The Preparation and Properties of Fluoroacrylate-Modified Polysiloxane as a Fabric Coating Agent

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
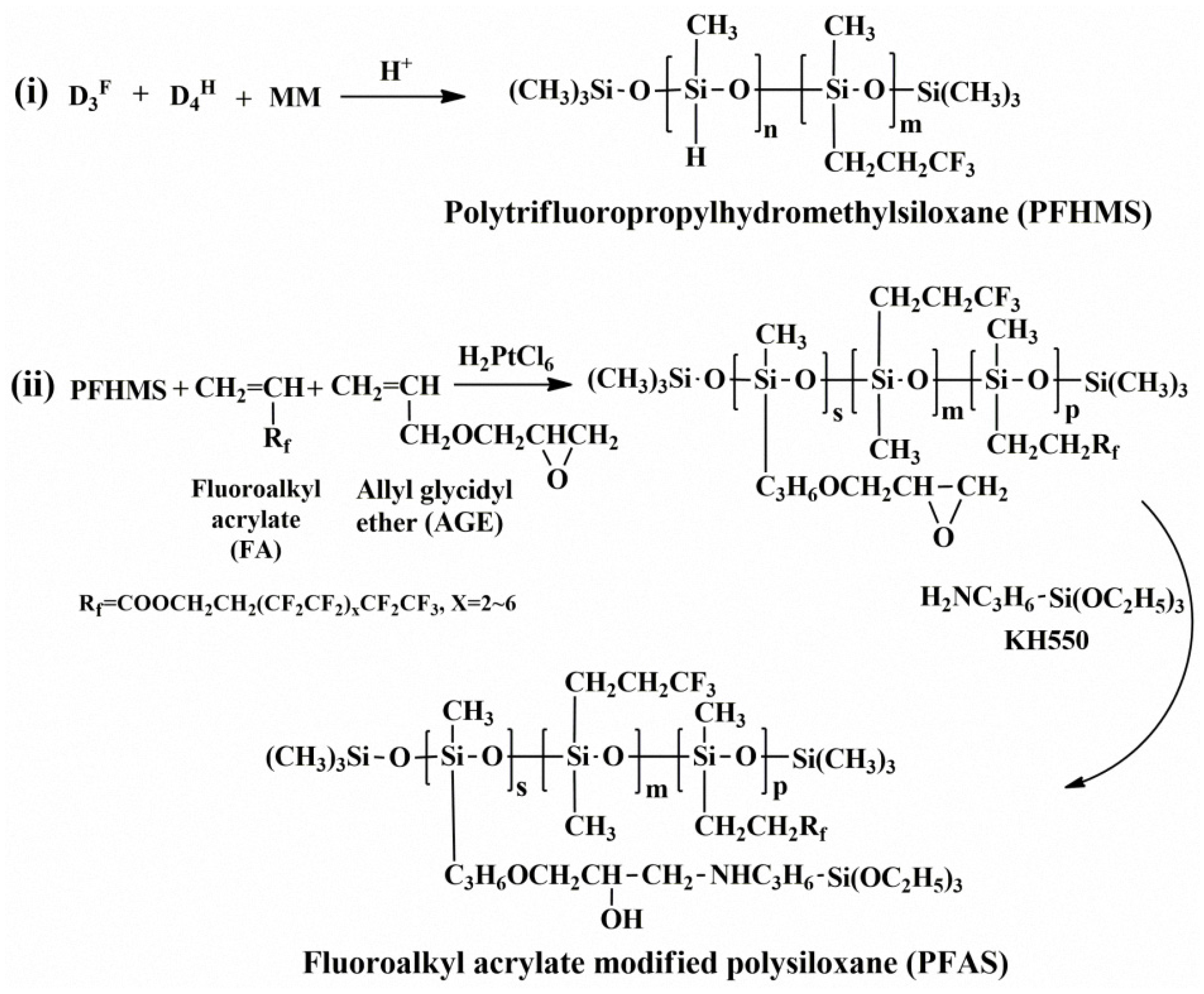
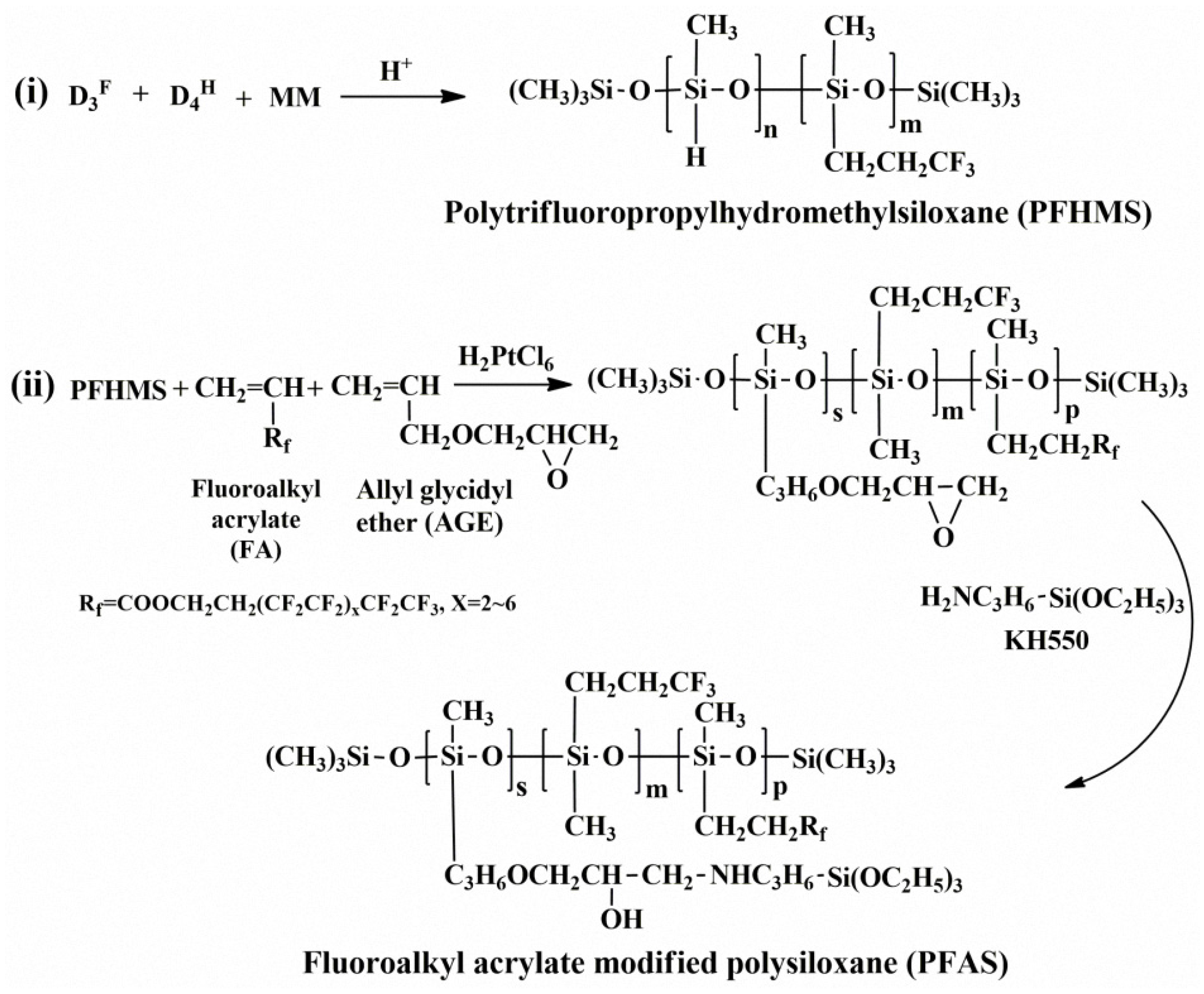
2.2. Preparation of Fluoroalkyl Acrylate Modified Polysiloxane (PFAS)
2.3. Coating of Cotton Fabric with PFAS
2.4. Characterization
3. Results and Discussion
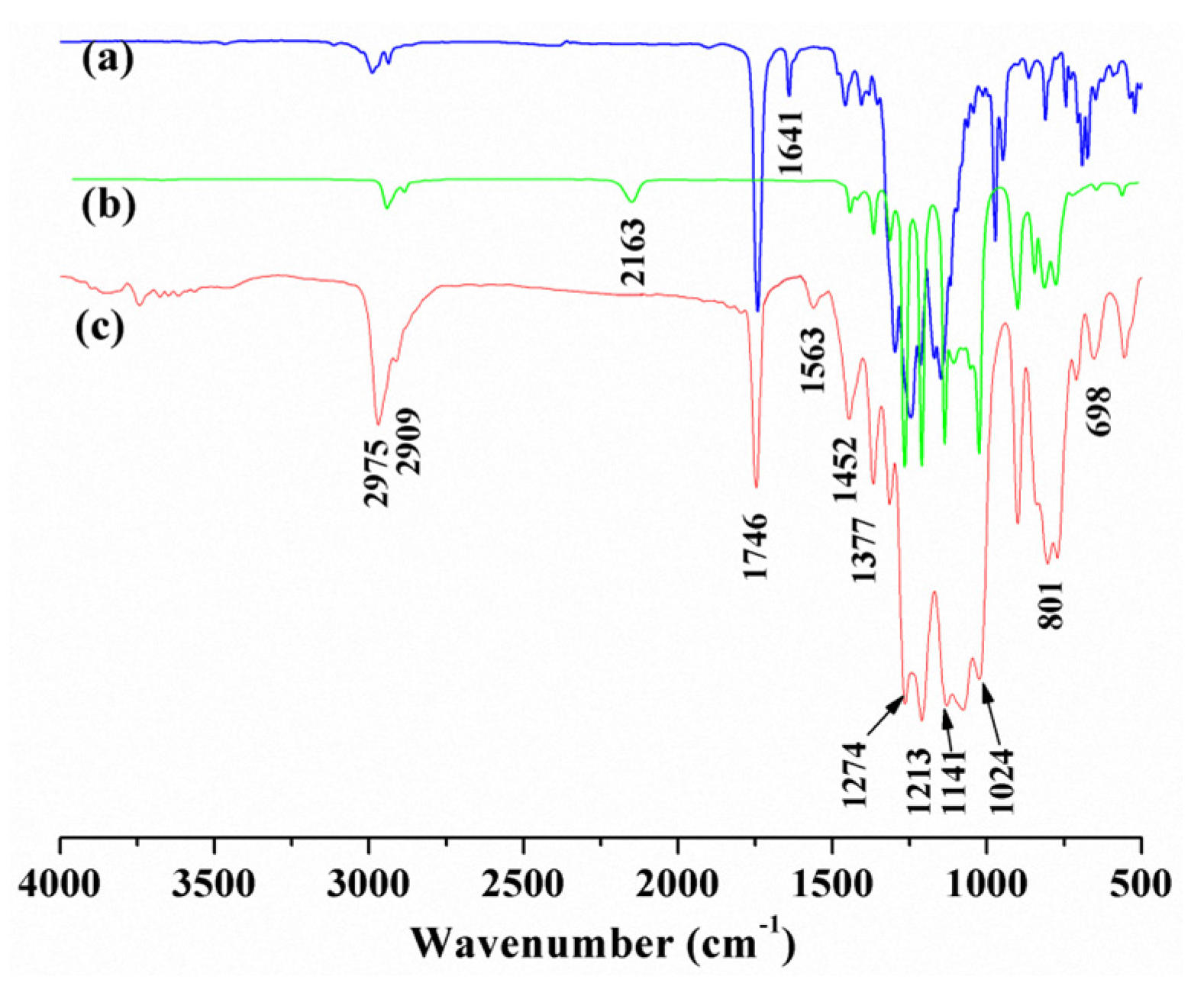
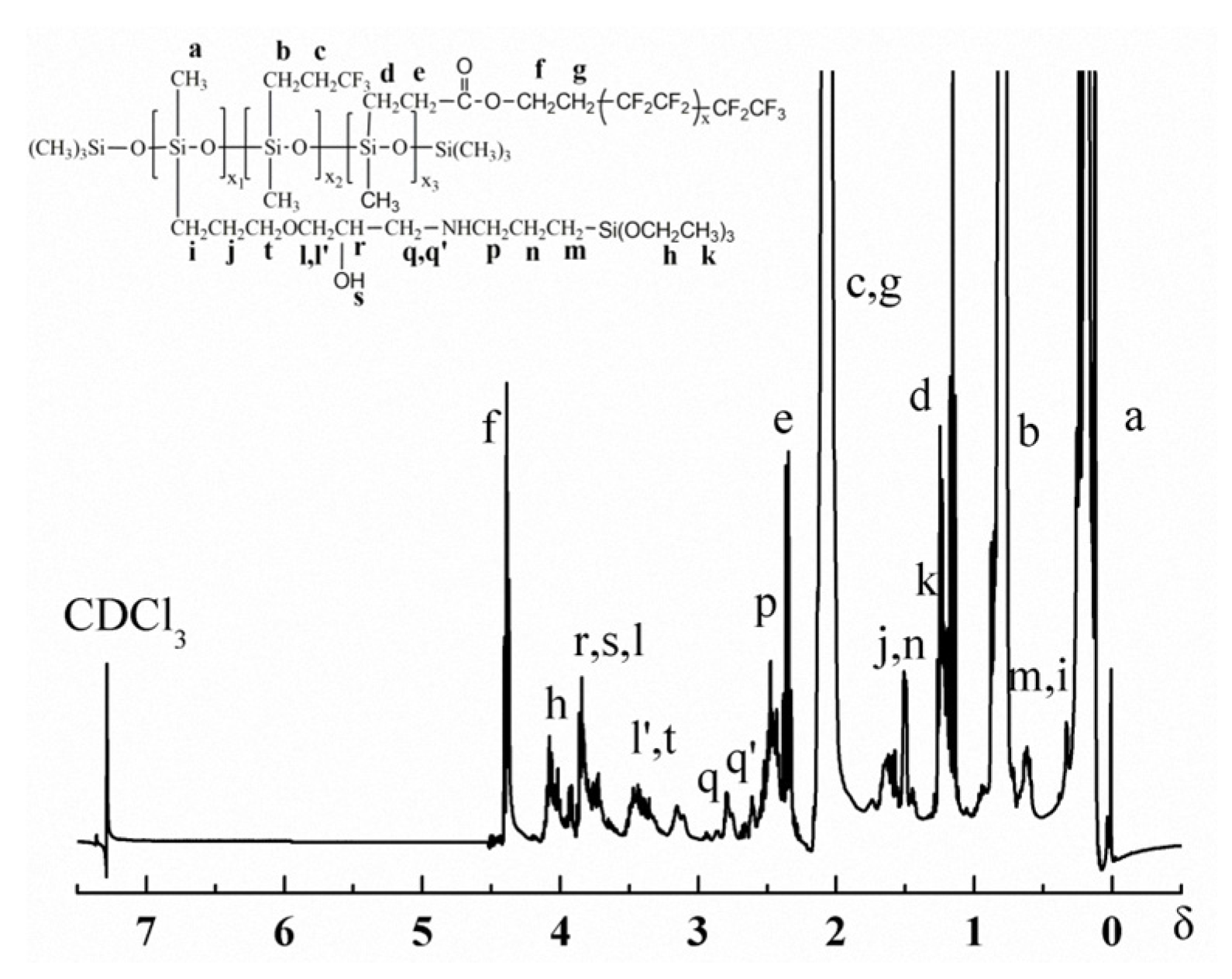
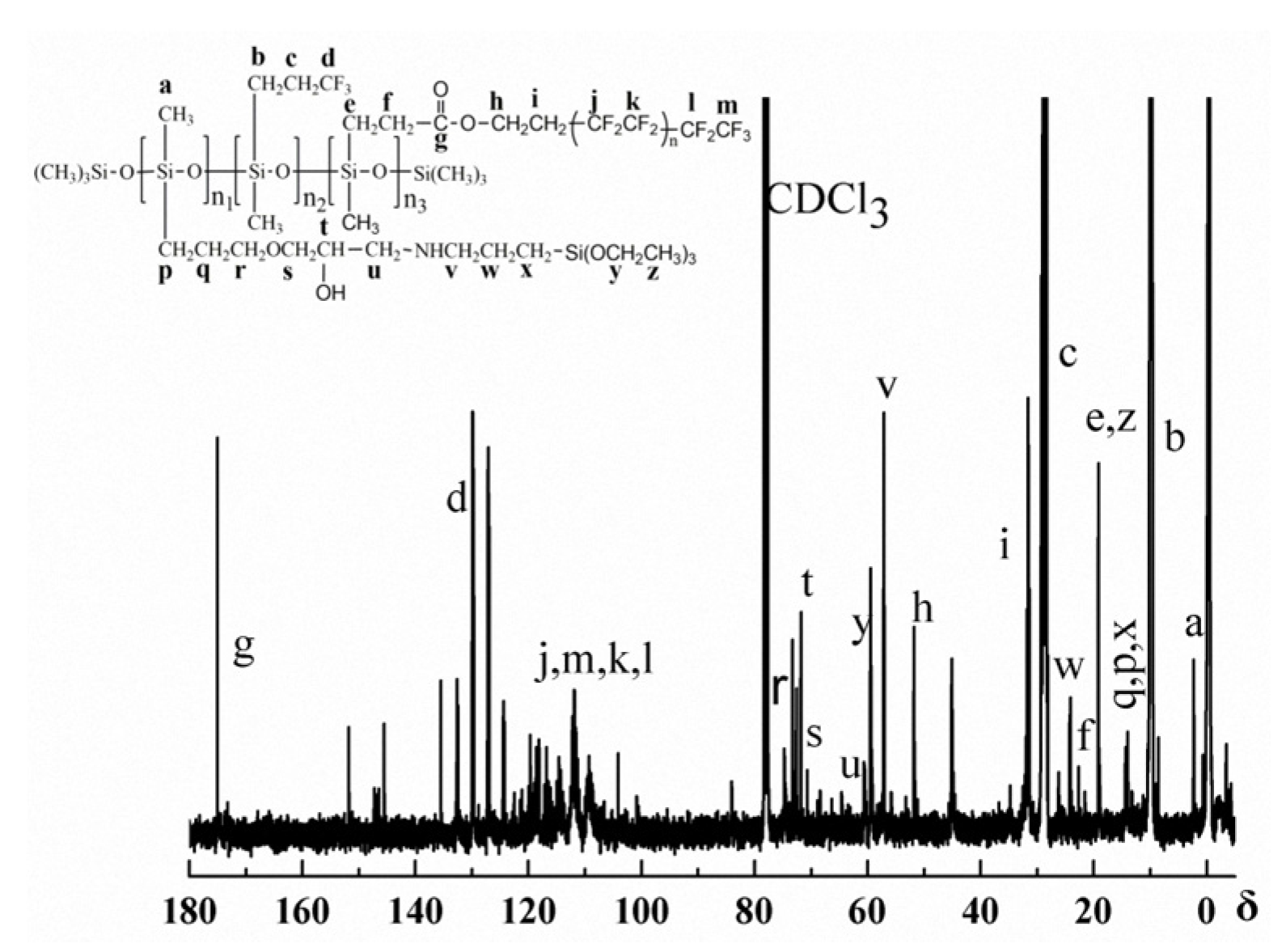
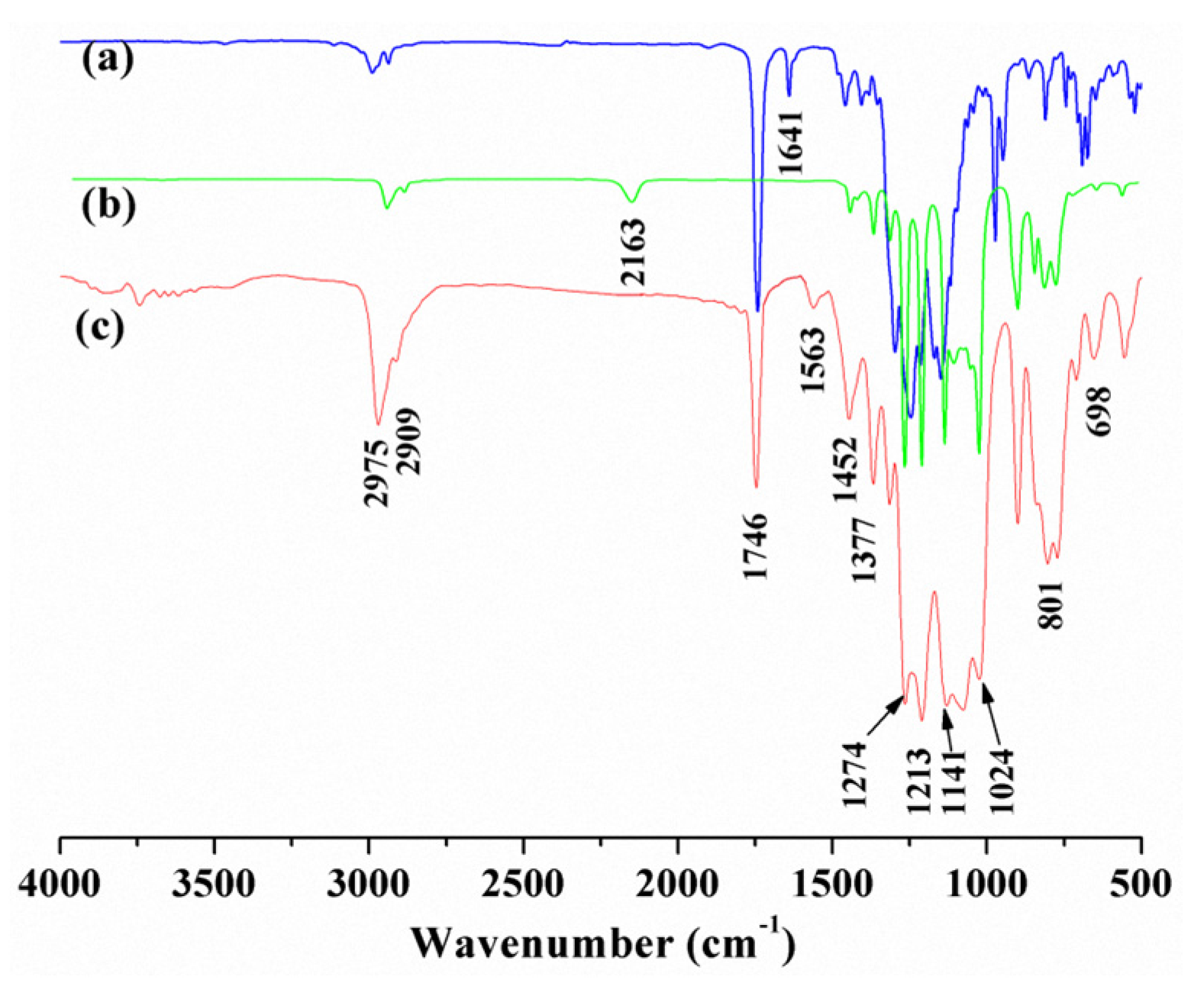
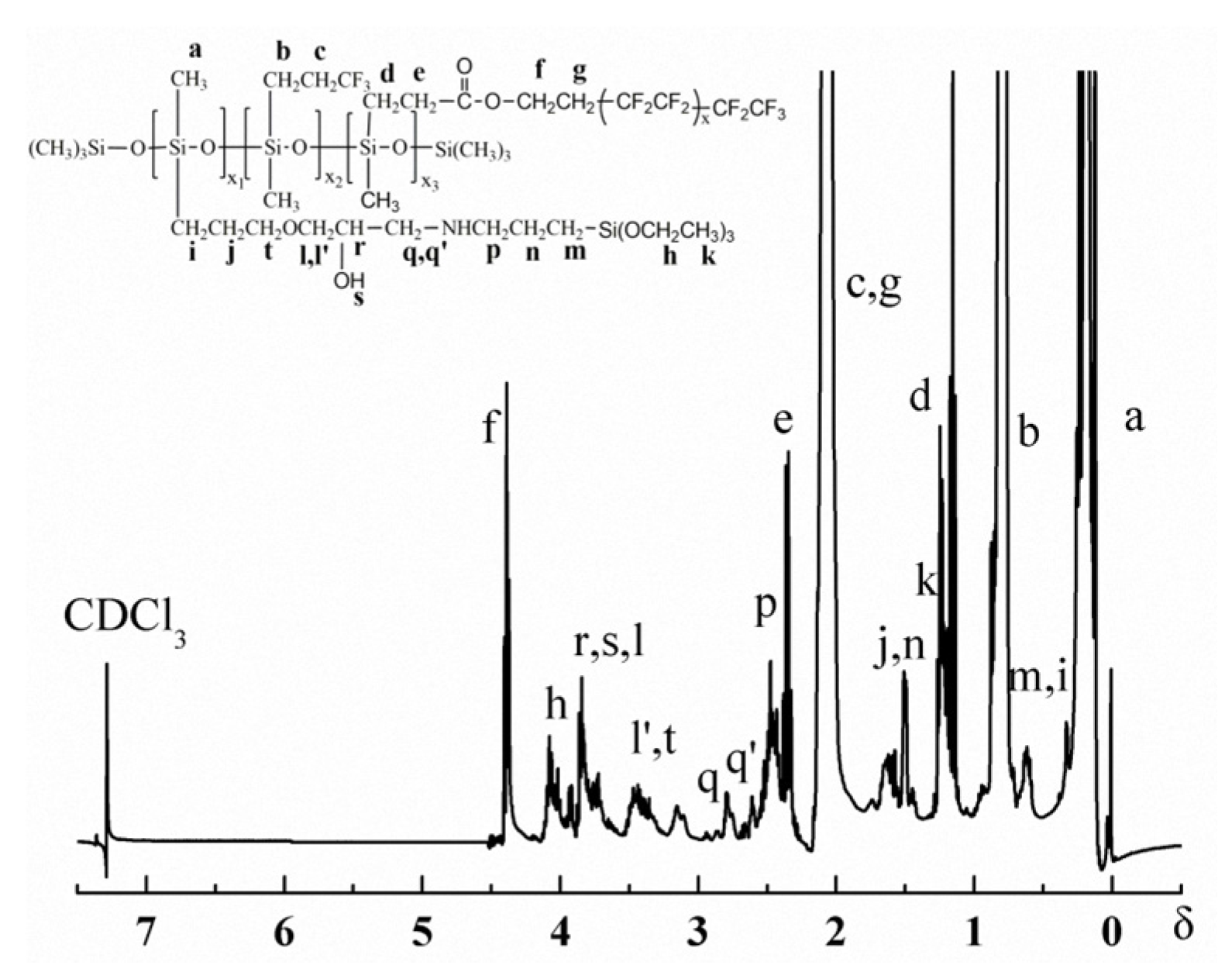
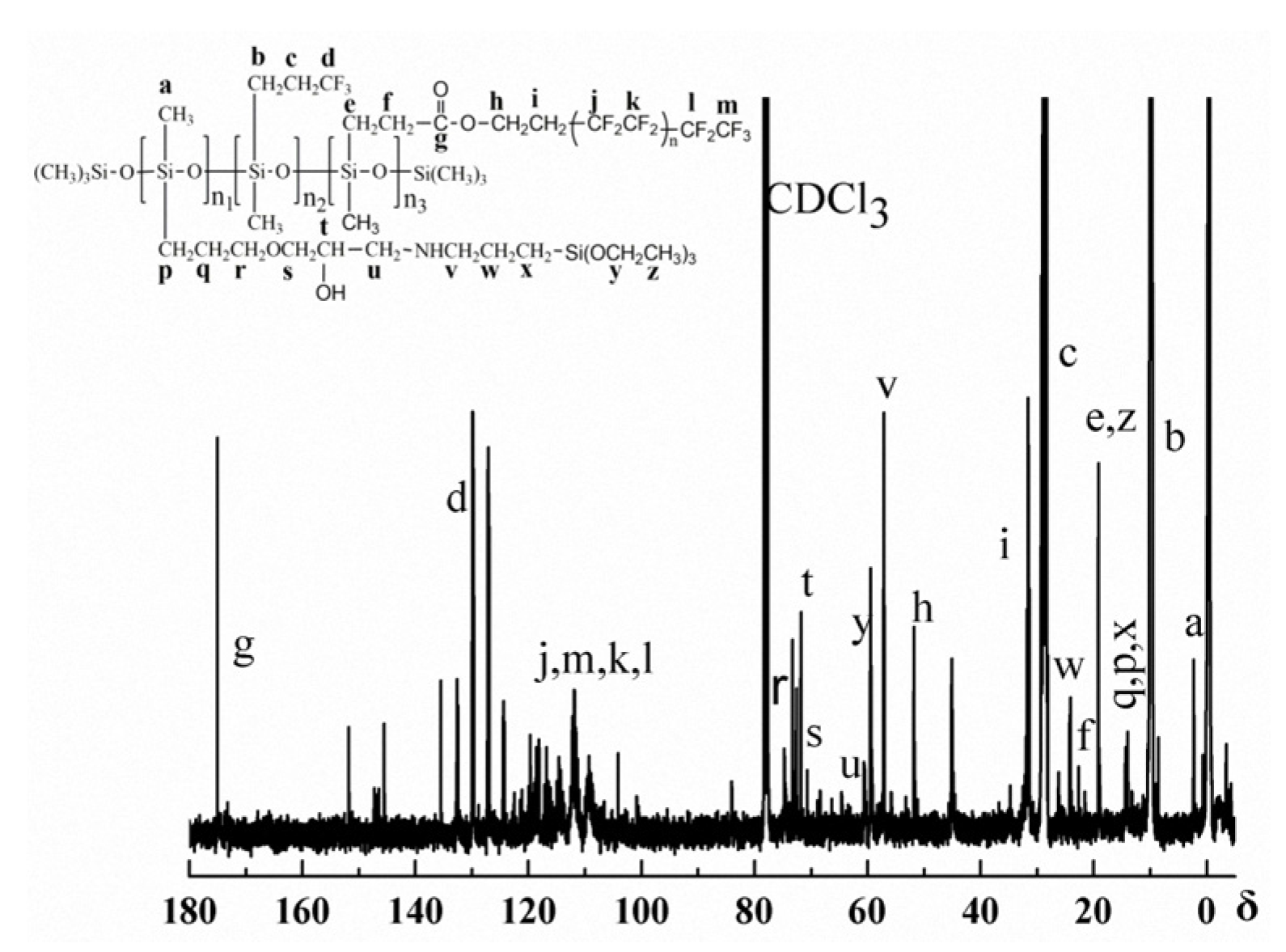
3.1. Structure Characterization
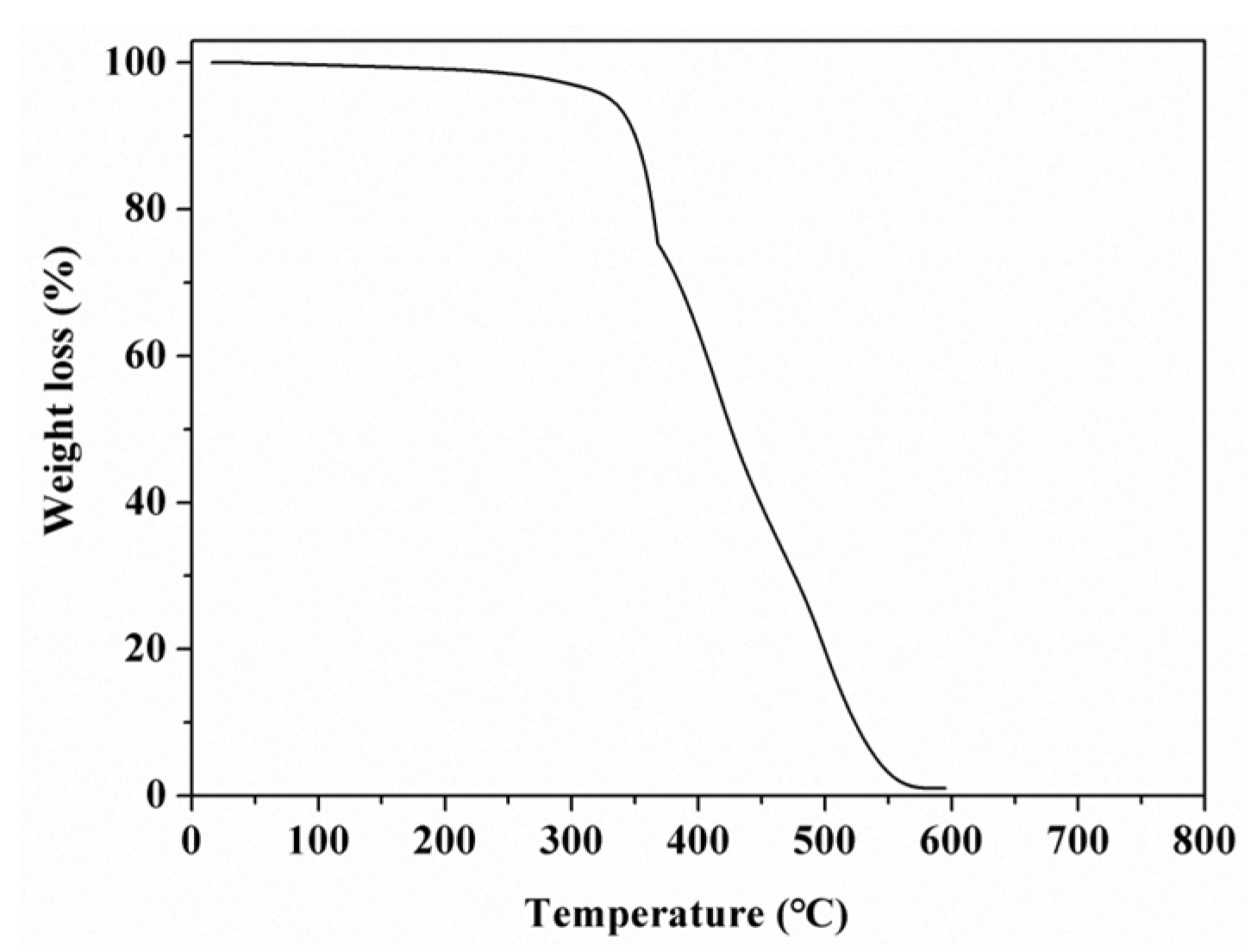
3.2. Thermal Stability
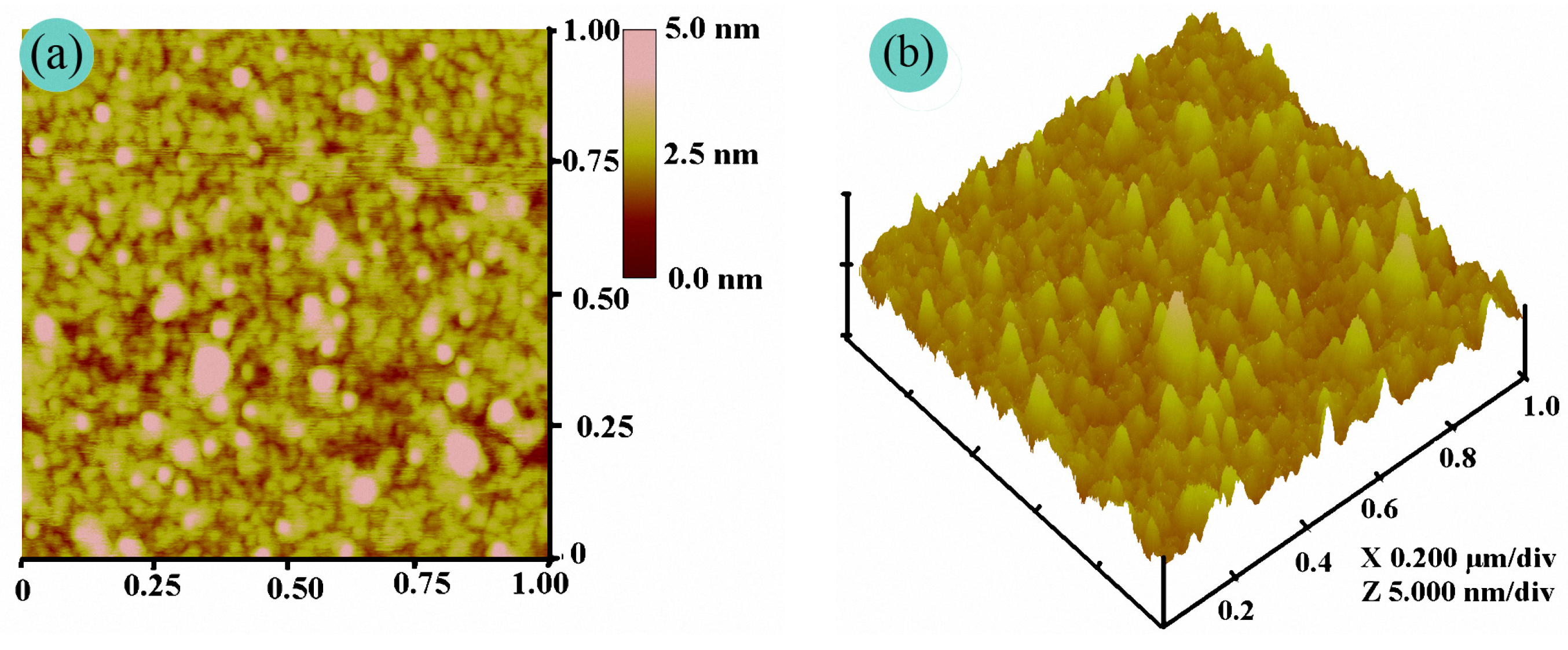
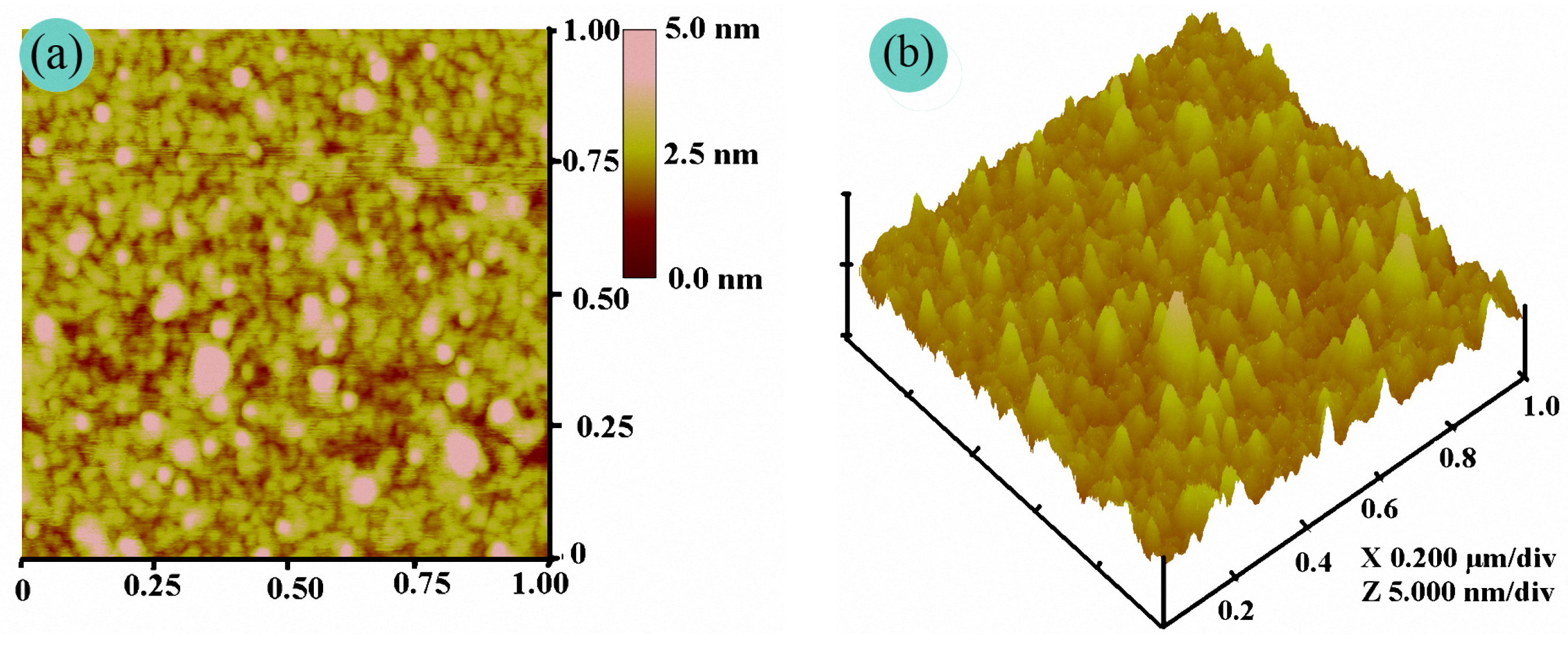
3.3. Film Morphology of the PFAS
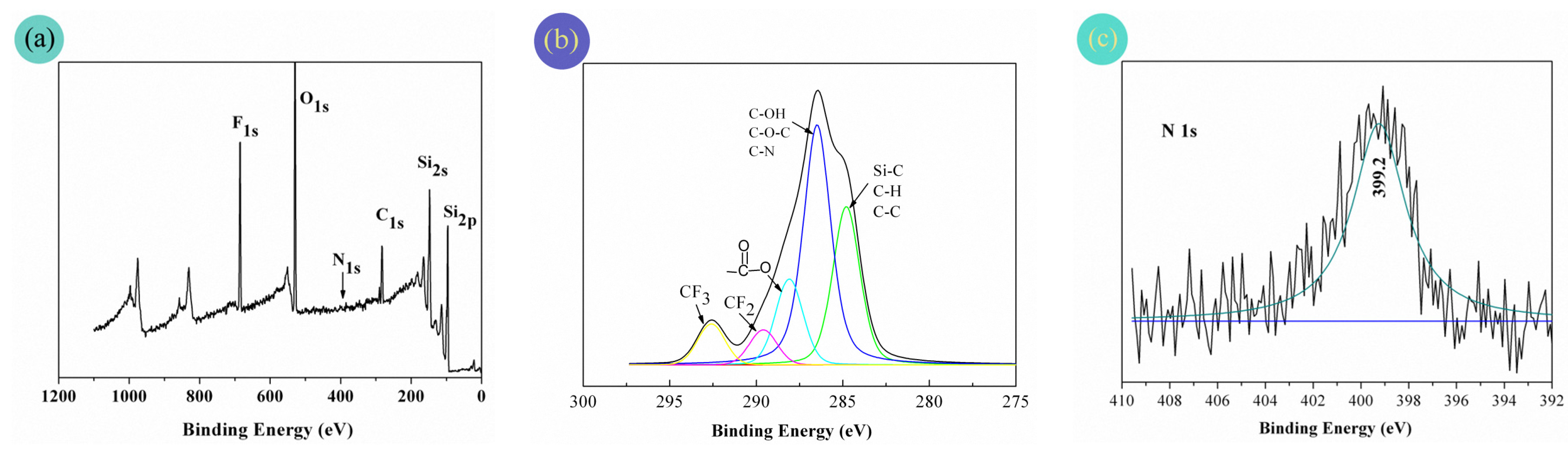
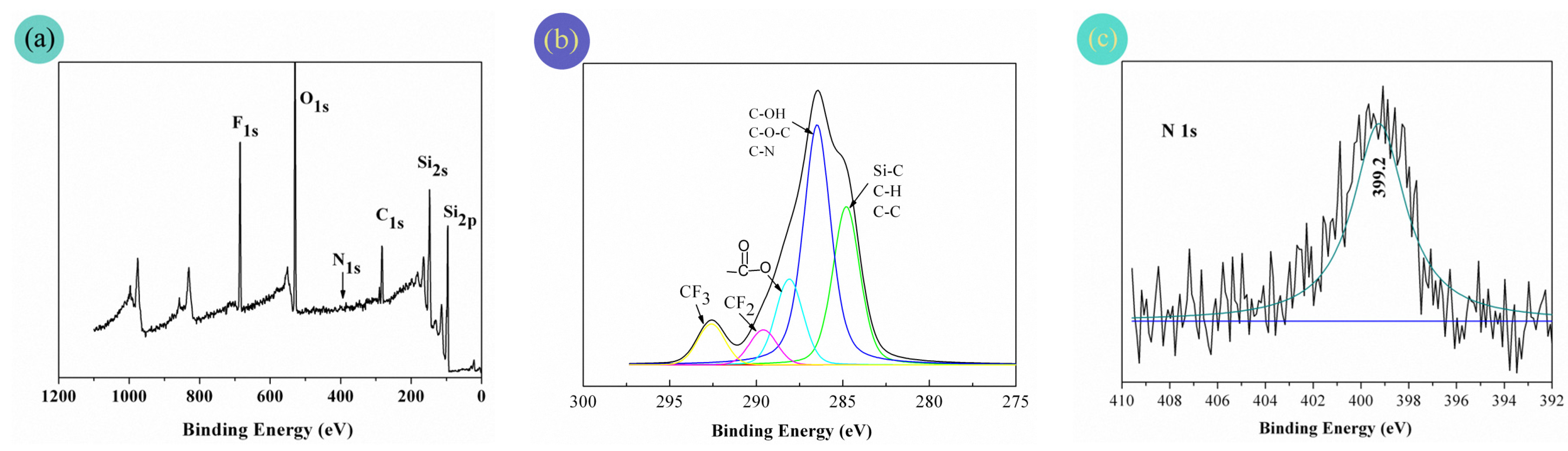
3.4. Chemical Compositions of the Fabric Coatings
3.5. Performance Properties of the Fabrics Finished with PFAS
4. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- An, Q.-F.; Li, L.-S.; Huang, L.-X.; Chen, K.-C. Film morphology and characterization of functional polysiloxane softeners. AATCC Rev. 2006, 6, 39–43. [Google Scholar]
- An, Q.-F.; Wang, K.-F.; Jia, Y. Film morphology, orientation and performance of dodecyl/carboxyl functional polysiloxane on cotton substrates. Appl. Surf. Sci. 2011, 257, 4569–4574. [Google Scholar] [CrossRef]
- Huang, J.-Q.; Meng, W.-D.; Qing, F.-L. Synthesis and repellent properties of vinylidene fluoride-containing polyacrylates. J. Fluorine Chem. 2007, 128, 1469–1477. [Google Scholar] [CrossRef]
- Chang, K.-C.; Chen, H.; Huang, C.-K.; Huang, S.-I. Preparation of super-hydrophobic film with fluorinated-copolymer. J. Appl. Polym. Sci. 2007, 104, 1646–1653. [Google Scholar] [CrossRef]
- Li, J.; Wang, Q.-J.; Su, C.-H.; Chen, Q.-M. Preparation and characterization of fluorine-containing acrylate copolymers by 60Co γ-ray radiation co-polymerization. Eur. Polym. J. 2007, 43, 2928–2934. [Google Scholar] [CrossRef]
