β-Lactam Antibiotics Renaissance

Abstract
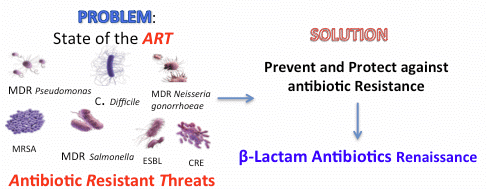
:
1. Introduction
- multidrug-resistant strains (MDR), which are non-susceptible to one or more drugs belonging to ≥3 antimicrobial classes;
- and extremely drug-resistant strains (XDR), which are non-susceptible (or nearly so) to all classes of antimicrobials [3].
2. Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
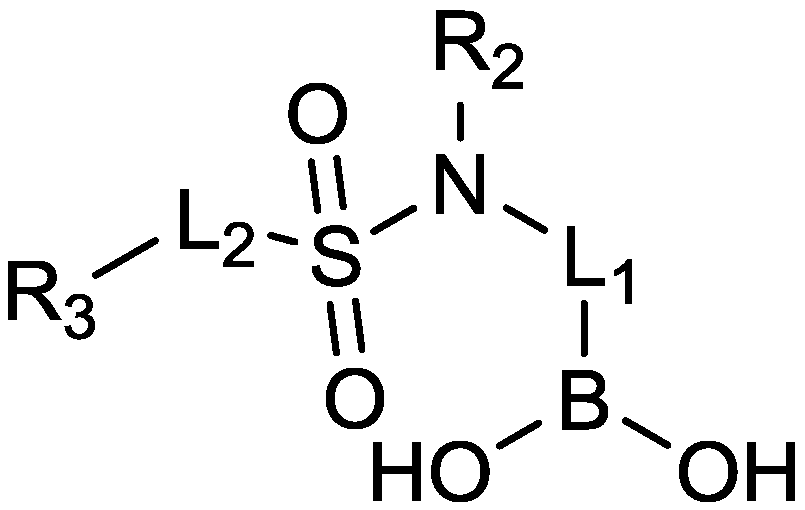{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year approved | Drug name | Chemical structure | Class | Bacterial profile | Special features |
|---|---|---|---|---|---|
| 2000 | Linezolid | 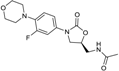 | oxazolidinone | G+ | MRSA |
