A DFT-Based Model on the Adsorption Behavior of H2O, H+, Cl−, and OH− on Clean and Cr-Doped Fe(110) Planes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Theoretical Calculation Methods
3. Results and Discussion
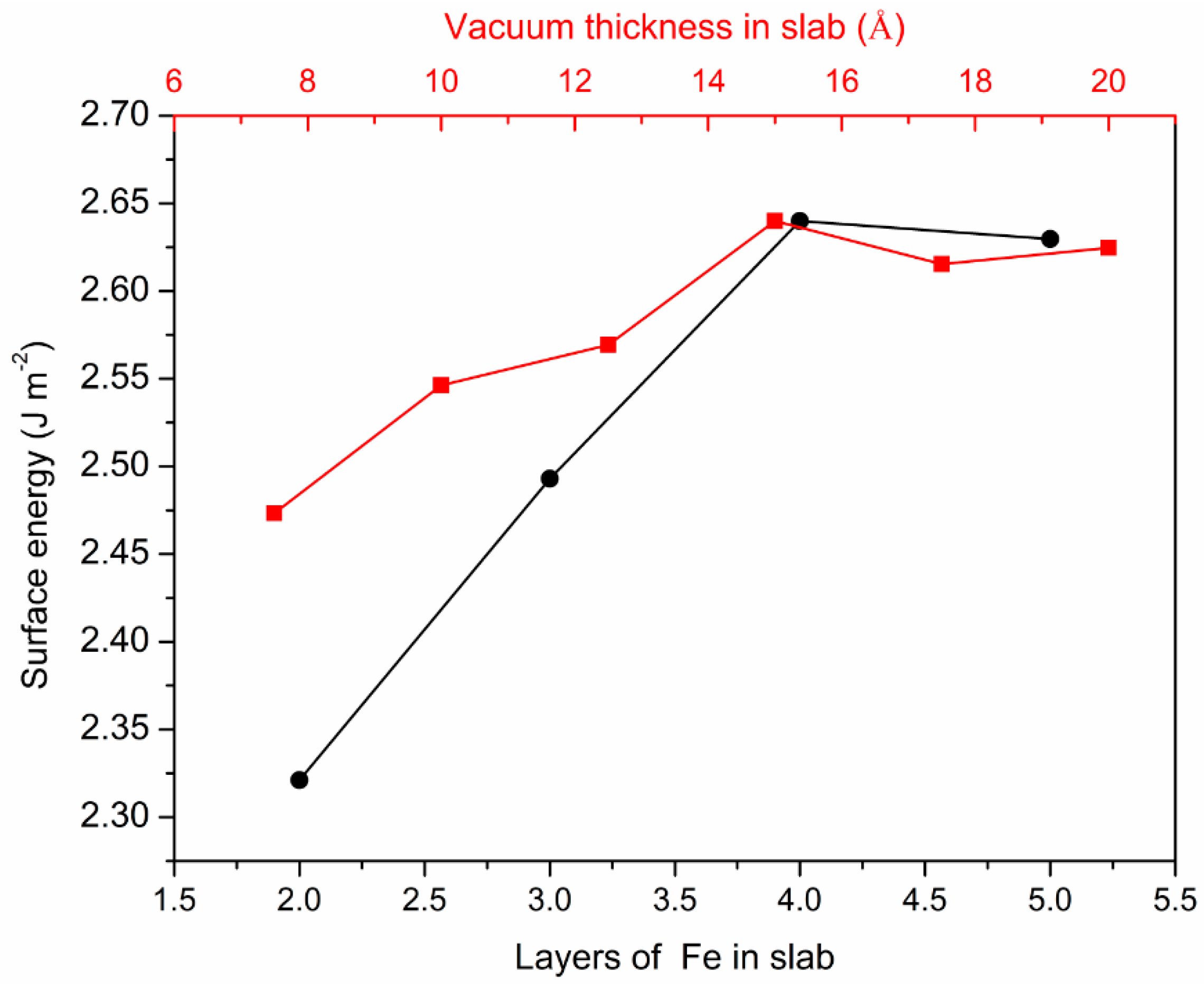
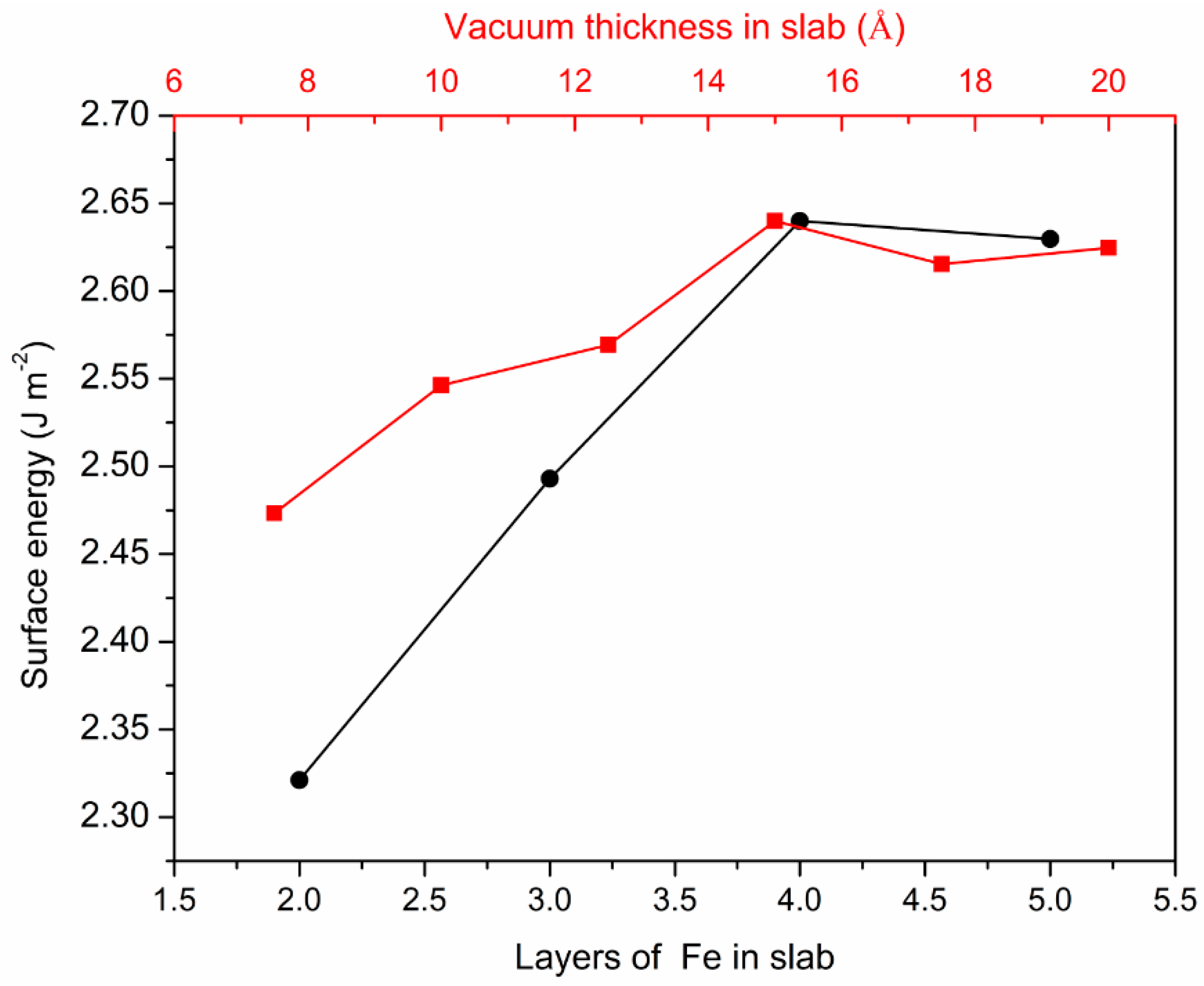
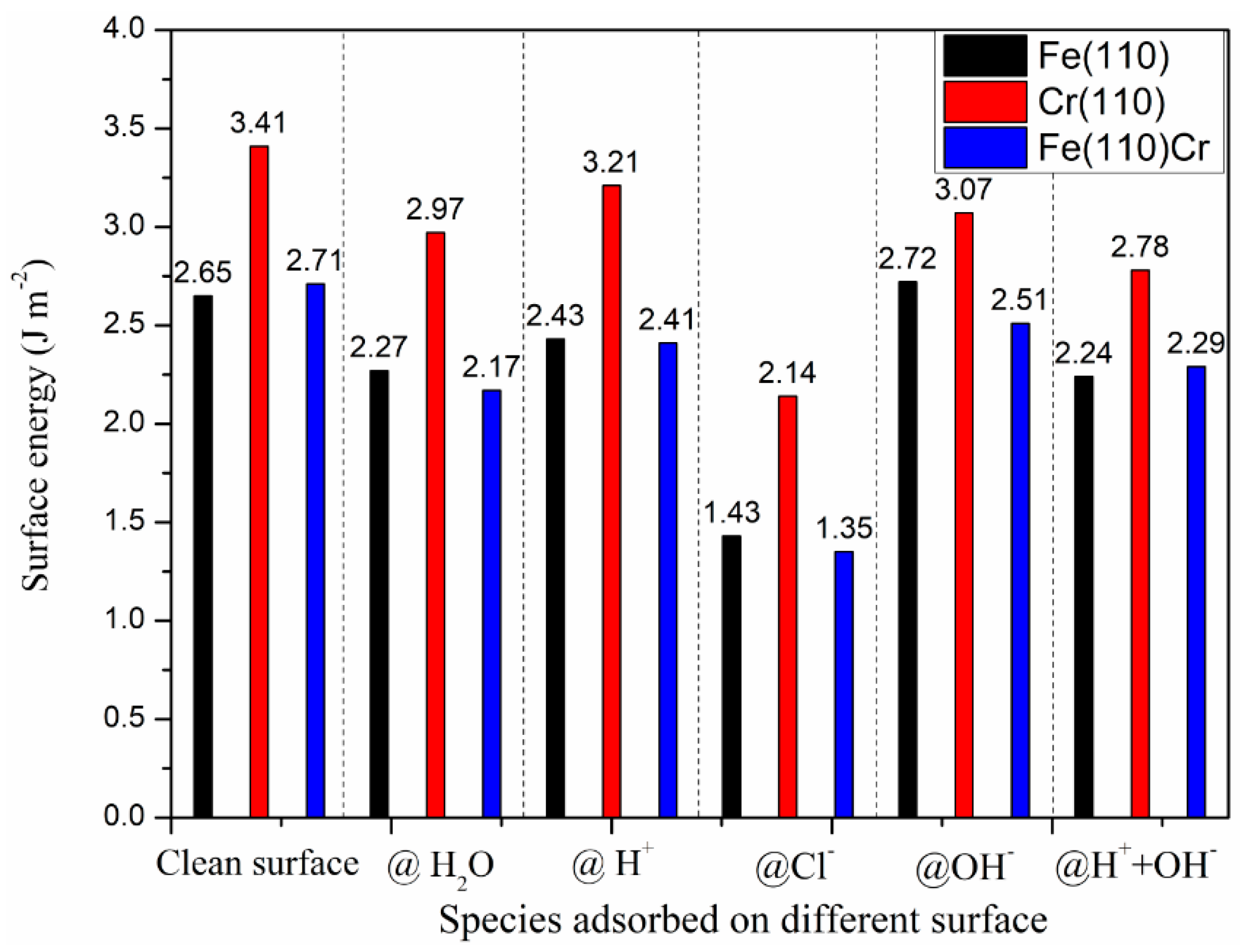
3.1. Surface Energies Calculation
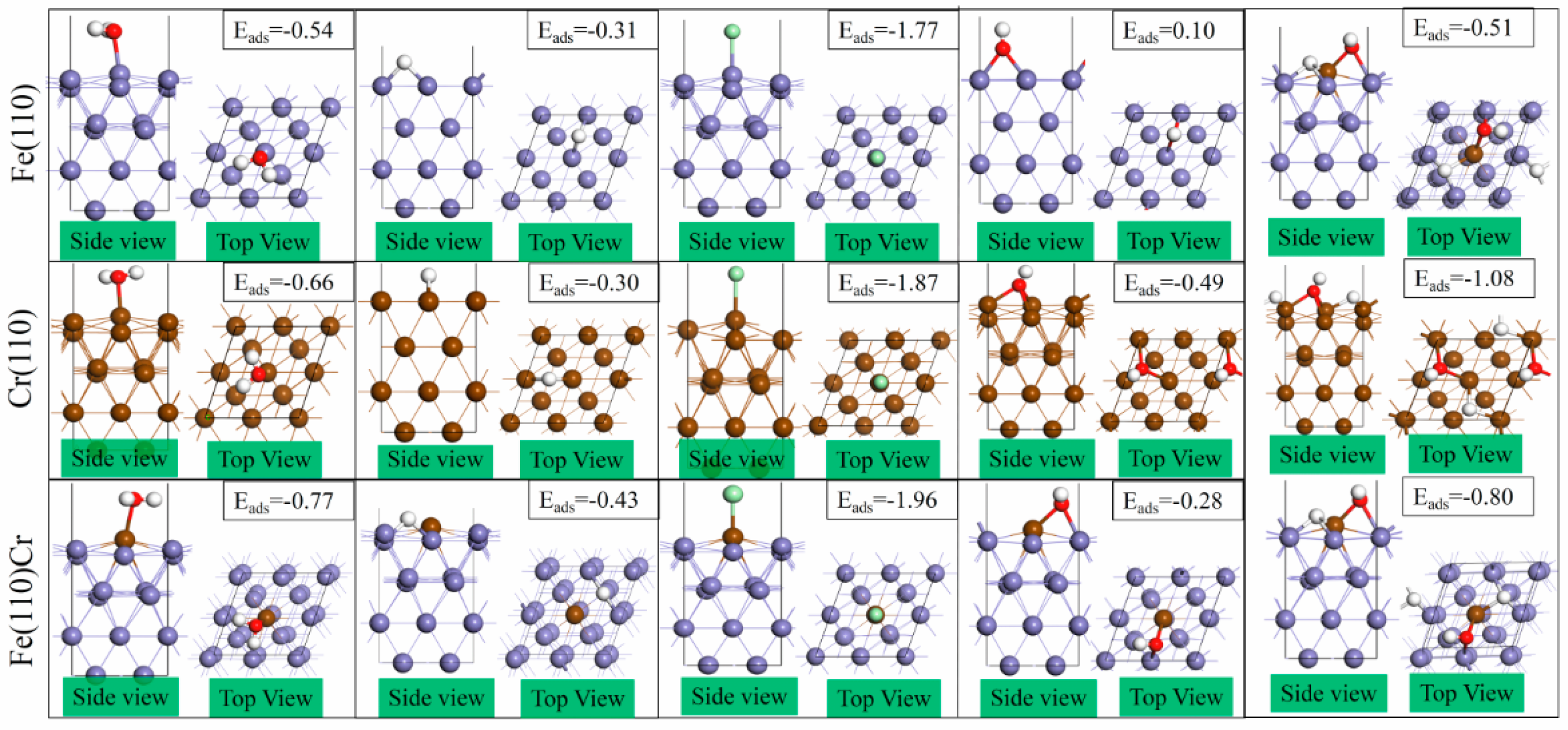
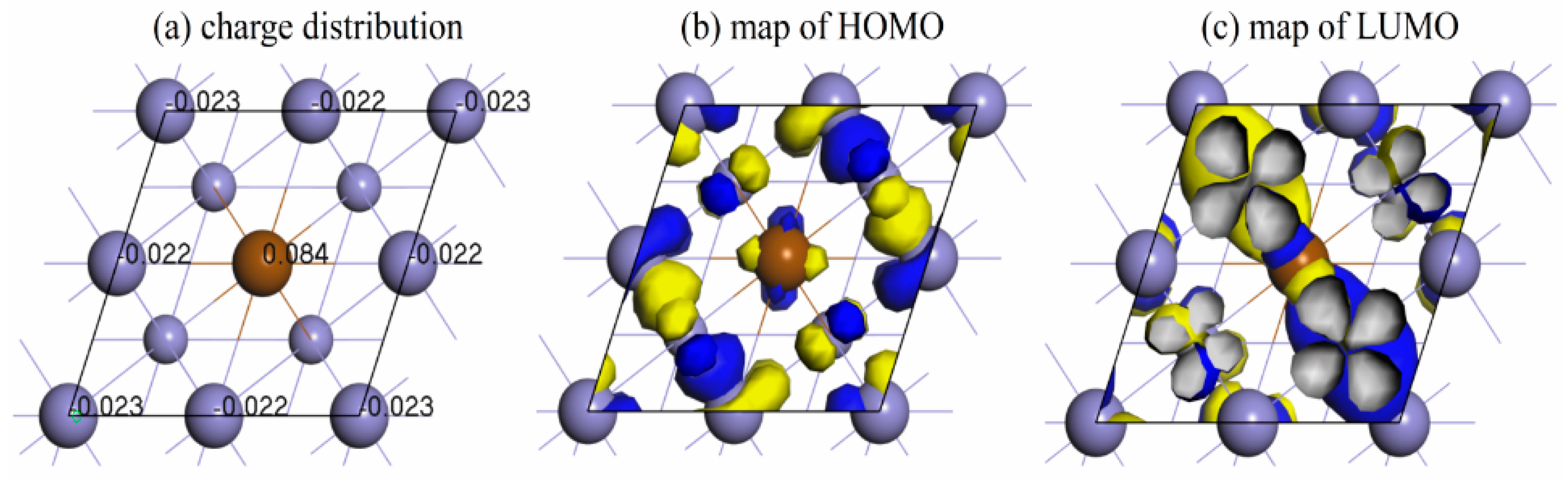
3.2. The Most Stable Adsorption Structure of Relevant Particles
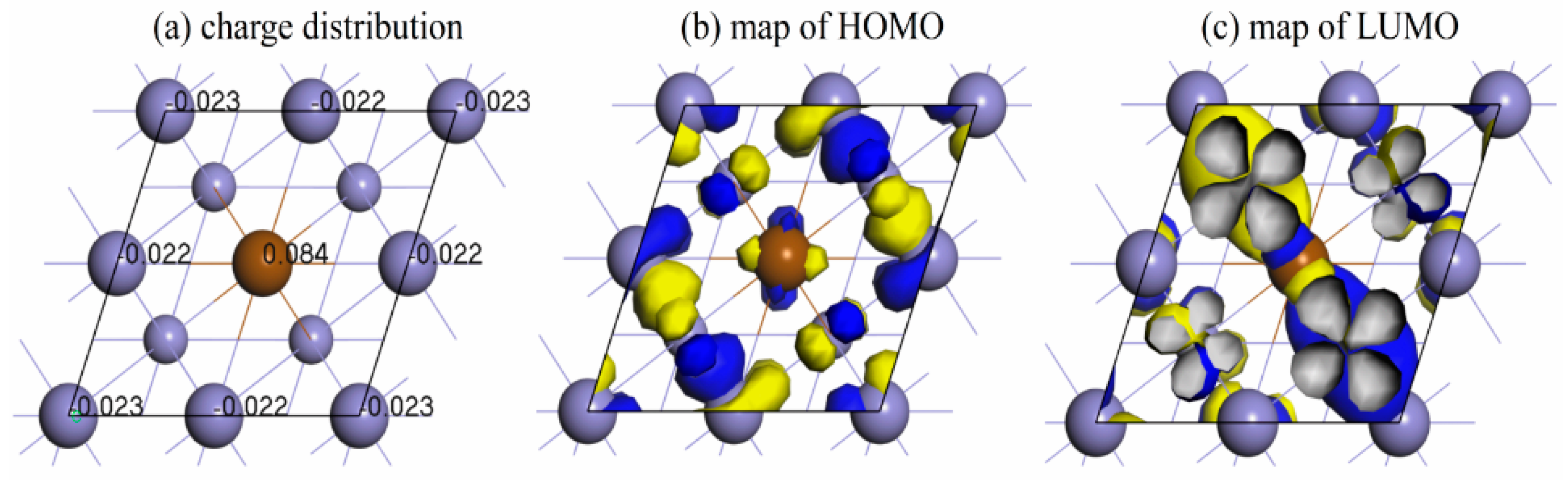
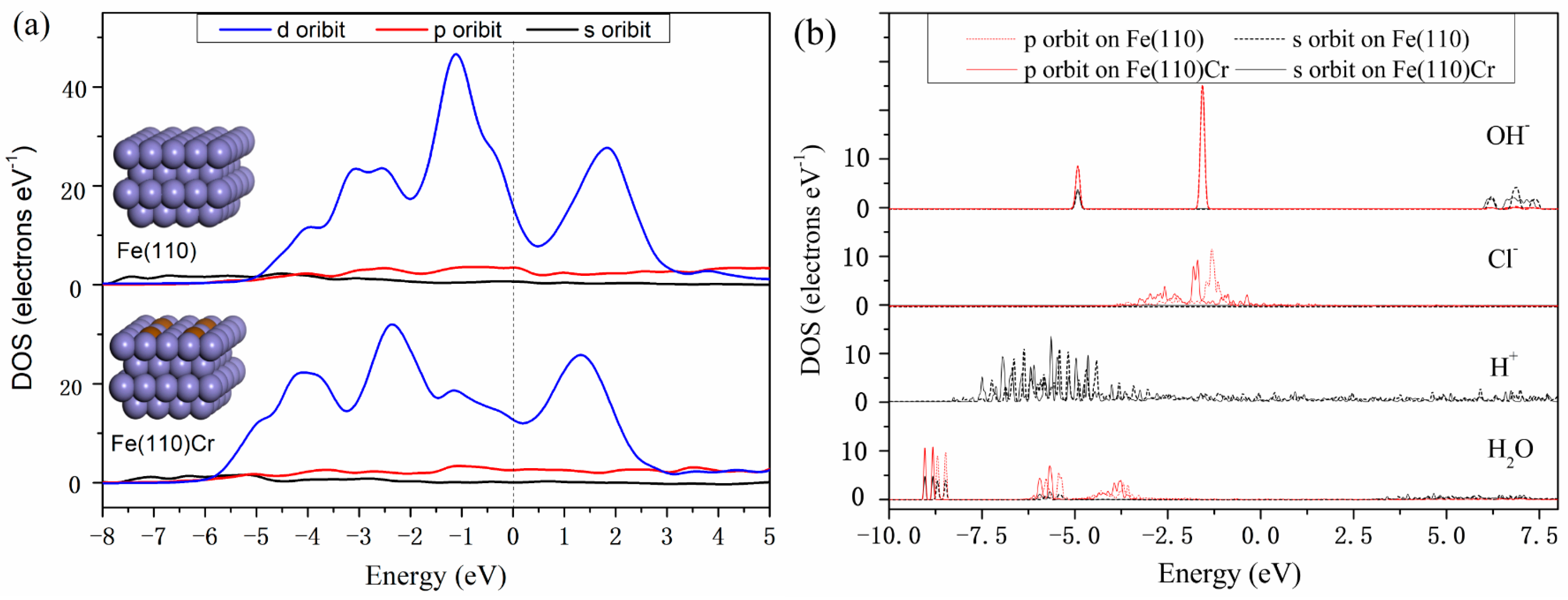
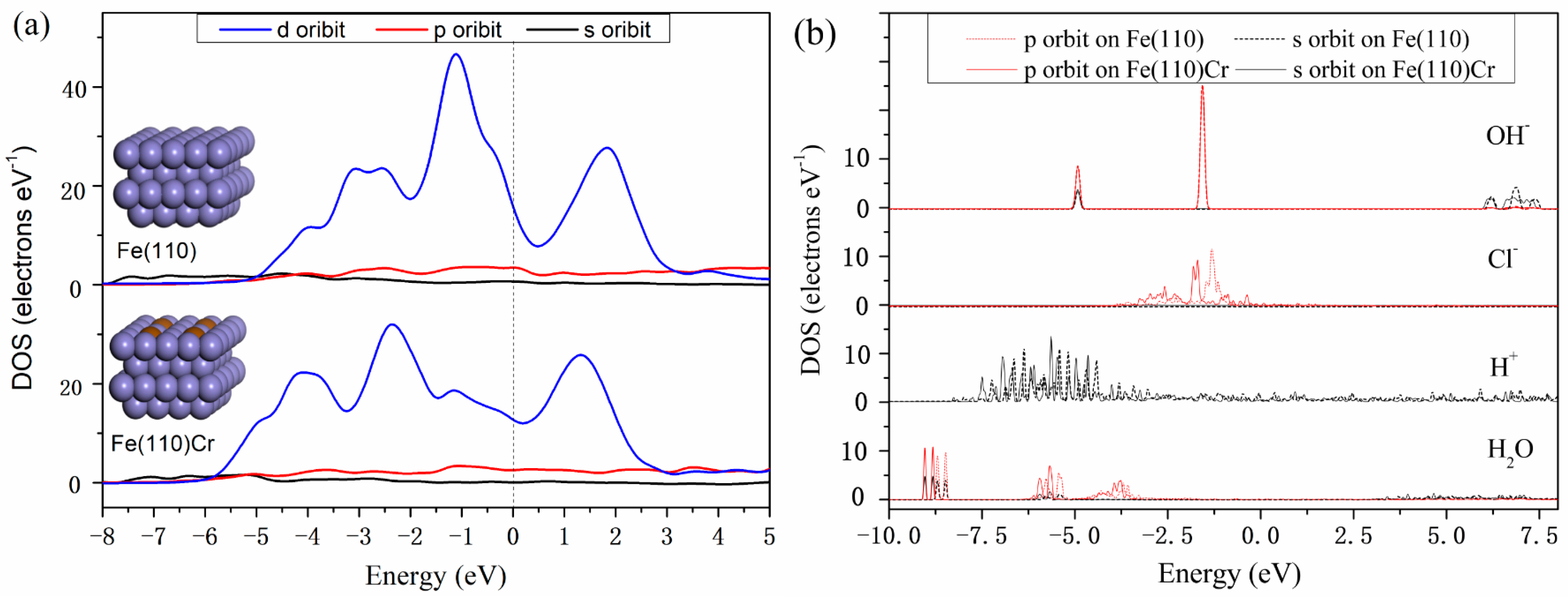
3.3. Density of States
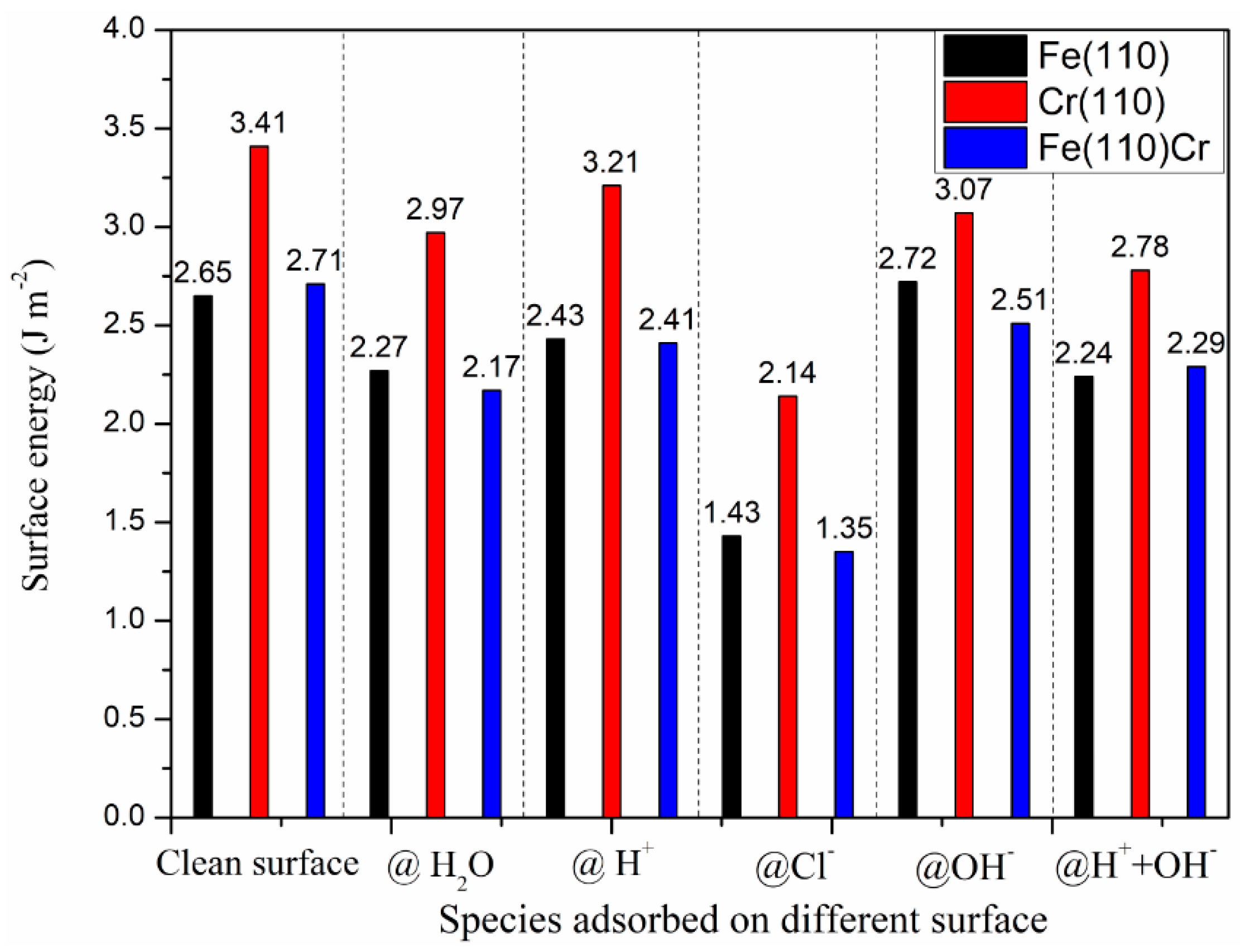
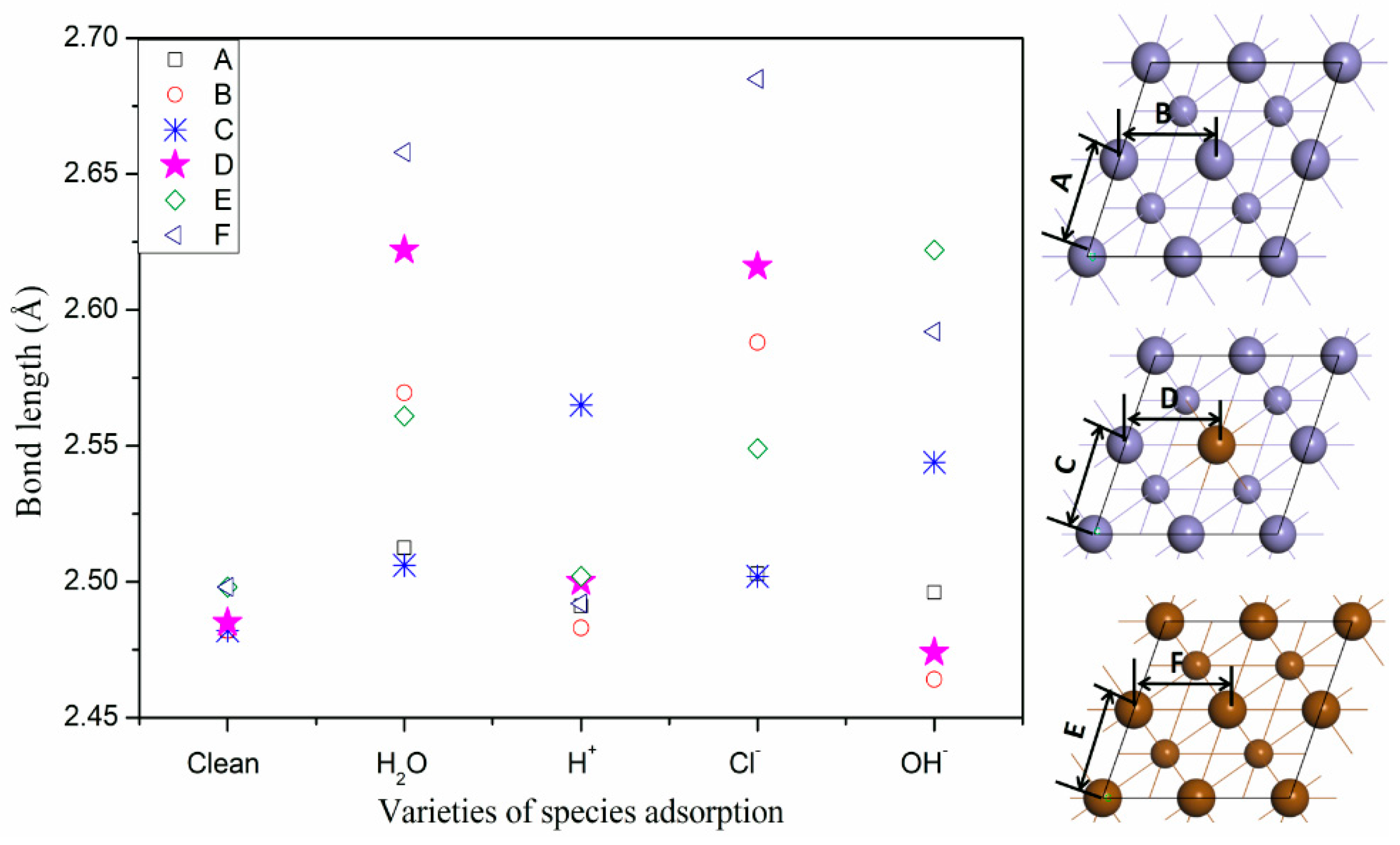
3.4. The Effect Adsorbed Species on Surface