- Furukawa, Y.; Kotera, M. Synthesis of fluorosilicone having highly fluorinated alkyl side chains based on the hydrosilylation of fluorinated olefins with polyhydromethylsiloxane. J. Polym. Sci. Polym. Chem. 2002, 40, 3120–3128. [Google Scholar] [CrossRef]
- Furukawa, Y.; Kotera, M. Water and oil repellency of polysiloxanes with highly fluorinated alkyl side chains. J. Appl. Polym. Sci. 2002, 87, 1085–1091. [Google Scholar] [CrossRef]
- Guo, L.-H.; Jiang, S.-J.; Qiu, T.; Zhang, S.-W.; He, L.-F.; Tan, J.; Li, X.-Y. Miniemulsion polymerization of fluorinated siloxane-acrylate latex and the application as waterborne textile finishing agent. J. Appl. Polym. Sci. 2014, 131. [Google Scholar] [CrossRef]
- Luo, Z.-H.; He, T.-Y.; Yu, H.-J.; Dai, L.-Z. A novel ABC triblock copolymer with very low surface energy: Poly(dimethylsiloxane)-block-poly(methyl methacrylate)-block-poly(2,2,3,3,4,4,4-heptafluorobutyl methacrylate). Macromol. React. Eng. 2008, 2, 398–406. [Google Scholar] [CrossRef]
