; Phang, J.M. MnSOD inhibits proline oxidase-induced apoptosis in colorectal cancer cells. Carcinogenesis 2005, 26, 1335–1342. [Google Scholar] [CrossRef]
- Natarajan, S.K.; Becker, D.F. Role of apoptosis-inducing factor, proline dehydrogenase, and NADPH oxidase in apoptosis and oxidative stress. Cell Health Cytoskelet. 2012, 2012, 11–27. [Google Scholar] [CrossRef]
- Surazynski, A.; Donald, S.P.; Cooper, S.K.; Whiteside, M.A.; Salnikow, K.; Liu, Y.; Phang, J.M. Extracellular matrix and HIF-1 signaling: The role of prolidase. Int. J. Cancer 2008, 122, 1435–1440. [Google Scholar] [CrossRef] [PubMed]
- Zareba, I.; Palka, J. Prolidase-proline dehydrogenase/proline oxidase-collagen biosynthesis axis as a potential interface of apoptosis/autophagy. Biofactors 2016, 42, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Vincent, E.E.; Griss, T.; Samborska, B.; Izreig, S.; Svensson, R.U.; Mamer, O.A.; Avizonis, D.; Shackelford, D.B.; Shaw, R.J.; et al. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1α. Proc. Natl. Acad. Sci. USA 2014, 111, 2554–2559. [Google Scholar] [CrossRef]
- Liu, W.; Phang, J.M. Proline dehydrogenase (oxidase), a mitochondrial tumor suppressor, and autophagy under the hypoxia microenvironment. Autophagy 2012, 8, 1407–1409. [Google Scholar] [CrossRef]
- Di Nardo, G.; Zhang, C.; Marcelli, A.G.; Gilardi, G. Molecular and structural evolution of cytochrome P450 aromatase. Int. J. Mol. Sci. 2021, 22, 631. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K. Tamoxifen in the treatment of breast cancer. N. Engl. J. Med. 1998, 339, 1609–1618. [Google Scholar] [CrossRef]
- Bailey, S.T.; Shin, H.; Westerling, T.; Liu, X.S.; Brown, M. Estrogen receptor prevents p53-dependent apoptosis in breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 18060–18065. [Google Scholar] [CrossRef] [PubMed]
- Konduri, S.D.; Medisetty, R.; Liu, W.; Kaipparettu, B.A.; Srivastava, P.; Brauch, H.; Fritz, P.; Swetzig, W.M.; Gardner, A.E.; Khan, S.A.; et al. Mechanisms of estrogen receptor antagonism toward p53 and its implications in breast cancer therapeutic response and stem cell regulation. Proc. Natl. Acad. Sci. USA 2010, 107, 15081–15086. [Google Scholar] [CrossRef]
- Liu, W.; Konduri, S.D.; Bansal, S.; Nayak, B.K.; Rajasekaran, S.A.; Karuppayil, S.M.; Rajasekaran, A.K.; Das, G.M. Estrogen receptor-alpha binds p53 tumor suppressor protein directly and represses its function. J. Biol. Chem. 2006, 281, 9837–9840. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Katzenellenbogen, B.S. Estrogen receptor-beta modulation of the ERalpha-p53 loop regulating gene expression, proliferation, and apoptosis in breast cancer. Horm. Cancer 2017, 8, 230–242. [Google Scholar] [CrossRef]
- Cechowska-Pasko, M.; Palka, J.; Wojtukiewicz, M.Z. Enhanced prolidase activity and decreased collagen content in breast cancer tissue. Int. J. Exp. Pathol. 2006, 87, 289–296. [Google Scholar] [CrossRef]
- Miltyk, W.; Anchim, T.; Wolczynski, S.; Palka, J. Estrogen-dependent regulation of prolidase activity in breast cancer MCF-7 cells. Gynecol. Endocrinol. 1999, 13, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Surazynski, A.; Jarzabek, K.; Haczynski, J.; Laudanski, P.; Palka, J.; Wolczynski, S. Differential effects of estradiol and raloxifene on collagen biosynthesis in cultured human skin fibroblasts. Int. J. Mol. Med. 2003, 12, 803–809. [Google Scholar] [CrossRef]
- Surazynski, A.; Miltyk, W.; Prokop, I.; Palka, J. The effect of estrogen on prolidase-dependent regulation of HIF-1α expression in breast cancer cells. Mol. Cell. Biochem. 2013, 379, 29–36. [Google Scholar] [CrossRef]