| 2001 | Telithromycin | 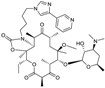 | Macrolide (ketolide) | G+/some G− | Safety concerns |
| 2002 | Biapenem |  | carbapenem | G+, G− | Broad spectrum, including many β-lactamase producers |
| 2002 | Ertapenem |  | carbapenem | G+, G− | Broad spectrum, including many β-lactamase producers |
| 2002 | Prulifloxacin | 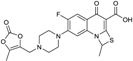 | fluoroquinolone | G+, G− | Broad spectrum |
| 2002 | Pazufloxacin | 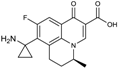 | fluoroquinolone | G+, G− | Broad spectrum |
| 2002 | Balofloxacin | 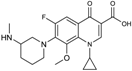 | fluoroquinolone | G+, G− | Broad spectrum |
| 2003 | Daptomycin | 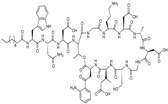 | lipopeptide | G+ | MRSA, VRE |
| 2004 | Gemifloxacin | 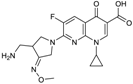 | fluoroquinolone | G+, G− | Broad spectrum |
| 2005 | Doripenem | 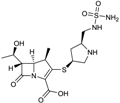 | carbapenem | G+, G− | Broad spectrum, including many β-lactamase producers |
| 2005 | Tigecycline |  | tetracycline (glycylglycine) | G+, G− | Broad spectrum |
| 2007 | Retapamulin |  | pleuromutilin | G+ | MRSA |
| 2007 | Garenoxacin | 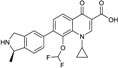 | Quinolone | G+, G− | Broad spectrum, |
| 2008 | Ceftobiprole medocaril | 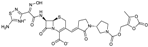 | cephalosporin | G+, G− | Broad spectrum, MRSA |