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Duy, D.V.; Aleksey, G.L.; Truong, K.N.; Thoi, T.N. Nitrogen trapping ability of hydrogen-induced vacancy and the effect on the formation of AlN in aluminum. Coatings 2017, 7, 79. [Google Scholar] [CrossRef]
- Menzel, D. Water on a metal surface. Science 2002, 295, 58–59. [Google Scholar] [CrossRef] [PubMed]
- Roman, O.; Elena, V.; Joachim, S. Water adsorption and O-defect formation on Fe2O3(0001) surfaces. Phys. Chem. Chem. Phys. 2016, 18, 25560–25568. [Google Scholar]
- Magali, B.; Nathalie, T.; Joseph, M. Adsorption energy of small molecules on core–shell Fe@Au nanoparticles: Tuning by shell thickness. Phys. Chem. Chem. Phys. 2016, 18, 9112–9123. [Google Scholar]
- Liu, X.; Kang, C.; Qiao, H.; Ren, Y.; Tan, X.; Sun, S. Theoretical studies of the adsorption and migration behavior of boron atoms on hydrogen-terminated diamond (001) surface. Coatings 2017, 7, 57. [Google Scholar] [CrossRef]
- Lakshmi, R.V.; Bharathidasan, T.; Basu, J. Superhydrophobicity of AA2024 by a simple solution immersion technique. Surf. Innov. 2015, 1, 241–247. [Google Scholar] [CrossRef]
- Wang, X.; Yin, X.; Nalaskowski, J.; Du, H.; Miller, J.D. Molecular features of water films created with bubbles at silica surfaces. Surf. Innov. 2015, 3, 20–26. [Google Scholar] [CrossRef]
- Freitas, R.R.Q.; Rivelino, R.; de Brito Mota, F.; de Castilho, C.M.C. Dissociative adsorption and aggregation of water on the Fe(100) Surface: A DFT Study. J. Phys. Chem. C 2012, 116, 20306–20314. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, J.D.; Liu, F.B.; Chen, D.R. First principles investigation of water adsorption on Fe (110) crystal surface containing N. Adv. Tribol. 2008, 1, 654–657. [Google Scholar]
- Bozsom, F.; Ertlm, G.; Grunze, M.; Weiss, M. Chemisorption of hydrogen on iron surfaces. Appl. Surf. Sci. 1977, 1, 103–119. [Google Scholar] [CrossRef]