- Younes, M.; Honma, N. Estrogen receptor beta. Arch. Pathol. Lab. Med. 2011, 135, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Messina, M. Soy and health update: Evaluation of the clinical and epidemiologic literature. Nutrients 2016, 8, 754. [Google Scholar] [CrossRef]
- Yahfoufi, N.; Alsadi, N.; Jambi, M.; Matar, C. The immunomodulatory and anti-inflammatory role of polyphenols. Nutrients 2018, 10, 1618. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, C.; Sanchez-Quesada, C.; Gaforio, J.J. Dietary flavonoids as cancer chemopreventive agents: An updated review of human studies. Antioxidants 2019, 8, 137. [Google Scholar] [CrossRef]
- Chirumbolo, S.; Bjorklund, G.; Lysiuk, R.; Vella, A.; Lenchyk, L.; Upyr, T. Targeting cancer with phytochemicals via their fine tuning of the cell survival signaling pathways. Int. J. Mol. Sci. 2018, 19, 3568. [Google Scholar] [CrossRef] [PubMed]
- Perez-Vizcaino, F.; Fraga, C.G. Research trends in flavonoids and health. Arch. Biochem. Biophys. 2018, 646, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Biersack, B.; Li, Y.; Kong, D.; Bao, B.; Schobert, R.; Padhye, S.B.; Sarkar, F.H. Targeted regulation of PI3K/Akt/mTOR/NF-kappaB signaling by indole compounds and their derivatives: Mechanistic details and biological implications for cancer therapy. Anticancer Agents Med. Chem. 2013, 13, 1002–1013. [Google Scholar] [CrossRef]
- El-Rayes, B.F.; Ali, S.; Ali, I.F.; Philip, P.A.; Abbruzzese, J.; Sarkar, F.H. Potentiation of the effect of erlotinib by genistein in pancreatic cancer: The role of Akt and nuclear factor-kappaB. Cancer Res. 2006, 66, 10553–10559. [Google Scholar] [CrossRef]
- Fahy, B.N.; Schlieman, M.G.; Mortenson, M.M.; Virudachalam, S.; Bold, R.J. Targeting BCL-2 overexpression in various human malignancies through NF-kappaB inhibition by the proteasome inhibitor bortezomib. Cancer Chemother. Pharmacol. 2005, 56, 46–54. [Google Scholar] [CrossRef]
- Wolczynski, S.; Surazynski, A.; Swiatecka, J.; Palka, J. Estrogenic and antiestrogenic effects of raloxifene on collagen metabolism in breast cancer MCF-7 cells. Gynecol. Endocrinol. 2001, 15, 225–233. [Google Scholar] [CrossRef]
- Kim, D.; Lee, A.S.; Jung, Y.J.; Yang, K.H.; Lee, S.; Park, S.K.; Kim, W.; Kang, K.P. Tamoxifen ameliorates renal tubulointerstitial fibrosis by modulation of estrogen receptor alpha-mediated transforming growth factor-beta1/Smad signaling pathway. Nephrol. Dial. Transplant. 2014, 29, 2043–2053. [Google Scholar] [CrossRef]
- Carthy, J.M.; Sundqvist, A.; Heldin, A.; van Dam, H.; Kletsas, D.; Heldin, C.H.; Moustakas, A. Tamoxifen Inhibits TGF-beta-Mediated Activation of Myofibroblasts by Blocking Non-Smad Signaling Through ERK1/2. J. Cell. Physiol. 2015, 230, 3084–3092. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, C.Z.; Du, G.J.; Qi, L.W.; Calway, T.; He, T.C.; Du, W.; Yuan, C.S. Genistein induces G2/M cell cycle arrest and apoptosis via ATM/p53-dependent pathway in human colon cancer cells. Int. J. Oncol. 2013, 43, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, C.; Lu, J.; Zhu, L.; Li, M. 2-Methoxyestradiol inhibits hypoxia-induced scleroderma fibroblast collagen synthesis by phosphatidylinositol 3-kinase/Akt/mTOR signalling. Rheumatology 2018, 57, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Cui, Y.; Zheng, S.; Huang, J.; Li, P.; Simoncini, T.; Zhang, Y.; Fu, X. 2-methoxyestradiol induces vasodilation by stimulating NO release via PPARgamma/PI3K/Akt pathway. PLoS ONE 2015, 10, e0118902. [Google Scholar] [CrossRef]
- Liu, W.; Phang, J.M. Proline dehydrogenase (oxidase) in cancer. Biofactors 2012, 38, 398–406. [Google Scholar] [CrossRef]