| 2008 | Sitafloxacin |  | fluoroquinolone | G+, G− | Broad spectrum |
| 2009 | Tebipenem pivoxil |  | carbapenem | G+, G− | Broad spectrum, including many β-lactamase producers |
| 2009 | Telavancin | 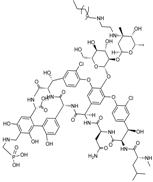 | glycopeptide | G+ | MRSA |
| 2009 | Antofloxacin |  | fluoroquinolone | G+, G− | Broad spectrum |
| 2009 | Besifloxacin |  | fluoroquinolone | G+, G− | Broad spectrum |
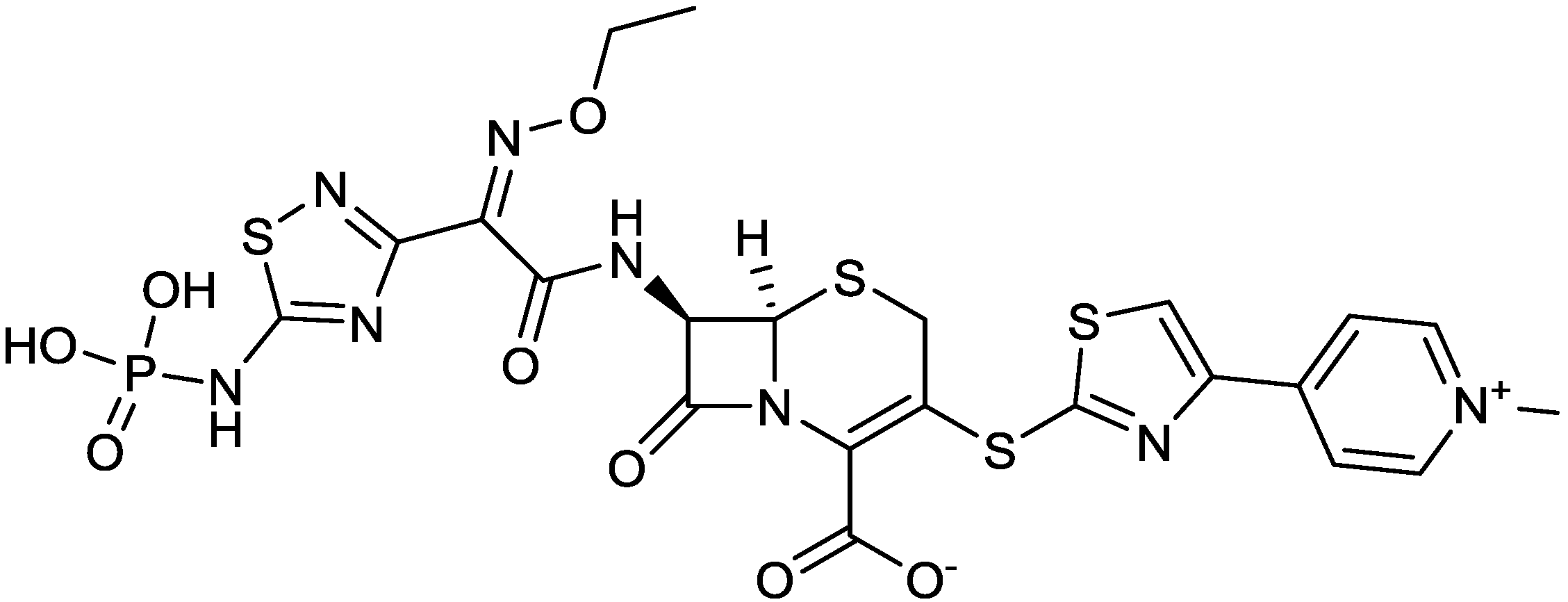
| 2010 | Ceftaroline fosaminyl | 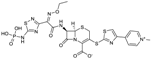 | cephalosporin | G+, G− | Broad spectrum |
| 2011 | Fidaxomycin | 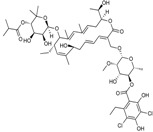 | macrocycle | G+ | C.difficile |
