
Fundamental Properties of Transition-Metals-Adsorbed Germanene: A DFT Study

Abstract
:1. Introduction
2. Materials and Methods
3. Results
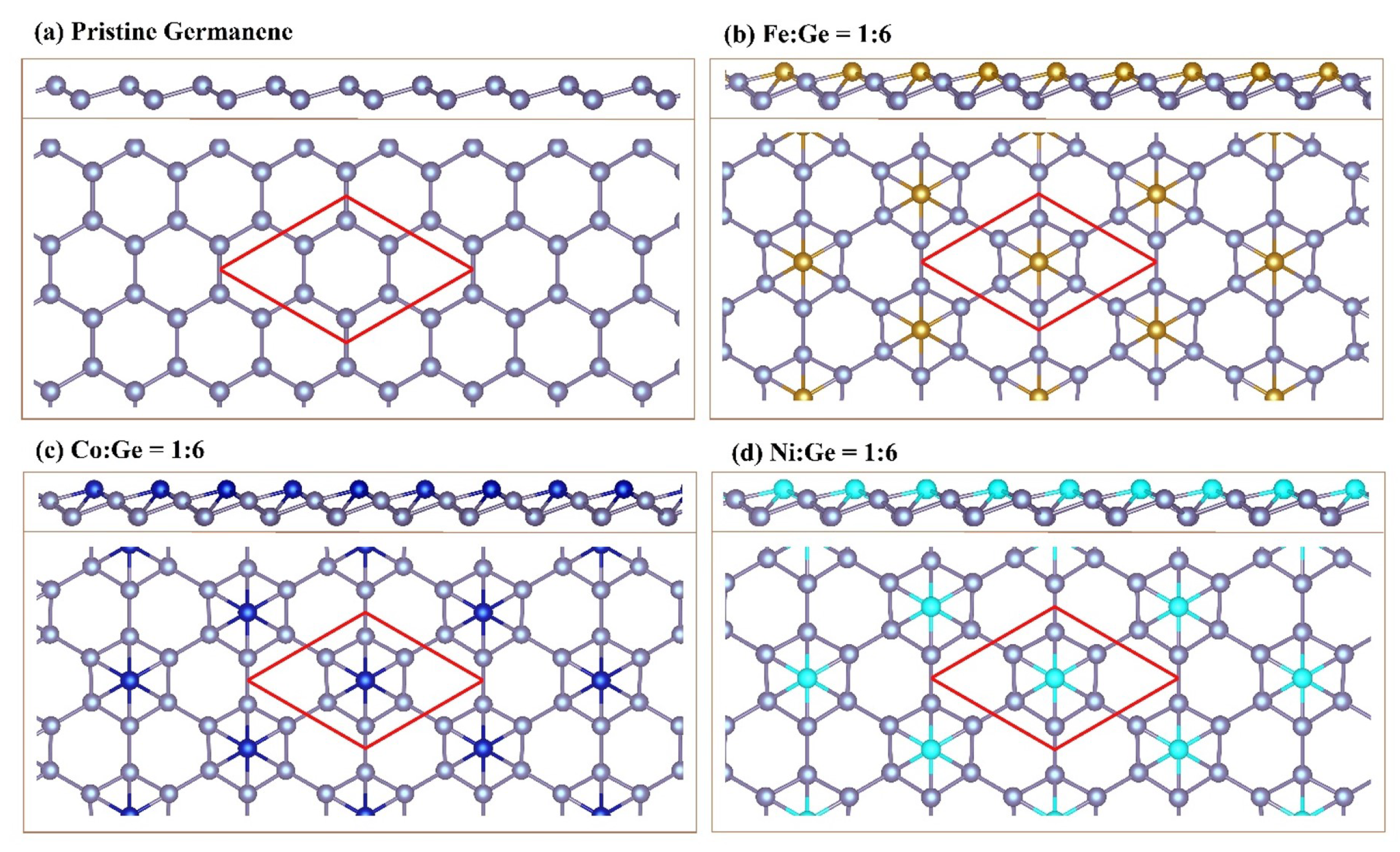
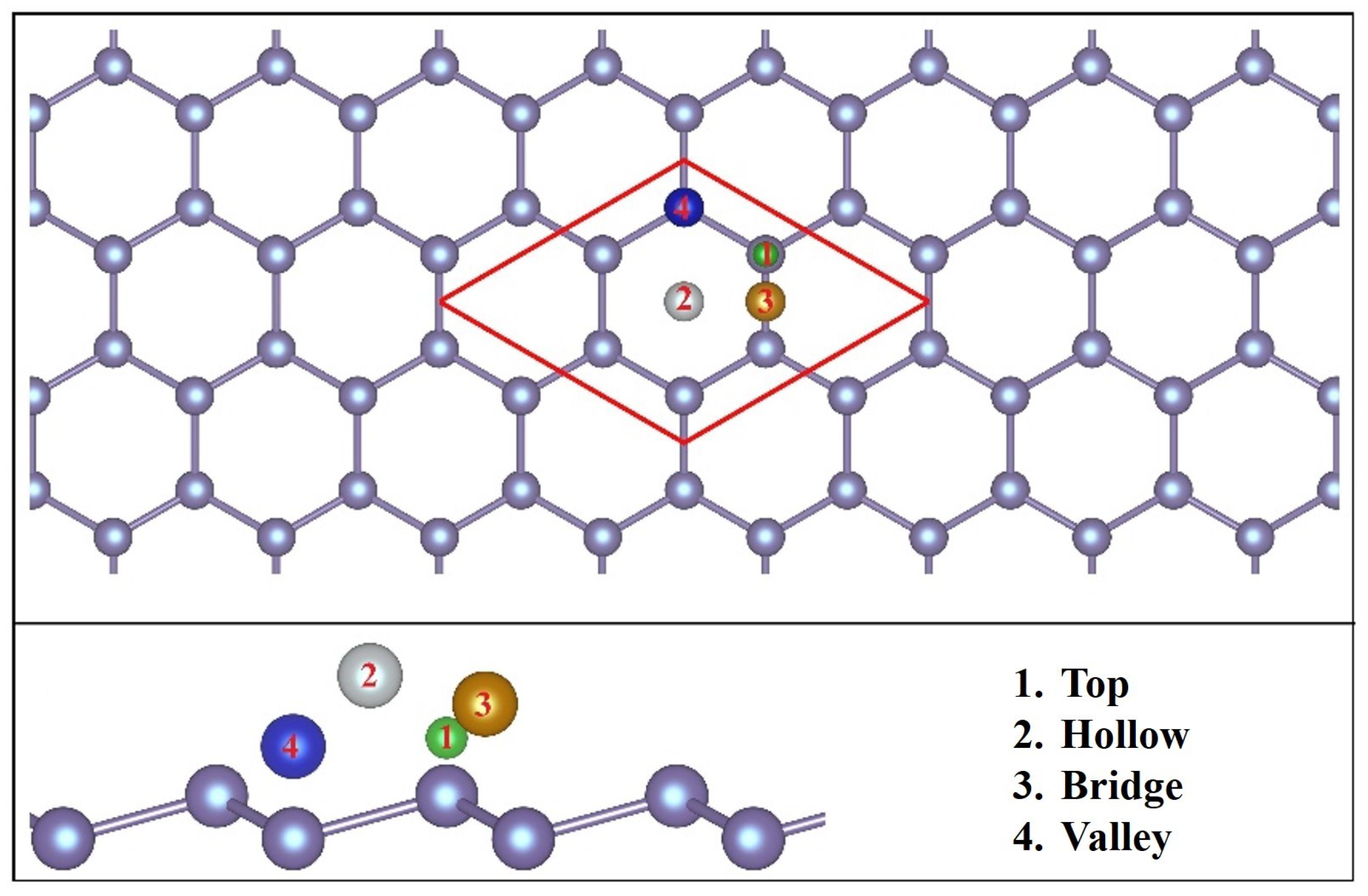
3.1. Geometric Structures
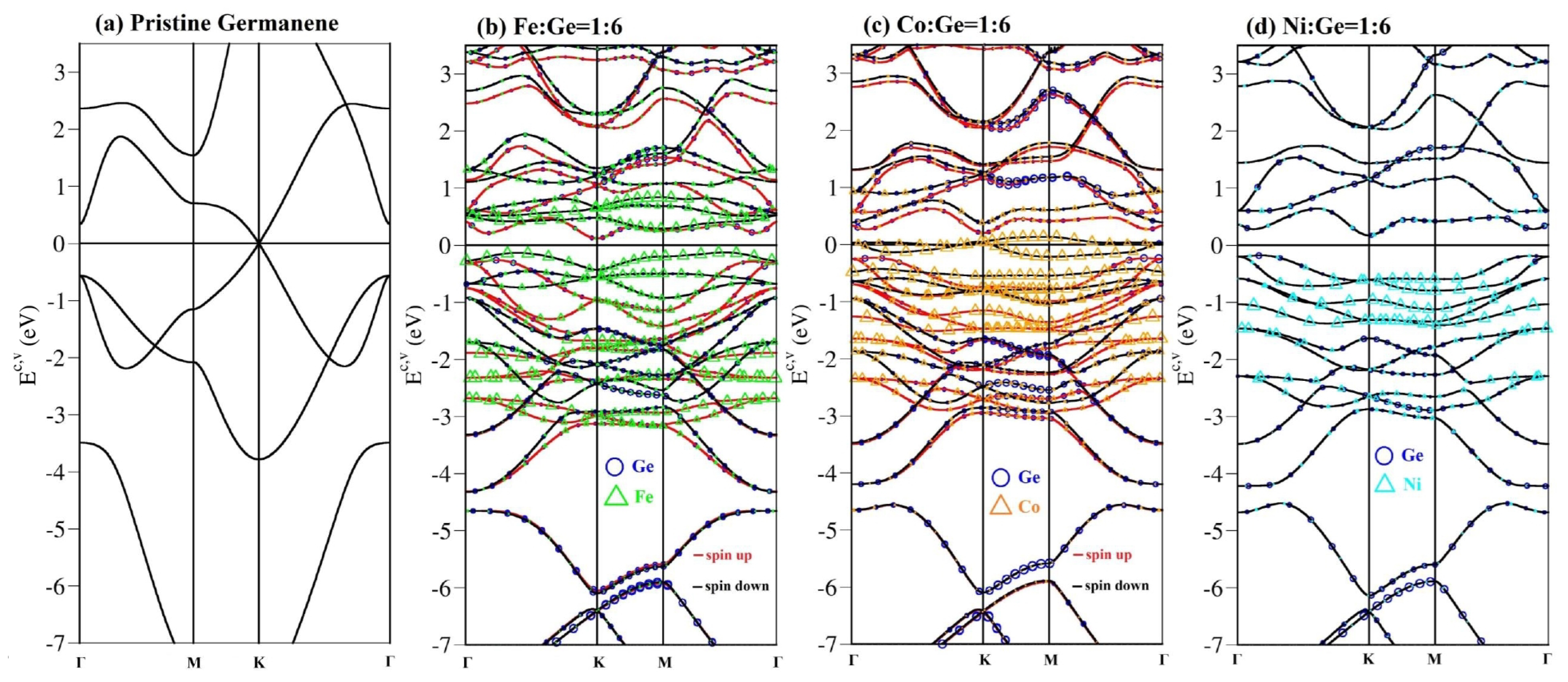
3.2. Energy Bands
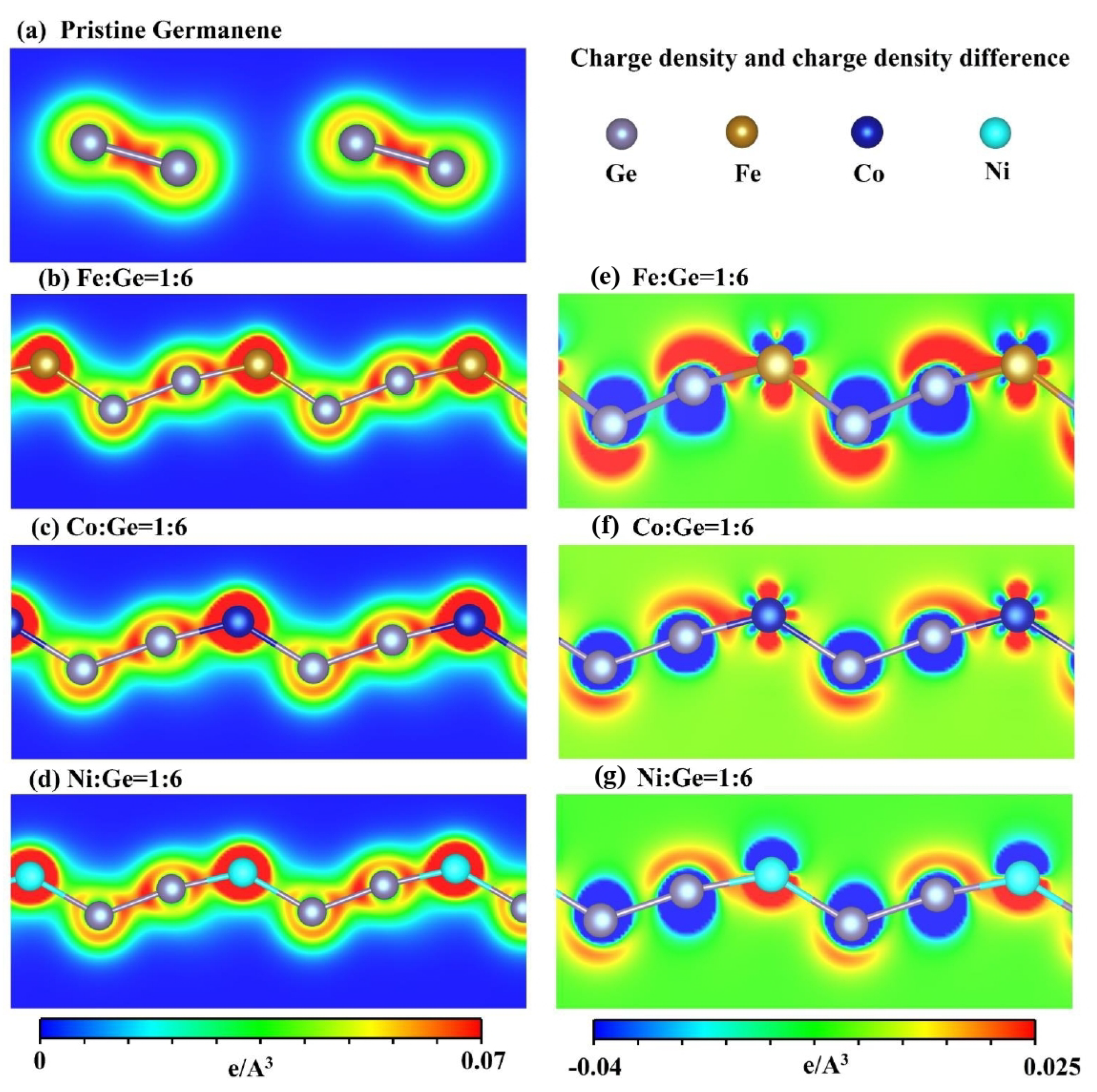
3.3. Spatial Charge Density Distribution
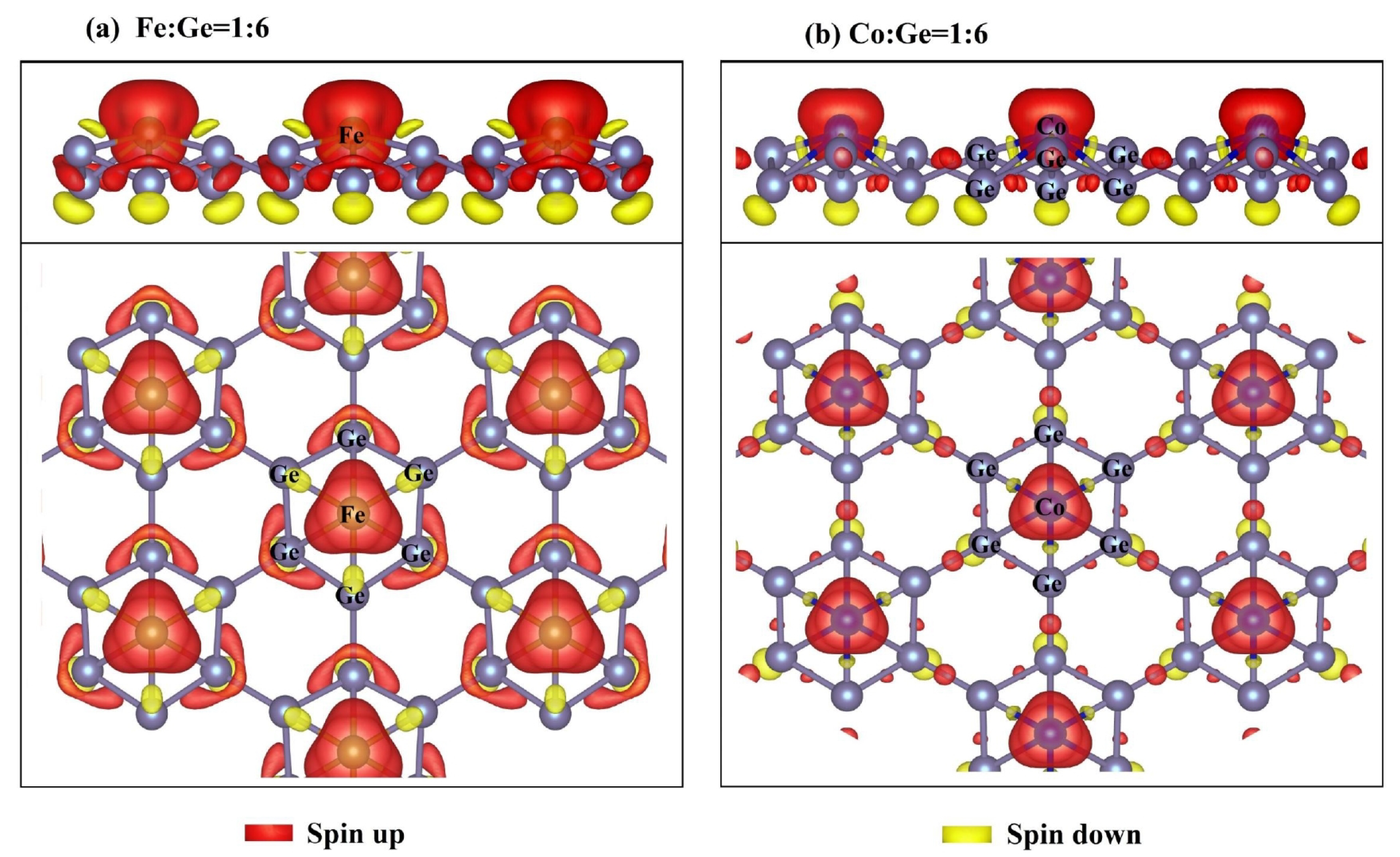
3.4. Spin Density
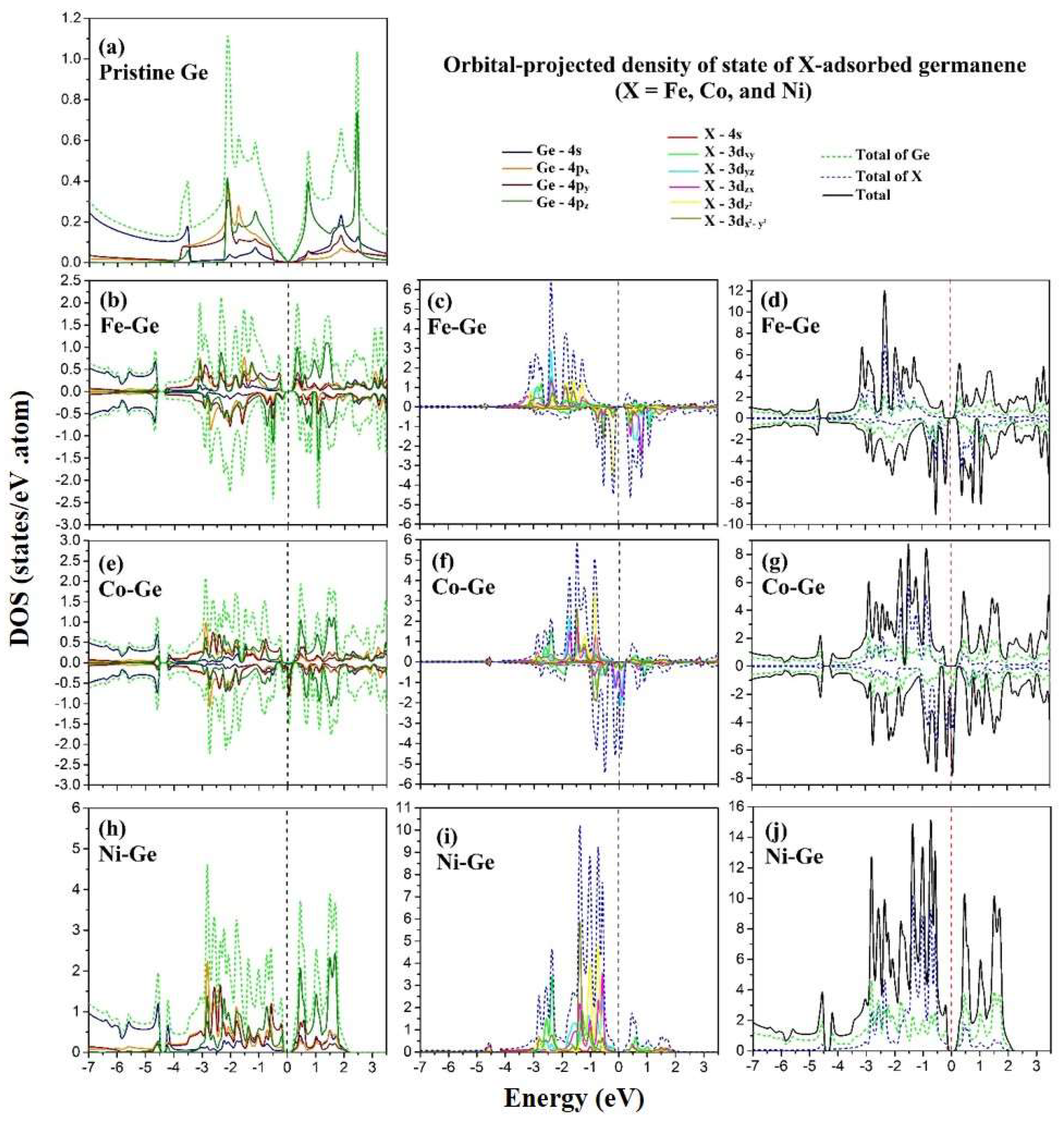
3.5. The Projected Density of States
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pham, H.D.; Gumbs, G.; Su, W.P.; Tran, N.T.T.; Lin, M.F. Unusual features of nitrogen substitutions in silicone. Rsc. Adv. 2020, 10, 32193–32201. [Google Scholar] [CrossRef] [PubMed]
- Pflugradt, P.; Matthes, L.; Bechstedt, F. Unexpected symmetry and AA stacking of bilayer silicene on Ag(111). Phys. Rev. B 2014, 89, 205428–205432. [Google Scholar] [CrossRef]
- Shen, M.; Zhang, Y.Y.; An, X.T.; Liu, J.J.; Li, S.S. Valley polarization in graphene-silicene-graphene heterojunction in zigzag nanoribbon. J. Appl. Phys. 2014, 115, 233702. [Google Scholar] [CrossRef] [Green Version]
- Ye, M.; Quhe, R.; Zheng, J.X.; Ni, Z.Y.; Wang, Y.Y.; Yuan, Y.K.; Tse, G.; Shi, J.J.; Gao, Z.X.; Lu, J. Tunable band gap in germanene by surface adsorption. Phys. E 2014, 59, 60–65. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.W.; Lee, C.E. Spin-orbit coupling-induced band inversion and spin Chern insulator phase in plumbene and stanine. Curr. Appl. Phys. 2020, 20, 413–418. [Google Scholar] [CrossRef]