- Kociecka, B.; Surazynski, A.; Miltyk, W.; Palka, J. The effect of Telmisartan on collagen biosynthesis depends on the status of estrogen activation in breast cancer cells. Eur. J. Pharmacol. 2010, 628, 51–56. [Google Scholar] [CrossRef]
- Kummer, S.; Jeruschke, S.; Wegerich, L.V.; Peters, A.; Lehmann, P.; Seibt, A.; Mueller, F.; Koleganova, N.; Halbenz, E.; Schmitt, C.P.; et al. Estrogen receptor alpha expression in podocytes mediates protection against apoptosis in-vitro and in-vivo. PLoS ONE 2011, 6, e27457. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Zilberman, Y.; Wassermann, K.; Bain, S.D.; Sadovsky, Y.; Gazit, D. Estrogen modulates estrogen receptor alpha and beta expression, osteogenic activity, and apoptosis in mesenchymal stem cells (MSCs) of osteoporotic mice. J. Cell. Biochem. Suppl. 2001, 81, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Dworatzek, E.; Mahmoodzadeh, S.; Schriever, C.; Kusumoto, K.; Kramer, L.; Santos, G.; Fliegner, D.; Leung, Y.K.; Ho, S.M.; Zimmermann, W.H.; et al. Sex-specific regulation of collagen I and III expression by 17beta-Estradiol in cardiac fibroblasts: Role of estrogen receptors. Cardiovasc. Res. 2019, 115, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Verdier-Sevrain, S.; Bonte, F.; Gilchrest, B. Biology of estrogens in skin: Implications for skin aging. Exp. Dermatol. 2006, 15, 83–94. [Google Scholar] [CrossRef]
- Mao, S.; Wang, Y.; Zhang, M.; Hinek, A. Phytoestrogen, tanshinone IIA diminishes collagen deposition and stimulates new elastogenesis in cultures of human cardiac fibroblasts. Exp. Cell Res. 2014, 323, 189–197. [Google Scholar] [CrossRef]
- Lewoniewska, S.; Oscilowska, I.; Huynh, T.Y.L.; Prokop, I.; Baszanowska, W.; Bielawska, K.; Palka, J. Troglitazone-induced PRODH/POX-dependent apoptosis occurs in the absence of estradiol or ERbeta in ER-negative breast cancer cells. J. Clin. Med. 2021, 10, 4641. [Google Scholar] [CrossRef]
- Surazynski, A.; Jarzabek, K.; Miltyk, W.; Wolczynski, S.; Palka, J. Estrogen-dependent regulation of PPAR-gamma signaling on collagen biosynthesis in adenocarcinoma endometrial cells. Neoplasma 2009, 56, 448–454. [Google Scholar] [CrossRef]
- Lee, C.Y.; Liu, X.; Smith, C.L.; Zhang, X.; Hsu, H.C.; Wang, D.Y.; Luo, Z.P. The combined regulation of estrogen and cyclic tension on fibroblast biosynthesis derived from anterior cruciate ligament. Matrix Biol. 2004, 23, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, S.; Reid, J.M.; Hawse, J.R.; Goetz, M.P. Endoxifen, an estrogen receptor targeted therapy: From bench to bedside. Endocrinology 2021, 162, bqab191. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewoniewska, S.; Oscilowska, I.; Forlino, A.; Palka, J. Understanding the Role of Estrogen Receptor Status in PRODH/POX-Dependent Apoptosis/Survival in Breast Cancer Cells. Biology 2021, 10, 1314. https://doi.org/10.3390/biology10121314
Lewoniewska S, Oscilowska I, Forlino A, Palka J. Understanding the Role of Estrogen Receptor Status in PRODH/POX-Dependent Apoptosis/Survival in Breast Cancer Cells. Biology. 2021; 10(12):1314. https://doi.org/10.3390/biology10121314
Chicago/Turabian StyleLewoniewska, Sylwia, Ilona Oscilowska, Antonella Forlino, and Jerzy Palka. 2021. "Understanding the Role of Estrogen Receptor Status in PRODH/POX-Dependent Apoptosis/Survival in Breast Cancer Cells" Biology 10, no. 12: 1314. https://doi.org/10.3390/biology10121314
APA StyleLewoniewska, S., Oscilowska, I., Forlino, A., & Palka, J. (2021). Understanding the Role of Estrogen Receptor Status in PRODH/POX-Dependent Apoptosis/Survival in Breast Cancer Cells. Biology, 10(12), 1314. https://doi.org/10.3390/biology10121314