- Yu, H.-J.; Luo, Z.-H. Novel superhydrophobic silica/poly(siloxane-fluoro-acrylate) hybrid nanoparticles prepared via two-step surface-initiated ATRP: Synthesis, characterization, and wettability. J. Polym. Sci. Polym. Chem. 2010, 48, 5570–5580. [Google Scholar] [CrossRef]
- Martinelli, E.; Suffredini, M.; Galli, G.; Glisenti, A.; Pettitt, M.-E.; Callow, M.-E.; Callow, J.-A.; Williams, D.; Lyall, G. Amphiphilic block copolymer/poly(dimethylsiloxane) (PDMS) blends and nanocomposites for improved fouling-release. Biofouling 2011, 27, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Galli, G.; Martinelli, E. Amphiphilic polymer platforms: Surface engineering of films for marine antibiofouling. Macromol. Rapid Commun. 2017, 38. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, E.; Moro, I.-D.; Galli, G.; Barbaglia, M.; Bibbiani, C.; Mennillo, E.; Oliva, M.; Pretti, C.; Antonioli, D.; Laus, M. Photopolymerized network polysiloxane films with dangling hydrophilic/hydrophobic chains for the biofouling release of invasive marine serpulid ficopomatus enigmaticus. ACS Appl. Mater. Interfaces 2015, 7, 8293–8301. [Google Scholar] [CrossRef] [PubMed]