- Kuzmany, H.; Shi, L.; Martinati, M.; Cambre, S.; Wenseleers, W.; Kurti, J.; Koltai, J.; Kukucska, G.; Cao, K.C.; Kaiser, U.; et al. Well-defined sub-nanometer graphene ribbons synthesized inside carbon nanotubes. Carbon 2021, 171, 221–229. [Google Scholar] [CrossRef]
- Usachov, D.Y.; Fedorov, A.V.; Vilkov, O.Y.; Senkovskiy, B.V.; Adamchuk, V.K.; Andryushechkin, B.V.; Vyalikh, D.V. Synthesis and electronic structure of nitrogen-doped graphene. Phys. Solid State 2013, 55, 1325–1332. [Google Scholar] [CrossRef]
- Ileri, N.; Goldman, N. Graphene and nano-diamond synthesis in expansions of molten liquid carbon. J. Chem. Phys. 2014, 141, 164709–164714. [Google Scholar] [CrossRef] [Green Version]
- Du, M.; Xu, C.H.; Sun, J.; Gao, L. One step synthesis of Fe2O3/nitrogen-doped graphene composite as anode materials for lithium ion batteries. Electrochim. Acta 2012, 80, 302–307. [Google Scholar] [CrossRef]
- Zhi-Gang Shao, Z.G.; Ye, X.S.; Yang, L.; Wang, C.L. First-principles calculation of intrinsic carrier mobility of silicene. J. Appl. Phys. 2013, 114, 93712–93715. [Google Scholar] [CrossRef]
- Cinquanta, E.; Fratesi, G.; Conte, S.D.; Grazianetti, C.; Scotognella, F.; Stagira, S.; Vozzi, C.; Onida, G.; Molle, A. Optical response and ultrafast carrier dynamics of the silicene-silver interface. Phys. Rev. B 2015, 92, 165427–165431. [Google Scholar] [CrossRef] [Green Version]