| 2012 | Bedaquiline |  | diarylquinoline | acid-fast bacteria | Mycobacterium tuberculosis |
2.1. β-Lactam and β-Lactamase Inhibitors in Development
| Compounds | Chemical classes | Bacteria profile | Indication (Company) | (Pre)-Clinical phase |
|---|---|---|---|---|
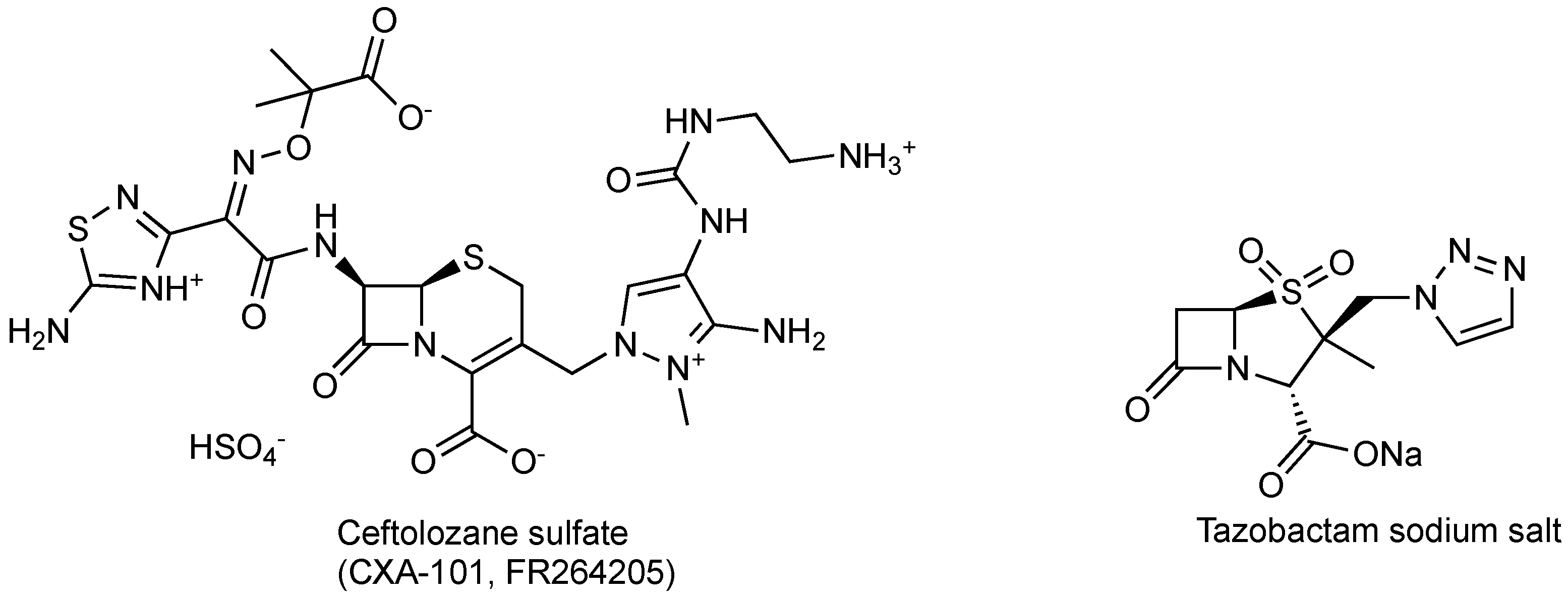
| CXA-201 (ceftolozane/tazobactam) | cephalosporin/sulfone penam | G+, G− | cUTI, cIAI; HABP/VABP (Cubist) | III, Completed II |
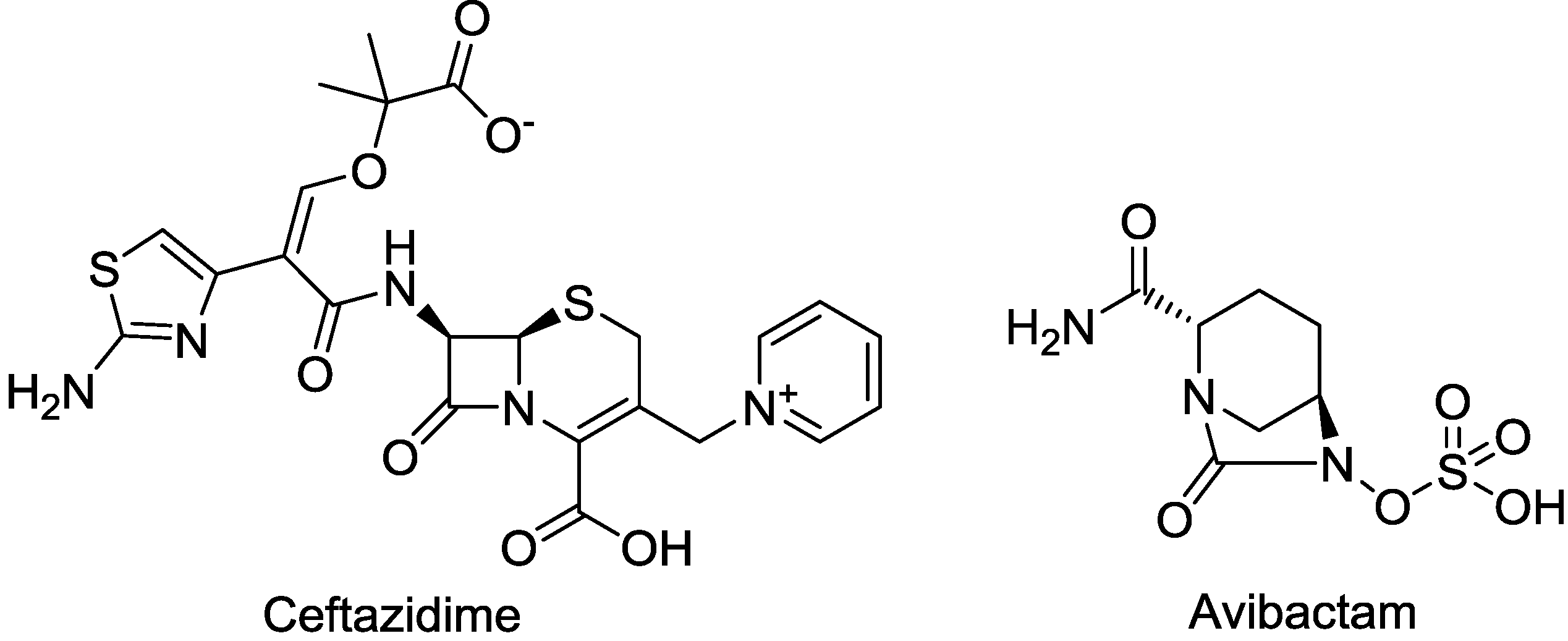
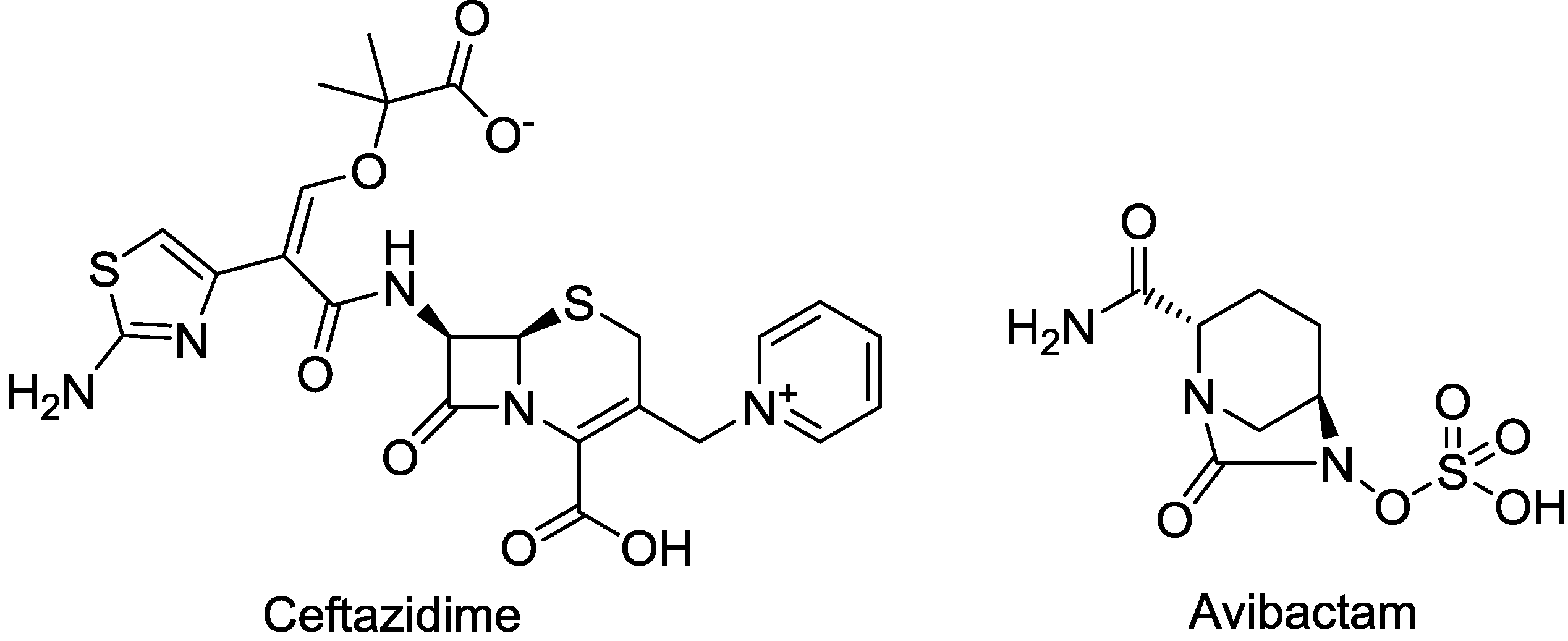
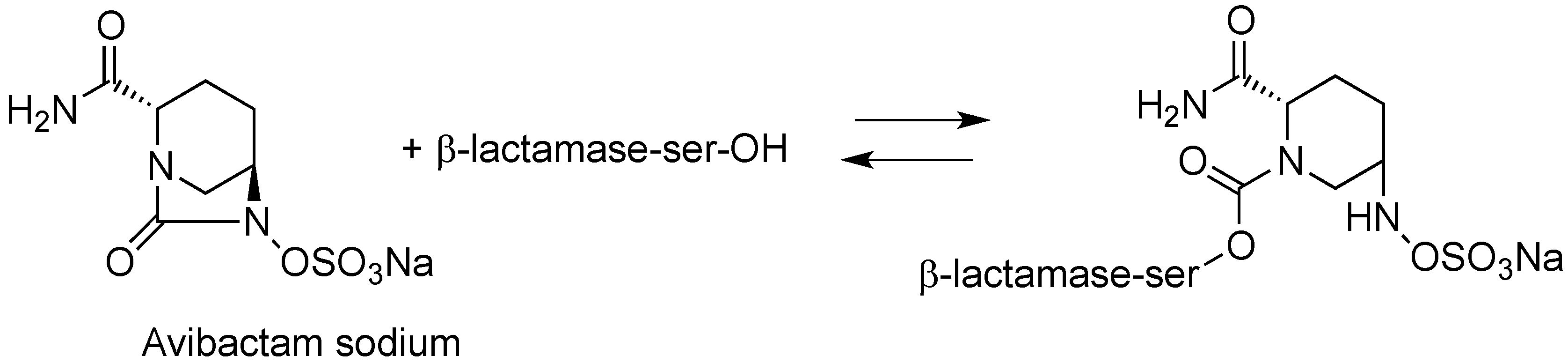
| CAZ104 (ceftazidime/avibactam) | cephalosporin/diazabicyclooctane | G+, G− | cIAI; UTI (AstraZeneca) | III |
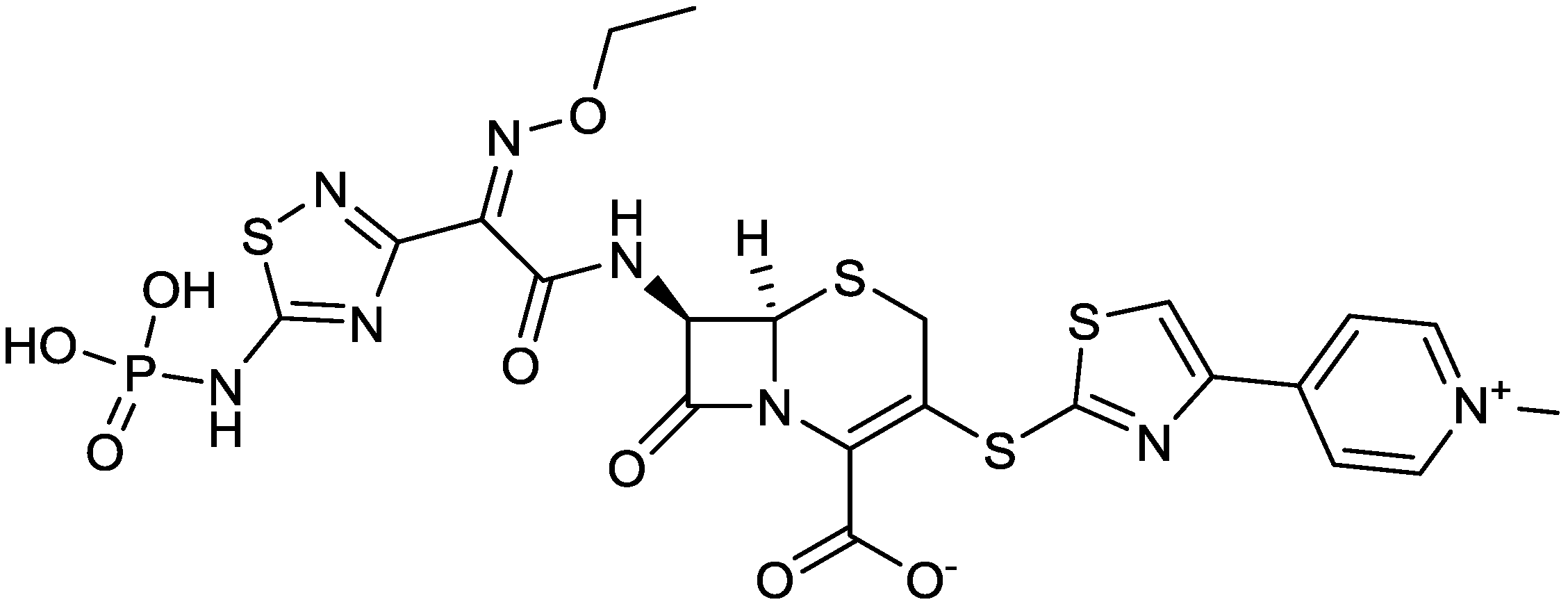