- Keisuke, T.; Shigehito, I.; Somei, O. Chemisorption of hydrogen on Fe clusters through hybrid bonding mechanisms. Appl. Phys. Lett. 2013, 102, 11308. [Google Scholar]
- Sun, C.; Hui, R.; Qu, W.; Yick, S. Progress in corrosion resistant materials for supercritical water reactors. Corros. Sci. 2009, 51, 2508–2523. [Google Scholar] [CrossRef]
- Wang, H.; Nie, X.; Guo, X.; Song, C. A computational study of adsorption and activation of CO2 and H2 over Fe(100) surface. J. CO2 Utilization 2016, 15, 107–114. [Google Scholar] [CrossRef]
- Cremaschi, P.; Yang, H.; Whitten, J.L. Ab initio chemisorption studies of H on Fe(110). Surf. Sci. 1995, 330, 255–264. [Google Scholar] [CrossRef]
- Wang, W.; Wang, G.; Shao, M. First-principles modeling of direct versus oxygen-assisted water dissociation on Fe(100) surfaces. Catalysts 2016, 6, 29. [Google Scholar] [CrossRef]
- Hu, J.; Zhao, X.; Chen, W.; Su, H.; Chen, Z. Theoretical insight into the mechanism of photoelectrochemical oxygen evolution reaction on BiVO4 anode with oxygen vacancy. J. Phys. Chem. C 2017, 121, 18702–18709. [Google Scholar] [CrossRef]
- Horányi, G. Investigation of the specific adsorption of HSO4−(SO42−) and Cl− ions on Co and Fe by radiotracer technique in the course of corrosion of the metals in perchlorate media. Corros. Sci. 2004, 46, 1741–1749. [Google Scholar] [CrossRef]
- Ghiasi, M.; Kamalinahad, S.; Arabieh, M.; Zahedi, M. Carbonic anhydrase inhibitors: A quantum mechanical study of interaction between some antiepileptic drugs with active center of carbonic anhydrase enzyme. Comput. Theor. Chem. 2012, 992, 59–69. [Google Scholar] [CrossRef]