- Cahangirov, S.; Topsakal, M.; Akturk, E.; Sahin, H.; Ciraci, S. Two- and One-Dimensional Honeycomb Structures of Silicon and Germanium. Phys. Rev. Lett. 2009, 102, 236804–236807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, A.; Kumar, R.; Mazher, J.; Balan, A. Graphene made easy: High quality; large-area samples. Solid State Commun. 2009, 149, 718–721. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Liang, J.J.; Huang, Y.; Ma, Y.F.; Wang, Y.; Chen, Y.S. Size-controlled synthesis of graphene oxide sheets on a large scale using chemical exfoliation. Carbon 2009, 47, 3365–3368. [Google Scholar] [CrossRef]
- Chen, L.; Hernandez, Y.; Feng, X.L.; Mullen, K. From Nanographene and Graphene Nanoribbons to Graphene Sheets: Chemical Synthesis. Angew. Chem. Int. Ed. 2012, 51, 7640–7654. [Google Scholar] [CrossRef]
- Ogikubo, T.; Shimazu, H.; Fujii, Y.; Ito, K.; Ohta, A.; Araidai, M.; Kurosawa, M.; Lay, G.L.; Yuhara, J. Continuous Growth of Germanene and Stanene Lateral Heterostructures. Adv. Mater. Interfaces 2020, 7, 1902132. [Google Scholar] [CrossRef]
- Ge, M.; Zong, M.; Xu, D.; Chen, Z.; Yang, J.; Yao, H.; Wei, C.; Chen, Y.; Lin, H.; Shi, J. Freestanding germanene nanosheets for rapid degradation and photothermal conversion. Mater. Today Nano 2021, 15, 100119. [Google Scholar] [CrossRef]
- Si, X.; She, W.; Xu, Q.; Yang, G.; Li, Z.; Wang, S.; Luan, J. First-Principles Density Functional Theory Study of Modified Germanene-Based Electrode Materials. Materials 2022, 15, 103. [Google Scholar] [CrossRef]