| CXL (ceftaroline/avibactam) | cephalosporin/diazabicyclooctane | G+, G− | MRSA (AstraZeneca) | III |
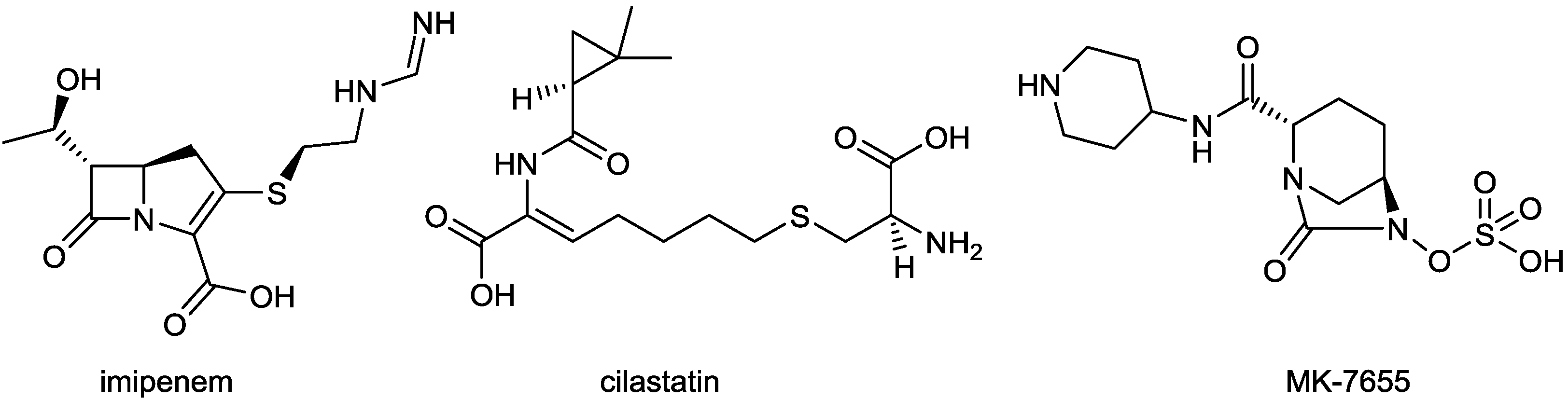
| Imipenem/cilastatin/MK-7655 | carbapenem/DHP-I inhibitor/diazabicyclooctane | G+, G− | UTI and cIAI (Merck) | II |
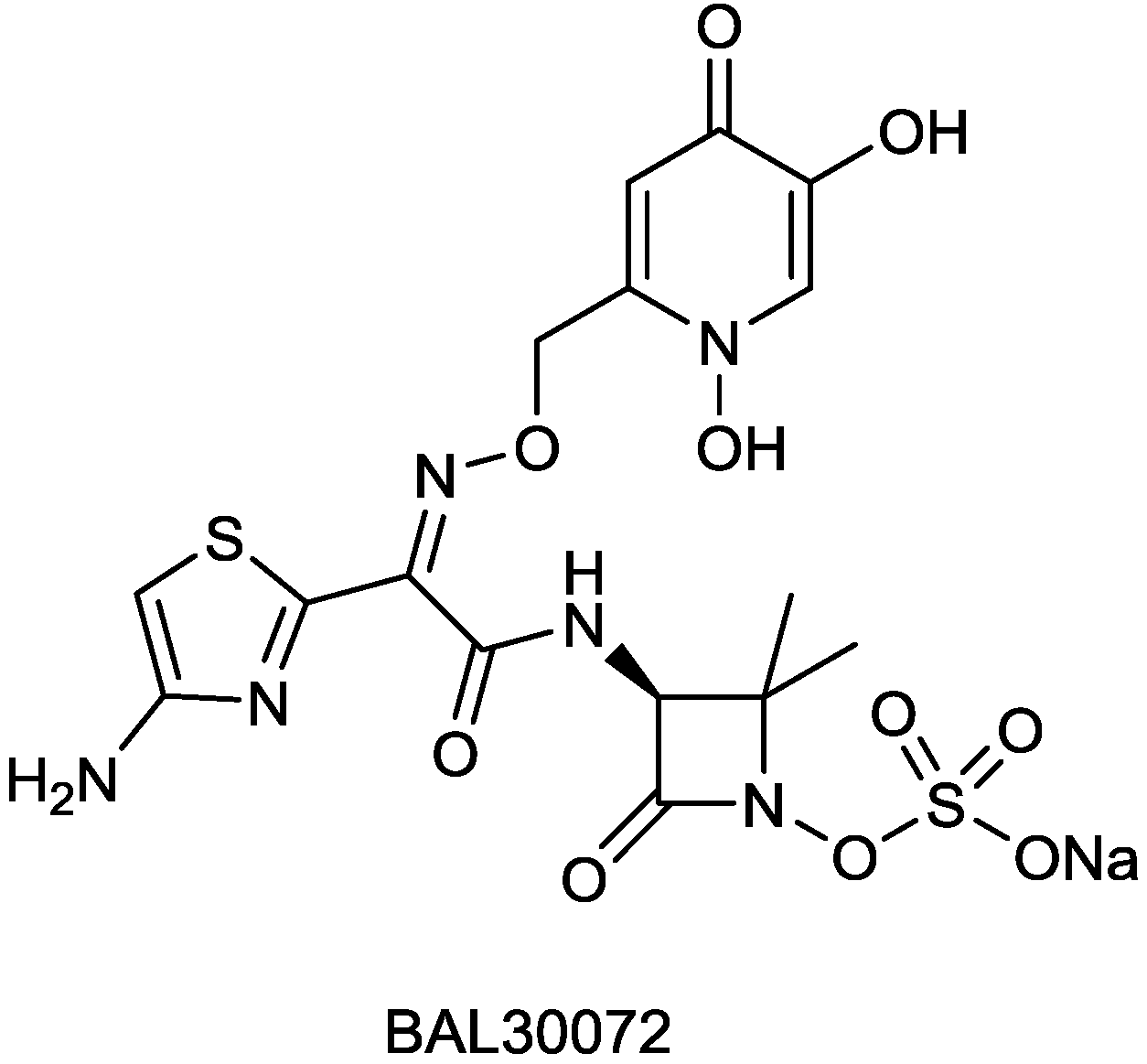
| BAL30072 | monobactam | G+, G− | Gram-negative (Basilea) | I |
| S-649266 (GSK-2696266) | cephalosporin | G+, G− | Gram-negative infections (Shionogi/GSK) | I |
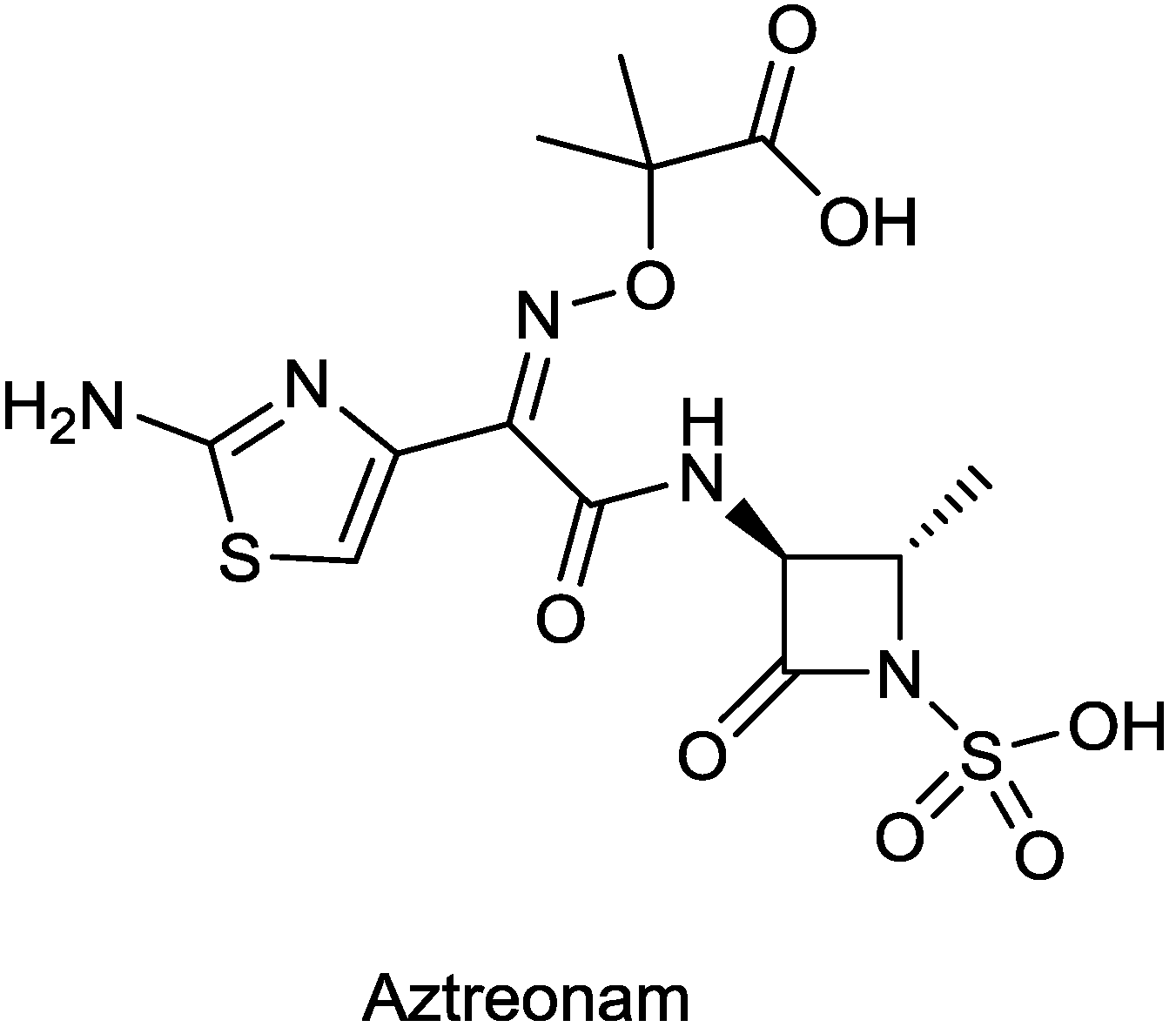
| ATM-AVI (aztreonam/avibactam) | monobactam/diazabicyclooctane | G+, G− | Metallo β-lactamase producers (AstraZeneca) | I |
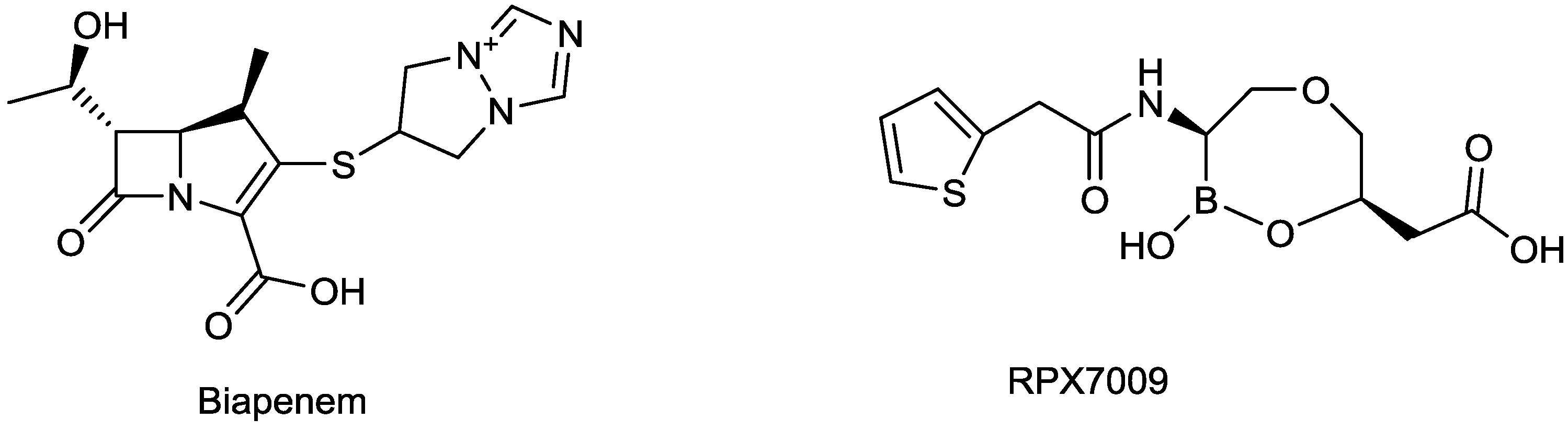
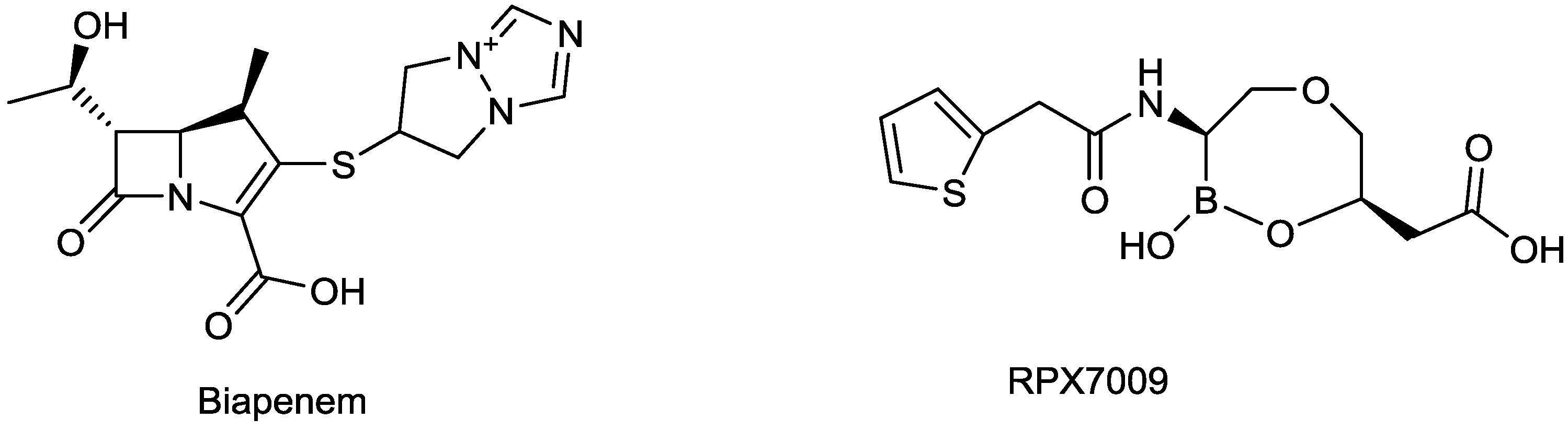