- Zhang, H. A DFT study on direct hydrogenation of amide catalyzed by a PNN Ru (II) pincer complex. Comput. Theor. Chem. 2015, 1066, 1–6. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- David, H.; Franz, D.F.; Dieter, V. Structure and surface energy of Au55 nanoparticles: An ab initio study. Comput. Mater. Sci. 2017, 134, 137–144. [Google Scholar]
- Ji, D.P.; Zhu, Q.X.; Wang, S.Q. Detailed first-principles studies on surface energy and work function of hexagonal metals. Surf. Sci. 2016, 651, 137–146. [Google Scholar] [CrossRef]
- Pople, J.A.; Gill, P.M.W.; Johnson, B.G. Kohn—Sham density-functional theory within a finite basis set. Chem. Phys. Lett. 1992, 199, 557–560. [Google Scholar] [CrossRef]
- Lanzani, G.; Laasonen, K. SO2 and its fragments on a Cu(110) surface. Surf. Sci. 2008, 602, 321–344. [Google Scholar] [CrossRef]
- Henderson, M.A. The interaction of water with solid surfaces: Fundamental aspects revisited. Surf. Sci. Rep. 2002, 46, 1–308. [Google Scholar] [CrossRef]
- Hodgson, A.; Haq, S. Water adsorption and the wetting of metal surfaces. Surf. Sci. Rep. 2009, 64, 381–451. [Google Scholar] [CrossRef]
- Xie, W.; Peng, L.; Peng, D.; Gu, F.L.; Liu, J. Processes of H2 adsorption on Fe(110) surface: A density functional theory study. Appl. Surf. Sci. 2014, 296, 47–52. [Google Scholar] [CrossRef]
- Michaelides, A.; Ranea, V.A.; de Andres, P.L.; King, D.A. General model for water monomer adsorption on close-packed transition and noble metal surfaces. Phys. Rev. Lett. 2003, 90. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Xie, K.; Liu, Y.; Chen, X.; Ma, W.; Liu, Z. Density functional theory study of leaching efficiency of acids on Si(110) surface with adsorbed boron. Hydrometallurgy 2015, 151, 84–90. [Google Scholar] [CrossRef]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, J.; Wang, C.; He, S.; Zhu, J.; Wei, L.; Zheng, S. A DFT-Based Model on the Adsorption Behavior of H2O, H+, Cl−, and OH− on Clean and Cr-Doped Fe(110) Planes. Coatings 2018, 8, 51. https://doi.org/10.3390/coatings8020051
Hu J, Wang C, He S, Zhu J, Wei L, Zheng S. A DFT-Based Model on the Adsorption Behavior of H2O, H+, Cl−, and OH− on Clean and Cr-Doped Fe(110) Planes. Coatings. 2018; 8(2):51. https://doi.org/10.3390/coatings8020051
Chicago/Turabian StyleHu, Jun, Chaoming Wang, Shijun He, Jianbo Zhu, Liping Wei, and Shunli Zheng. 2018. "A DFT-Based Model on the Adsorption Behavior of H2O, H+, Cl−, and OH− on Clean and Cr-Doped Fe(110) Planes" Coatings 8, no. 2: 51. https://doi.org/10.3390/coatings8020051
APA StyleHu, J., Wang, C., He, S., Zhu, J., Wei, L., & Zheng, S. (2018). A DFT-Based Model on the Adsorption Behavior of H2O, H+, Cl−, and OH− on Clean and Cr-Doped Fe(110) Planes. Coatings, 8(2), 51. https://doi.org/10.3390/coatings8020051