- Hussain, T.; Kaewmaraya, T.; Chakraborty, S.; Vovusha, H.; Amornkitbamrung, V.; Ahuja, R. Defected and Functionalized Germanene-based Nanosensors under Sulfur Comprising Gas Exposure. ACS Sens. 2018, 3, 867–874. [Google Scholar] [CrossRef] [Green Version]
- Ullah, F.; Irshad, S.; Khan, S.; Hashmi, M.A.; Ludwig, R.; Mahmood, T.; Ayub, K. Nonlinear optical response of first-row transition metal doped Al12P12 nanoclusters; a first-principles study. J. Phys. Chem. Solids 2021, 151, 109914. [Google Scholar] [CrossRef]
- Irshada, S.; Ullaha, F.; Khana, S.; Ludwigb, R.; Mahmooda, T.; Ayub, K. First row transition metals decorated boron phosphide nanoclusters as nonlinear optical materials with high thermodynamic stability and enhanced electronic properties; A detailed quantum chemical study. Opt. Laser Technol. 2021, 134, 106570. [Google Scholar] [CrossRef]
- Ullah, F.; Ayub, K.; Mahmood, T. High performance SACs for HER process using late first-row transition metals anchored on graphyne support: A DFT insight. Int. J. Hydrogen Energy 2021, 46, 37814–37823. [Google Scholar] [CrossRef]
- Sun, M.; Ren, Q.; Wang, S.; Zhang, Y.; Du, Y.; Yu, J.; Tang, W. Magnetism in transition-metal-doped germanene: A first-principles study. Comput. Mater. Sci. 2016, 118, 112–116. [Google Scholar] [CrossRef]
- Zhou, P.; Sun, L.Z. 3d Transition Metal Adsorption Induced the valley-polarized Anomalous Hall Effect in Germanene. Sci. Rep. 2016, 6, 27830. [Google Scholar] [CrossRef]
- Lin, Z.Z.; Chen, X. Transition-metal-decorated germanene as promising catalyst for removing CO contamination in H2. Mater. Des. 2016, 107, 82–89. [Google Scholar] [CrossRef]