- Baradie, B.; Lai, P.H.M.; Shoichet, M.-S. Synthesis and characterization of novel polysiloxane-grafted fluoropolymers. Can. J. Chem. 2005, 83, 553–558. [Google Scholar] [CrossRef]
- Xu, W.; An, Q.-F.; Hao, L.-F.; Zhang, D.; Zhang, M. Synthesis and characterization of self-crosslinking fluorinated polyacrylate soap-free latices with core-shell structure. Appl. Surf. Sci. 2013, 268, 373–380. [Google Scholar] [CrossRef]
- Xu, W.; An, Q.-F.; Hao, L.-F.; Huang, L.-X. Synthesis, film morphology and performance of cationic fluorinated polyacrylate emulsion with core-shell structure. J. Appl. Polym. Sci. 2012, 125, 2376–2383. [Google Scholar] [CrossRef]
- Hao, L.-F.; An, Q.-F.; Xu, W.; Huang, L.-X. Synthesis, film morphology and hydrophobicity of novel fluorinated polyacrylate emulsion and solution on silicon wafer. Colloids Surf. A 2012, 396, 83–89. [Google Scholar] [CrossRef]
- An, Q.-F.; Cheng, G.-W.; Li, L.-S. Synthesis, characterization, and film morphology of dodecylpolysiloxane. J. Appl. Polym. Sci. 2006, 101, 4480–4486. [Google Scholar] [CrossRef]