| Carbavance (biapenem/RPX7009) | carbapenem/Boronic acid | G+, G− | KPC, CRE (The Medicines Company, previously Rempex) | II |
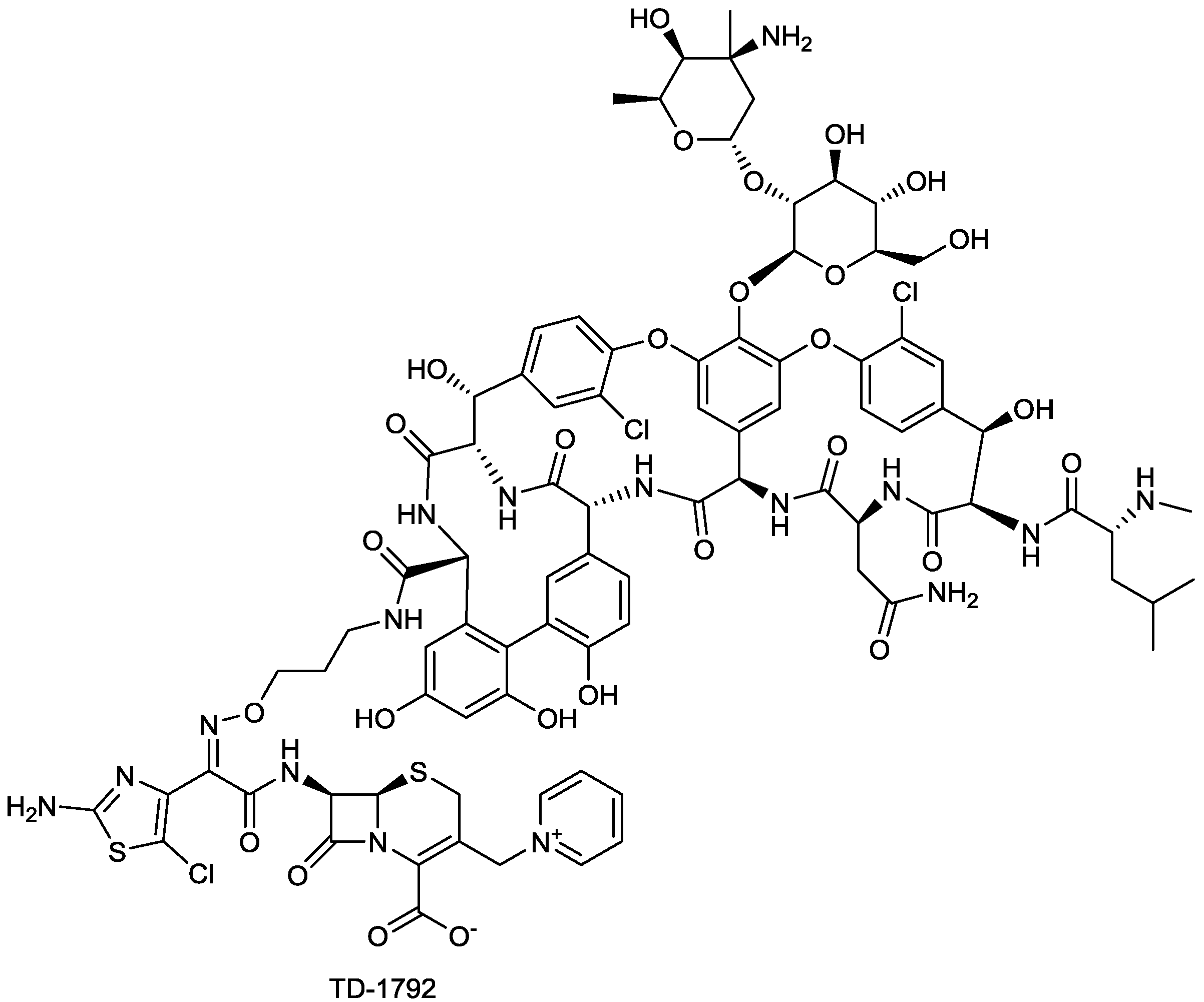
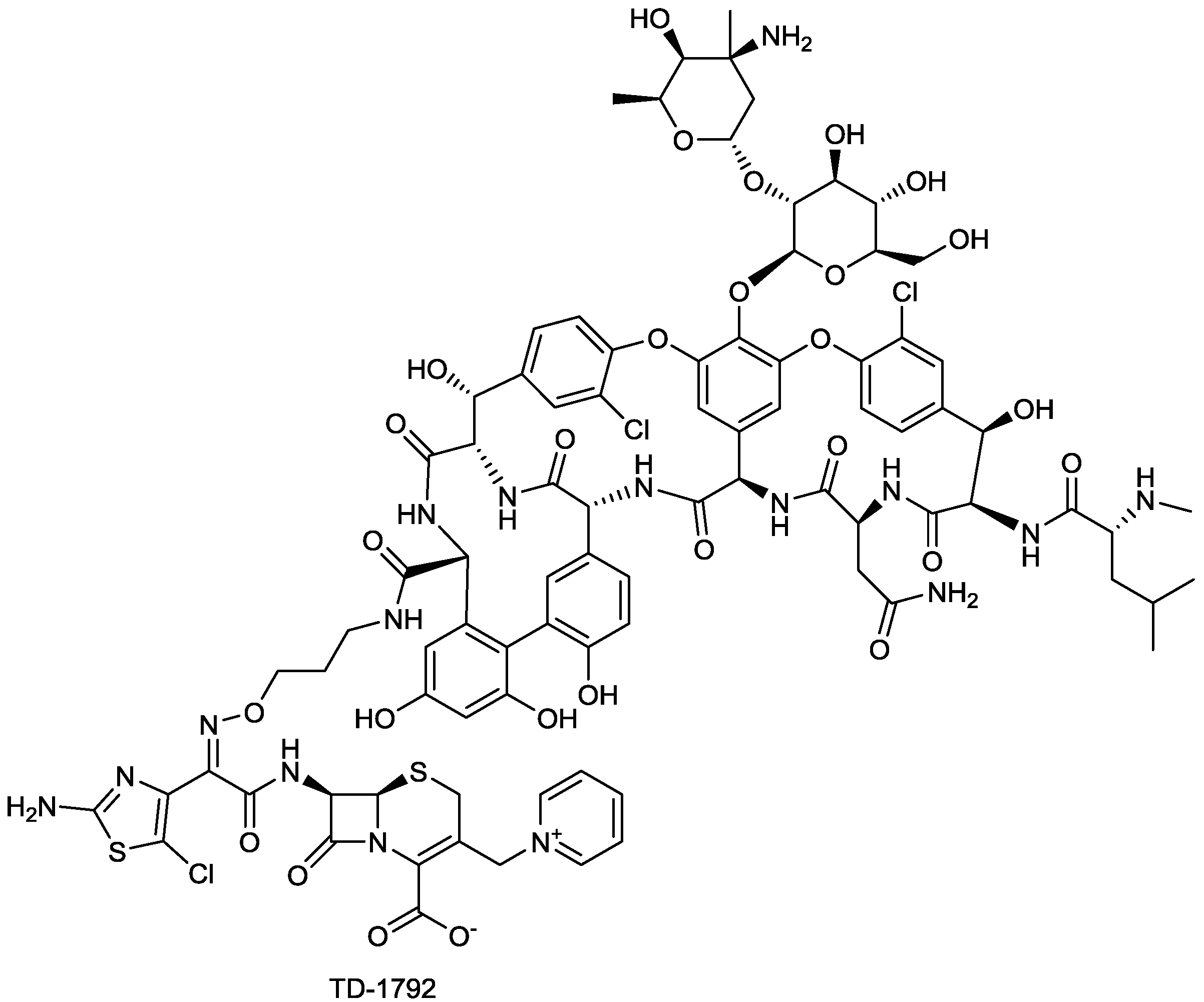
| TD-1792 | glycopeptide-cephalosporin hybrid | G+ | (Theravance) | II-III |
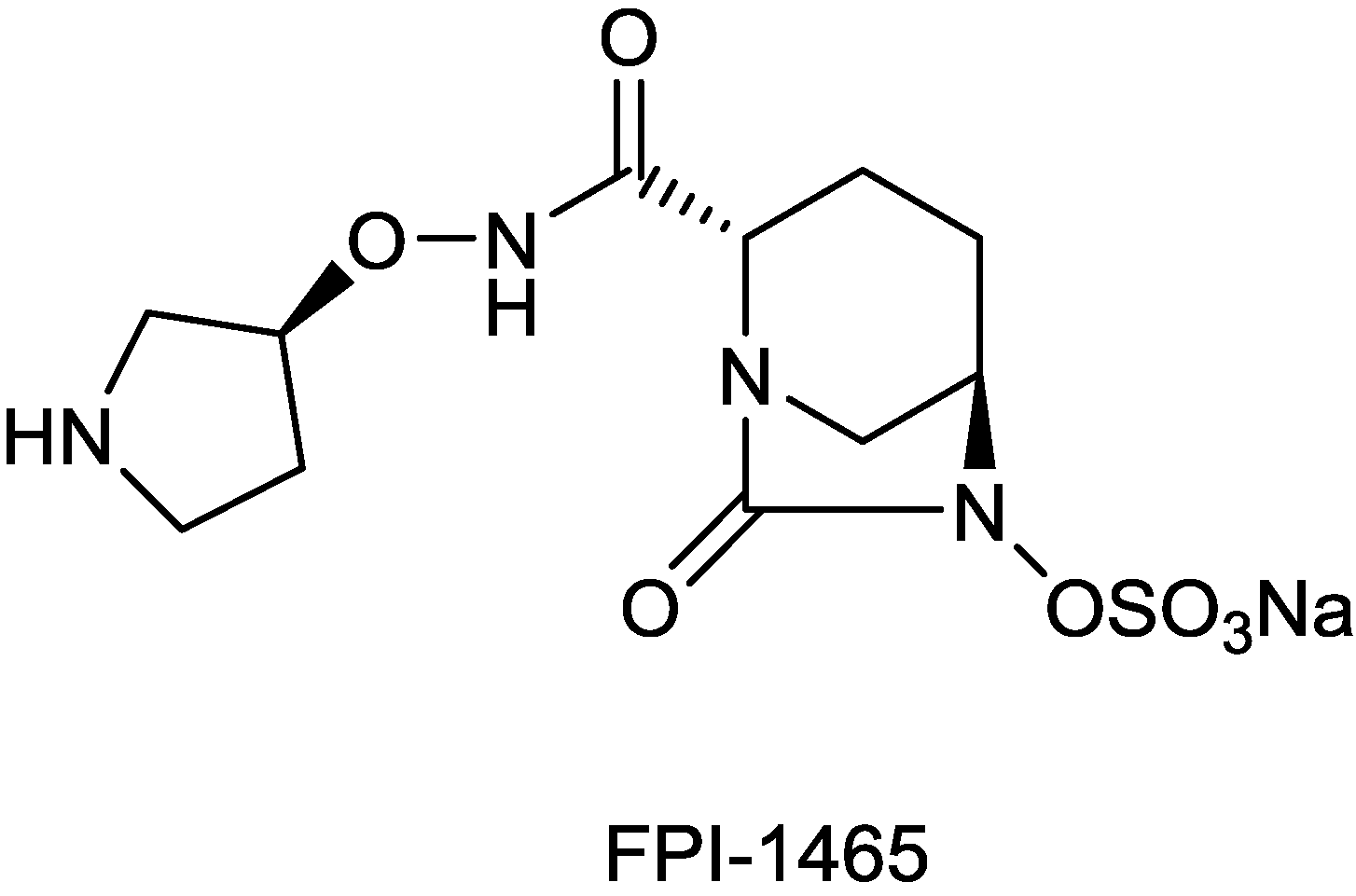
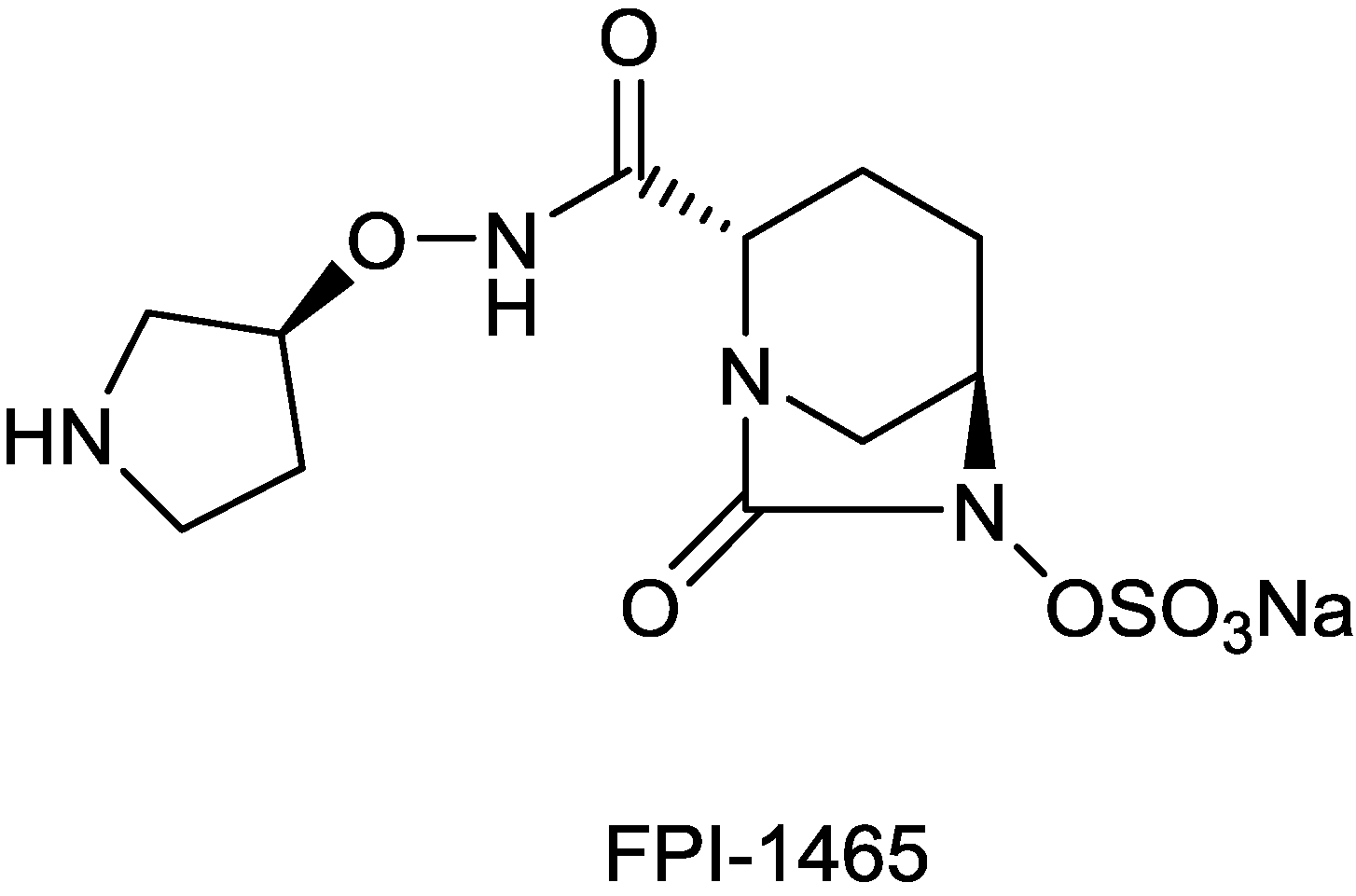
| FPI-1465 | diazabicyclooctane | Fedora | Discovery | |
| Novel β-lactamase inhibitors | sulphonamides | (John Hopkins Un.) | Discovery | |
| Novel β-lactamase inhibitors | Boronic acid | (John Hopkins Un.) | Discovery | |
| Novel β-lactamase inhibitors | Boronic acid | (Therabor Pharmaceuticals) | Discovery | |