- Kaloni, T.P. Tuning the Structural, Electronic, and Magnetic Properties of Germanene by the Adsorption of 3d Transition Metal Atoms. J. Phys. Chem. C 2014, 118, 25200–25208. [Google Scholar] [CrossRef] [Green Version]
- Pang, Q.; Li, L.; Zhang, C.L.; Wei, X.M.; Song, Y.L. Structural, electronic and magnetic properties of 3d transition metal atom adsorbed germanene: A first-principles study. Mater. Chem. Phys. 2015, 160, 96–104. [Google Scholar] [CrossRef]
- Nemoto, R.; Kruger, P.; Hartini, A.N.P.; Hosokai, T.; Horie, M.; Kera, S.; Yamada, T.K. Well-Ordered Monolayer Growth of Crown-Ether Ring Molecules on Cu(111) in Ultra-High Vacuum: An STM; UPS; and DFT Study. J. Phys. Chem. C 2019, 123, 18939–18950. [Google Scholar] [CrossRef]
- Heidarzadeh, A. Tensile behavior; microstructure; and substructure of the friction stir welded 70/30 brass joints: RSM; EBSD; and TEM study. Arch. Civ. Mech. Eng. 2019, 19, 137–146. [Google Scholar] [CrossRef]
- Cheng, Z.W.; Zhang, Z.Y.; Sun, H.G.; Li, S.J.; Yuan, H.; Wang, Z.J.; Cao, Y.; Shao, Z.B.; Bian, Q.; Zhang, X.; et al. Visualizing Dirac nodal-line band structure of topological semimetal ZrGeSe by ARPES. Appl. Mater. 2019, 7, 051105. [Google Scholar] [CrossRef] [Green Version]
- Ridene, M.; Girard, J.C.; Travers, L.; David, C.; Ouerghi, A. STM/STS investigation of edge structure in epitaxial graphene. Surf. Sci. 2012, 606, 1289–1292. [Google Scholar] [CrossRef]