- Chen, L.-J.; Wu, F.-Q. BA-MMA-POMA copolymer latexes prepared by using HMPS polymerizable emulsifier. J. Appl. Polym. Sci. 2011, 122, 819–826. [Google Scholar] [CrossRef]
- Hao, L.-F.; Xu, W.; An, Q.-F. Synthesis, film morphology and performance of novel crosslinked polysiloxane with end-capped epoxy groups on cotton substrates. Fibers Polym. 2014, 15, 1567–1574. [Google Scholar] [CrossRef]
- Dong, F.-Y.; Sun, X.-R.; Feng, S.-Y. Thermal degradation kinetics of functional polysiloxanes containing chloromethyl groups. Thermochim. Acta 2016, 639, 14–19. [Google Scholar] [CrossRef]
- Xu, Y.-S.; Liu, M.-Y. Corrosion behaviors of polysiloxane-ferroferric oxide coating coated on carbon steel in NaCl solution and geothermal water. Geothermics 2017, 70, 339–350. [Google Scholar] [CrossRef]
- Jiang, B.; Zhang, T.; Zhao, L.-W.; Xu, Z.-M.; Huang, Y.-D. Effect of polymerizable photoinitiators on the UV-polymerization behaviors of photosensitive polysiloxane. J. Polym. Sci. Pol. Chem. 2017, 55, 1696–1705. [Google Scholar] [CrossRef]
- Song, X.-Y.; Zhai, J.; Wang, Y.-L.; Jiang, L. Fabrication of superhydrophobic surfaces by self-assembly and their water-adhesion properties. J. Phys. Chem. B 2005, 109, 4048–4052. [Google Scholar] [CrossRef] [PubMed]
- Corpart, J.-M.; Girault, S.; Juhué, D. Structure and surface properties of liquid crystalline fluoroalkyl polyacrylates: Role of the spacer. Langmuir 2001, 17, 7237–7244. [Google Scholar] [CrossRef]
- Montazer, M.; Hashemikia, S. Application of polyurethane/citric acid/silicone softener composite on cotton/polyester knitted fabric producing durable soft and smooth surface. J. Appl. Polym. Sci. 2012, 124, 4141–4148. [Google Scholar] [CrossRef]
- Fouda, M.M.G.; Fahmy, H.M. Multifunctional finish and cotton cellulose fabric. Carbohydr. Polym. 2011, 86, 625–629. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fabric Weight | Yarn Count | Fabric Density | ||
|---|---|---|---|---|
| (g/m2) | Warp | Weft | End/cm | Picks/cm |
| 119 | 40 | 40 | 133 | 72 |
| Dose of the Finishing Agent | WCA (°) | Whiteness | BR (mN) | |
|---|---|---|---|---|
| (g/100 g Ethyl Acetate) | (°) | w | f | |