- Liu, X.; Wang, C.Z.; Yao, Y.X.; Lu, W.C.; Hupalo, M.; Tringides, M.C.; Ho, K.M. Bonding and charge transfer by metal adatom adsorption on graphene. Phys. Rev. B 2011, 83, 235411. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Blanc, X.; Cances, E.; Dupuy, M.S. Variational projector augmented-wave method. Comptes Rendus Math. 2017, 355, 665–670. [Google Scholar] [CrossRef]
- Li, S.S.; Zhang, C.W.; Ji, W.X.; Li, F.; Wang, P.J.; Hu, S.J.; Yan, S.S.; Liu, Y.S. Tunable electronic and magnetic properties in germanene by alkali, alkaline-earth, group III and 3d transition metal atom adsorption. Phys. Chem. Chem. Phys. 2014, 16, 15968–15978. [Google Scholar] [CrossRef]
- Naji, S.; Belhaj, A.; Labrim, H.; Bhihi, M.; Benyoussef, A.; Kenz, A.E. Adsorption of Co and Ni on Graphene with a Double Hexagonal Symmetry: Electronic and Magnetic Properties. J. Phys. Chem. C 2014, 118, 4924–4929. [Google Scholar] [CrossRef]
- Perdewa, J.P.; Yangc, W.; Burked, K.; Yanga, Z.; Grossf, E.K.U.; Schefflerg, M.; Scuseria, G.E.; Hendersonj, T.M.; Zhangg, I.Y.; Ruzsinszkya, A.; et al. Understanding band gaps of solids in generalized Kohn–Sham theory. Proc. Natl. Acad. Sci. USA 2017, 114, 2801–2806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudenko, A.N.; Katsnelson, M.I.; Roldan, R. Electronic properties of single-layer antimony: Tight-binding model, spin-orbit coupling, and the strength of effective Coulomb interactions. Phys. Rev. B 2017, 95, 081407. [Google Scholar] [CrossRef] [Green Version]
- Phark, S.H.; Sander, D. Spin-polarized scanning tunneling microscopy with quantitative insights into magnetic probes. Nano Converg. 2017, 4, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuyama, H.; Nara, D.; Kageyama, R.; Honda, K.; Sato, T.; Kusanagi, K.; Srinivasan, E.; Koike, K. Spin-polarized scanning tunneling microscopy experiments on the rough surface of a polycrystalline NiFe film with a fine magnetic tip sensitive to a well-defined magnetization component. AIP Adv. 2016, 6, 035122. [Google Scholar] [CrossRef] [Green Version]
- Farrar, L.S.; Nevill, A.; Lim, Z.J.; Balakrishnan, G.; Dale, S.; Bending, S.J. Superconducting Quantum Interference in Twisted van der Waals Heterostructures. Nano Lett. 2021, 21, 6725–6731. [Google Scholar] [CrossRef]
- Giazotto, F.; Peltonen, J.T.; Meschke, M.; Pekola, J.P. Superconducting quantum interference proximity transistor. Nat. Phys. 2010, 6, 254–260. [Google Scholar] [CrossRef] [Green Version]
- Krenner, W.; Kuhne, D.; Klappenberger, F.; Barth, J.V. Assessment of Scanning Tunneling Spectroscopy Modes Inspecting Electron Confinement in Surface-Confined Supramolecular Networks. Sci. Rep. 2013, 3, 1454. [Google Scholar] [CrossRef] [Green Version]
- Ting, W.; Egi, T.; Itti, R.; Kuroda, K.; Koshizuka, N.; Tanaka, S. STM/STS measurements on clean and stable surfaces of as-prepared Nd1Ba2Cu3Oy single crystals. Phys. C Supercond. 1996, 263, 185–188. [Google Scholar] [CrossRef]
- Phark, S.H.; Borme, J.; Vanegas, A.L.; Corbetta, M.; Sander, D.; Kirschner, J. Scanning tunneling spectroscopy of epitaxial graphene nanoisland on Ir(111). Nanoscale Res. Lett. 2012, 7, 255–258. [Google Scholar] [CrossRef]