| 0 | 0 | 76.89 | 239 | 175 |
| 0.1 | 139.3 ± 1.2 | 76.80 | 234 | 153 |
| 0.2 | 141.4 ± 1.8 | 76.31 | 230 | 152 |
| 0.3 | 143.9 ± 1.3 | 76.82 | 224 | 149 |
| 0.4 | 144.7 ± 1.1 | 76.75 | 218 | 141 |
| 0.5 | 144.9 ± 1.9 | 76.88 | 215 | 137 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, H.; Xu, W. The Preparation and Properties of Fluoroacrylate-Modified Polysiloxane as a Fabric Coating Agent. Coatings 2018, 8, 31. https://doi.org/10.3390/coatings8010031
Jin H, Xu W. The Preparation and Properties of Fluoroacrylate-Modified Polysiloxane as a Fabric Coating Agent. Coatings. 2018; 8(1):31. https://doi.org/10.3390/coatings8010031
Chicago/Turabian StyleJin, Hua, and Wei Xu. 2018. "The Preparation and Properties of Fluoroacrylate-Modified Polysiloxane as a Fabric Coating Agent" Coatings 8, no. 1: 31. https://doi.org/10.3390/coatings8010031
APA StyleJin, H., & Xu, W. (2018). The Preparation and Properties of Fluoroacrylate-Modified Polysiloxane as a Fabric Coating Agent. Coatings, 8(1), 31. https://doi.org/10.3390/coatings8010031