- Klusek, Z.; Dabrowski, P.; Kowalczyk, P.; Kozlowski, W.; Olejniczak, W.; Blake, P.; Szybowicz, M.; Runka, T. Graphene on gold: Electron density of states studies by scanning tunneling spectroscopy. Appl. Phys. Lett. 2009, 95, 113114. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adatoms | Sites | (eV) | h (Å) | X–Ge (Å) | Ge–Ge (Å) | Magnetic Moment () | Buckling Height (Å) |
|---|---|---|---|---|---|---|---|
| Pristine | 2.42 | 0.66 | |||||
| Valley | 3.06 | ||||||
| Iron | Hollow | 3.43 | 1.02 | 2.454, 2.710 | 2.651 | 2.73 | 1.13 |
| Top | 2.65 | ||||||
| Valley | 3.60 | ||||||
| Cobalt | Hollow | 4.20 | 1.05 | 2.437, 2.718 | 2.595 | 1.75 | 1.12 |
| Top | 3.75 | ||||||
| Valley | 1.19 | ||||||
| Nickel | Hollow | 1.85 | 0.95 | 2.436, 2.649 | 2.552 | 0 | 1.35 |
| Top | 1.50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.-Y.; Wu, J.-Y. Fundamental Properties of Transition-Metals-Adsorbed Germanene: A DFT Study. Coatings 2022, 12, 948. https://doi.org/10.3390/coatings12070948
Liu H-Y, Wu J-Y. Fundamental Properties of Transition-Metals-Adsorbed Germanene: A DFT Study. Coatings. 2022; 12(7):948. https://doi.org/10.3390/coatings12070948
Chicago/Turabian StyleLiu, Hsin-Yi, and Jhao-Ying Wu. 2022. "Fundamental Properties of Transition-Metals-Adsorbed Germanene: A DFT Study" Coatings 12, no. 7: 948. https://doi.org/10.3390/coatings12070948
APA StyleLiu, H.-Y., & Wu, J.-Y. (2022). Fundamental Properties of Transition-Metals-Adsorbed Germanene: A DFT Study. Coatings, 12(7), 948. https://doi.org/10.3390/coatings12070948