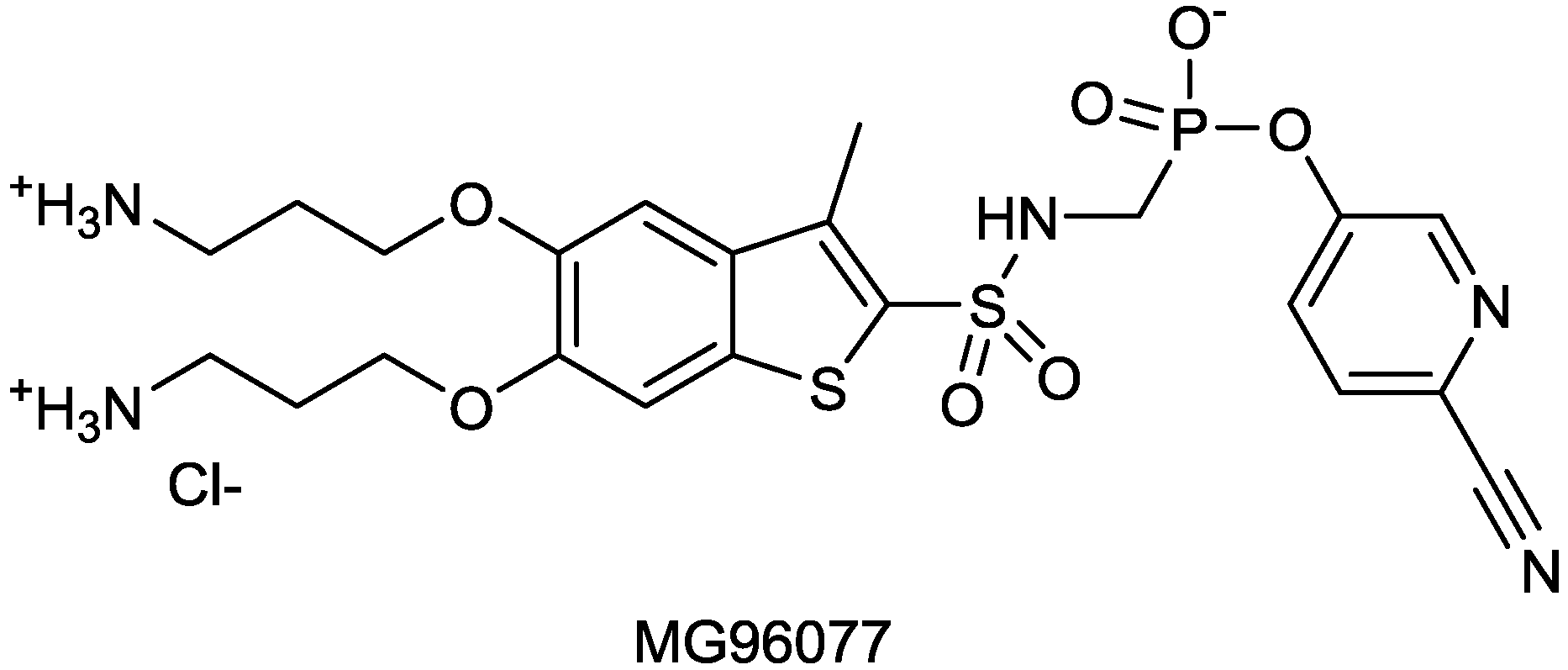
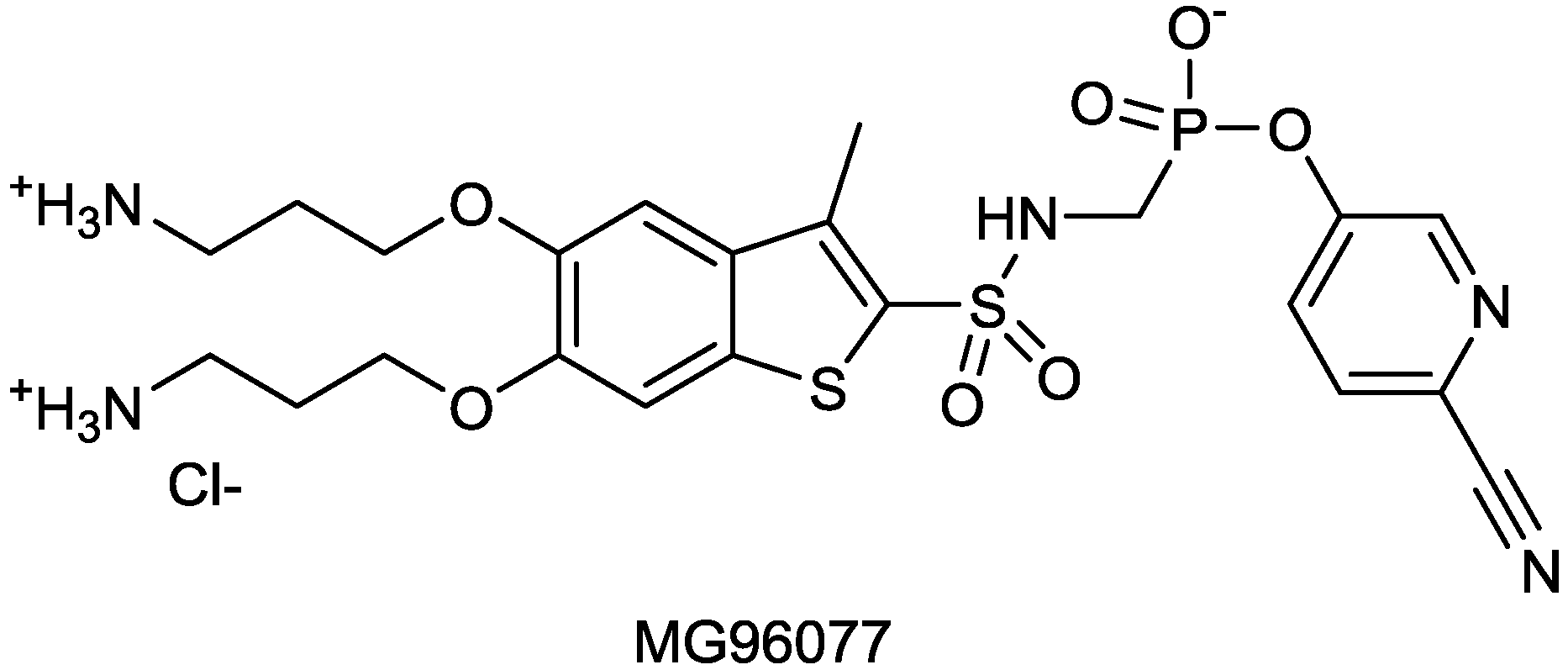
| MG96077 | phosphonate-based β-lactamase inhibitor | (Mirati Therapeutics) | Discovery | |
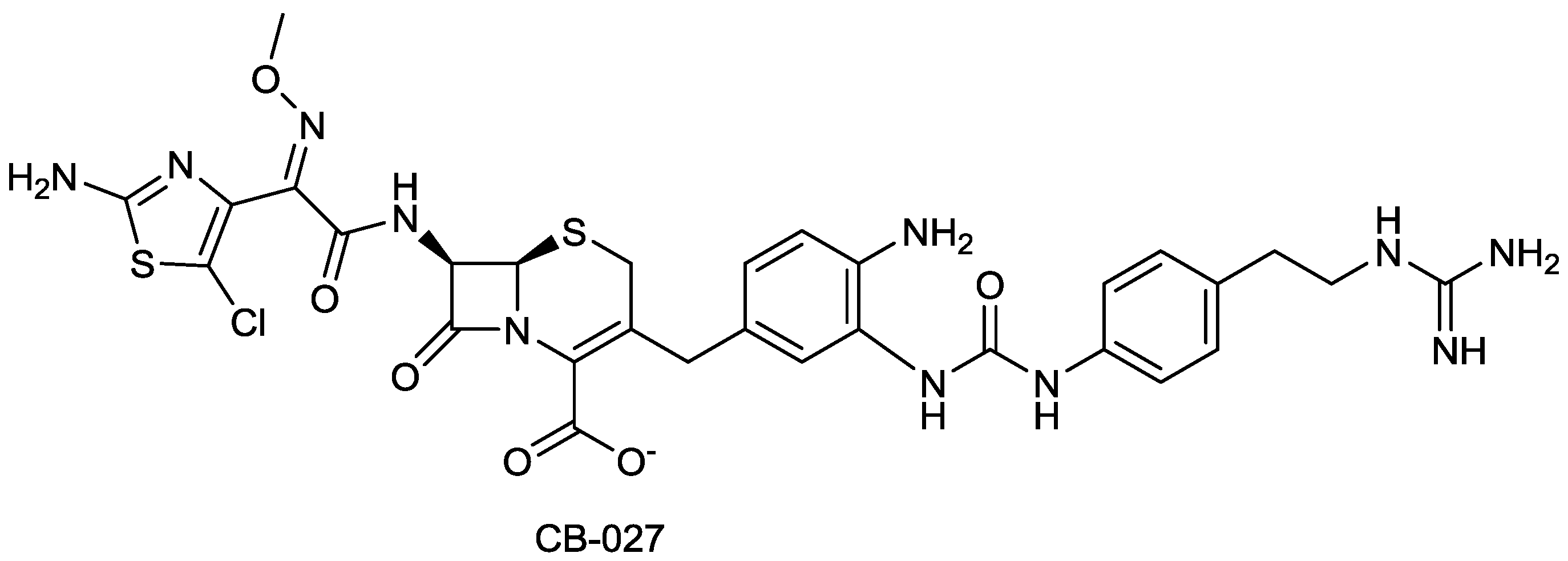
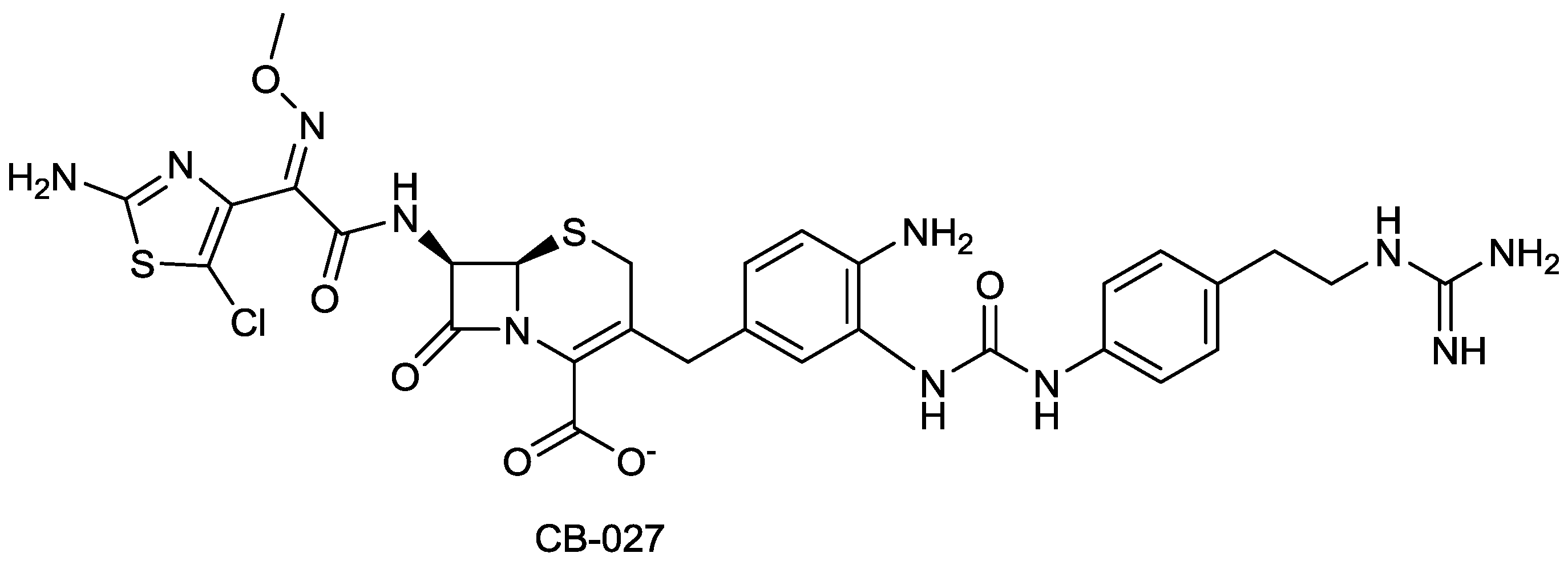
| CB-027 | cephalosporin | G+, G− | MRSA, P. aeruginosa (Cubist) | Discovery |
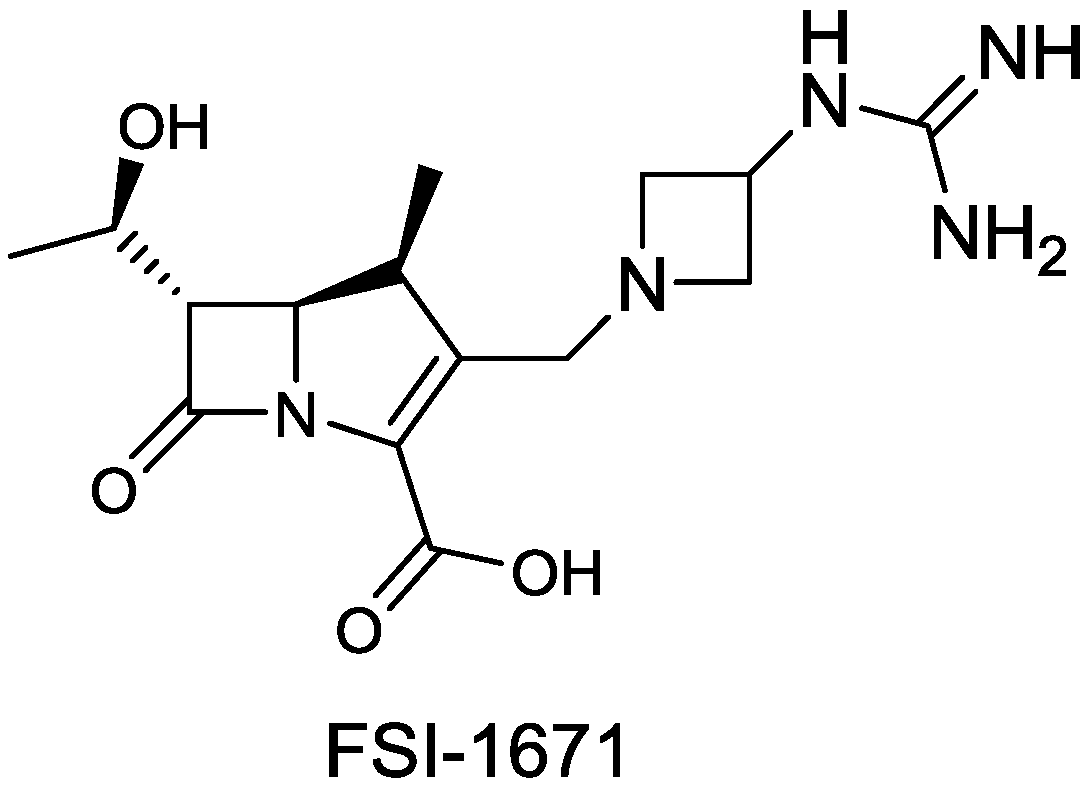
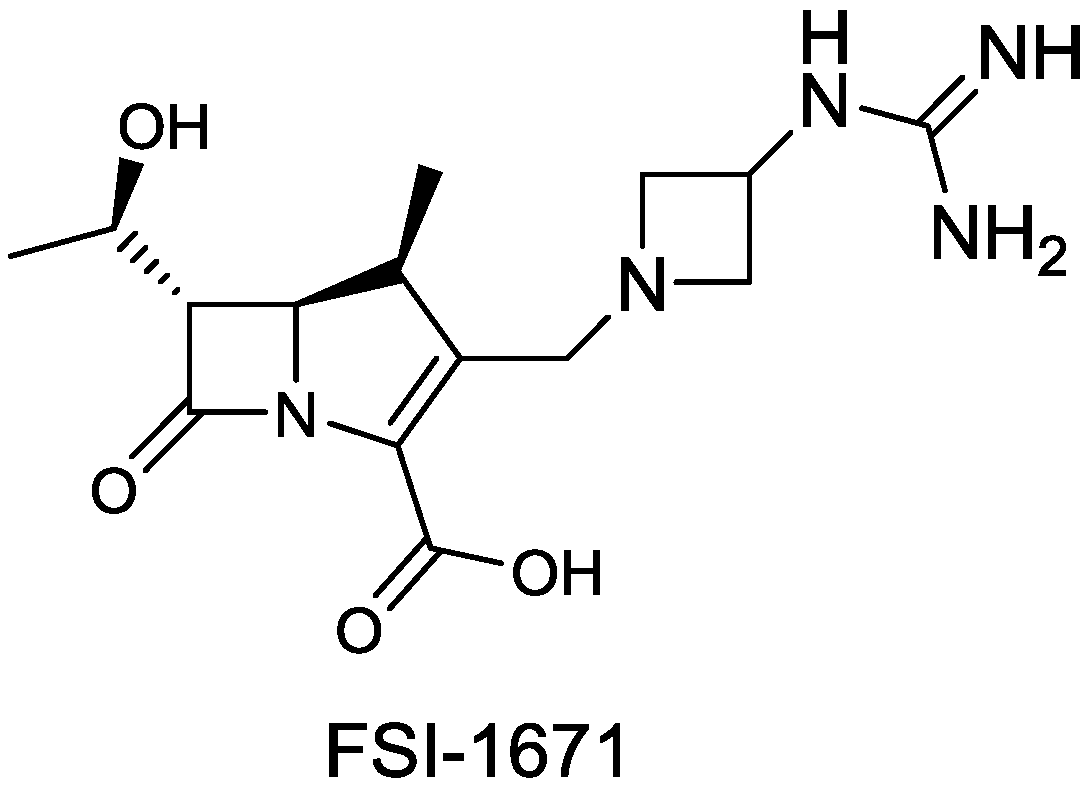
| FSI-1671 | carbapenem | G− | (FOB Synthesis Inc.) | Discovery |
2.1.1. CXA-201
2.1.2. CAZ104

2.1.3. CPT (Ceftaroline)
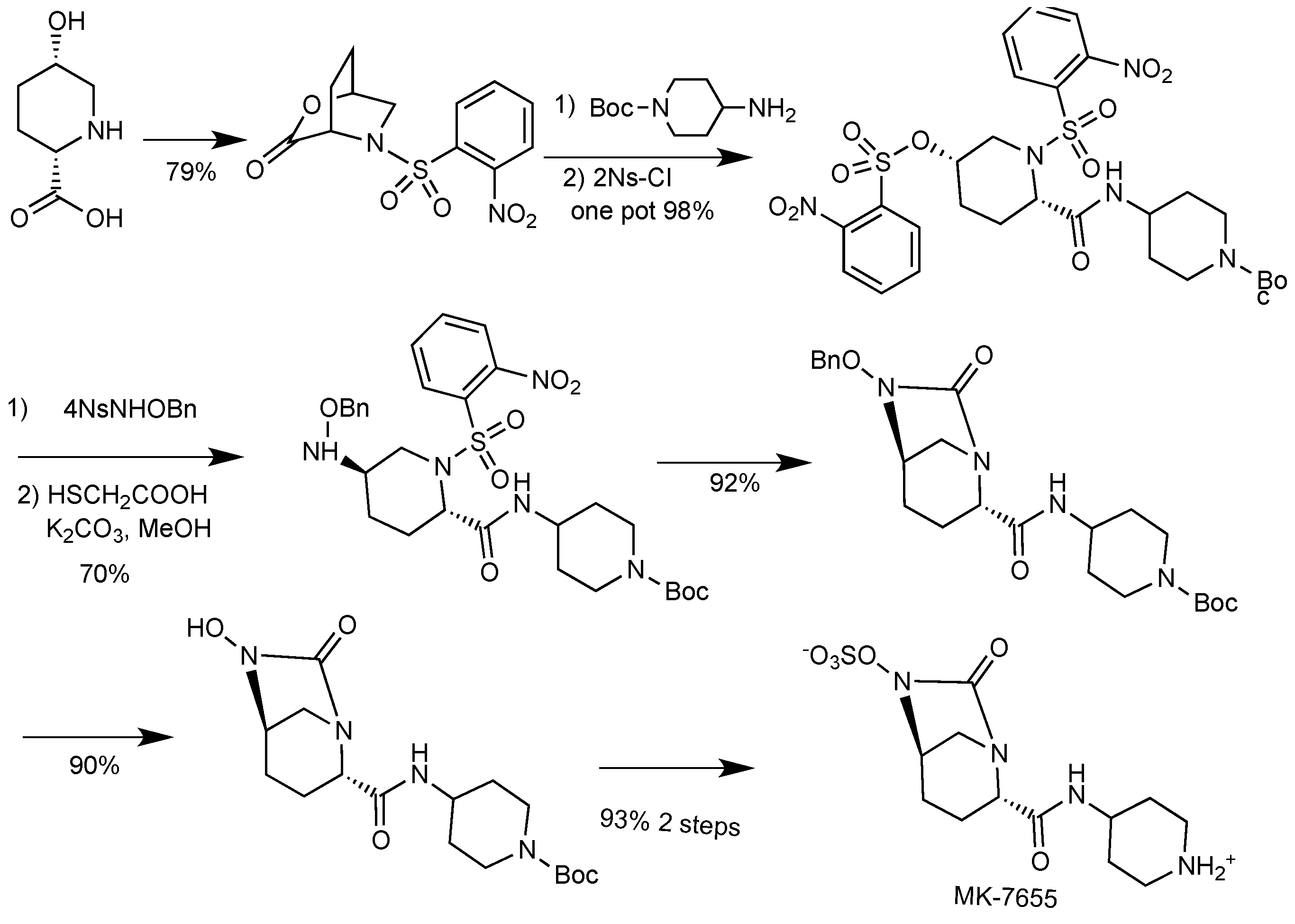
2.1.4. Imipenem/Cilastatin/MK-7655
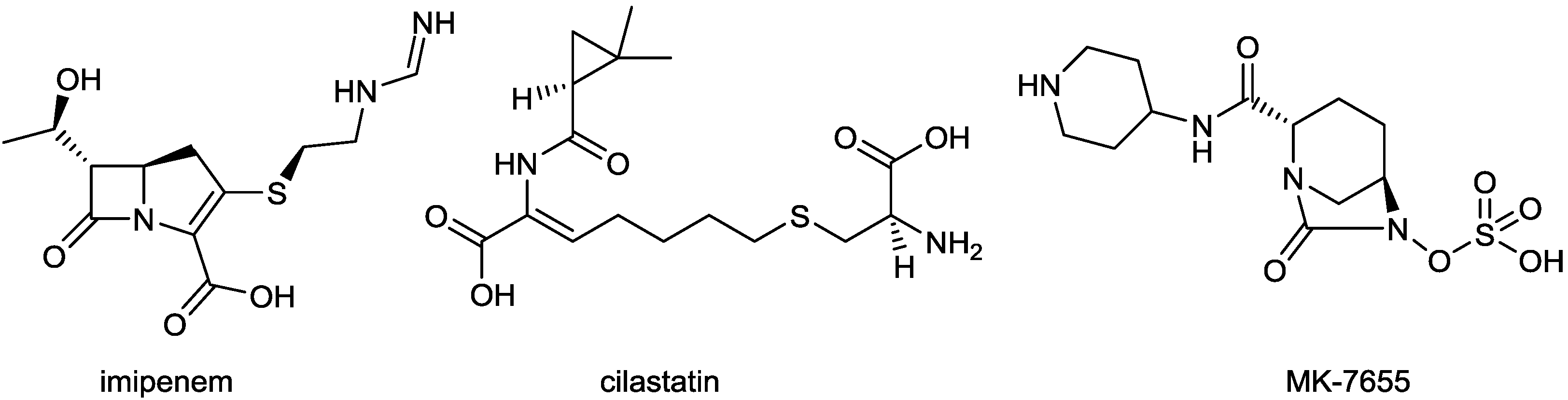
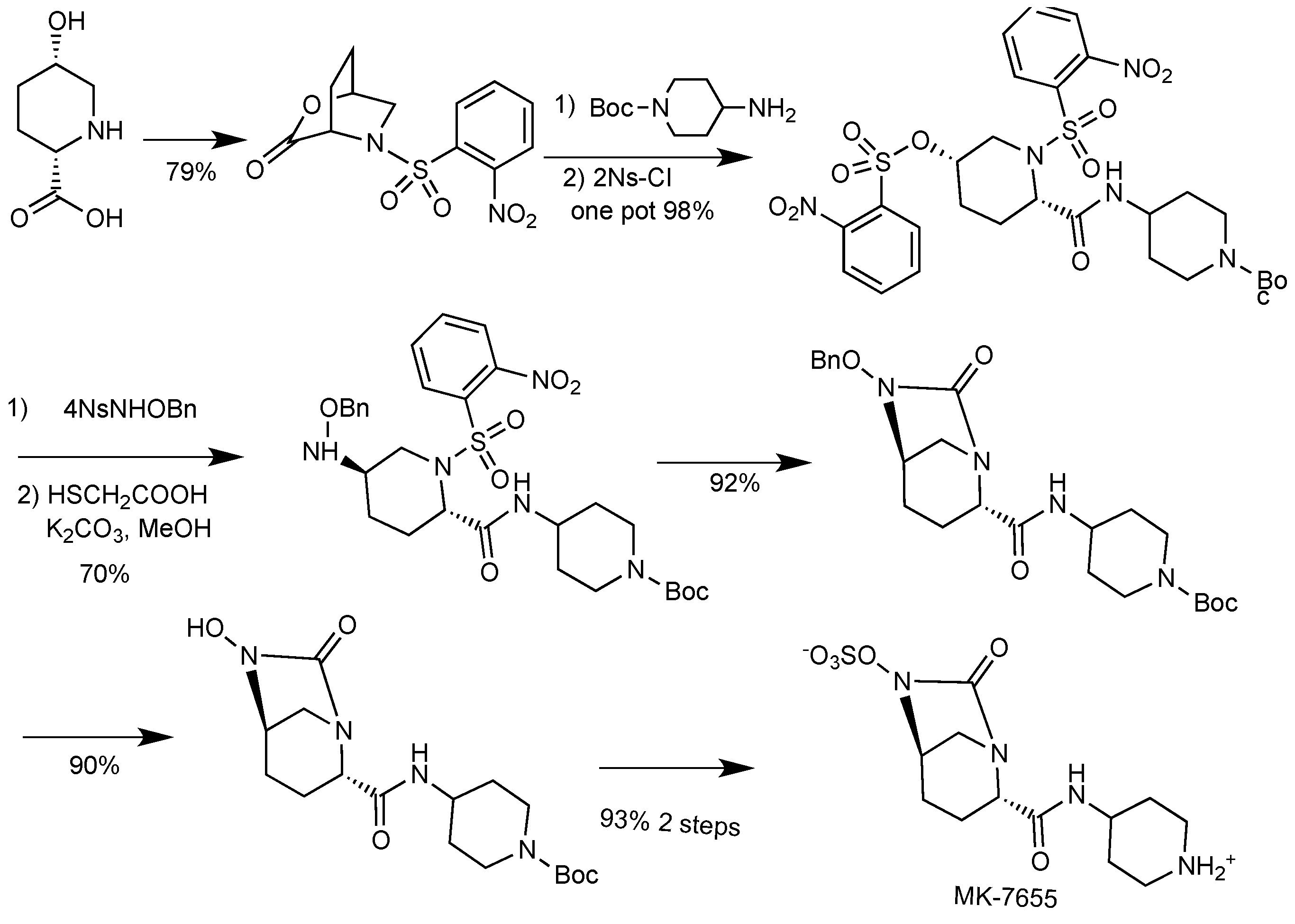
2.1.5. BAL30072
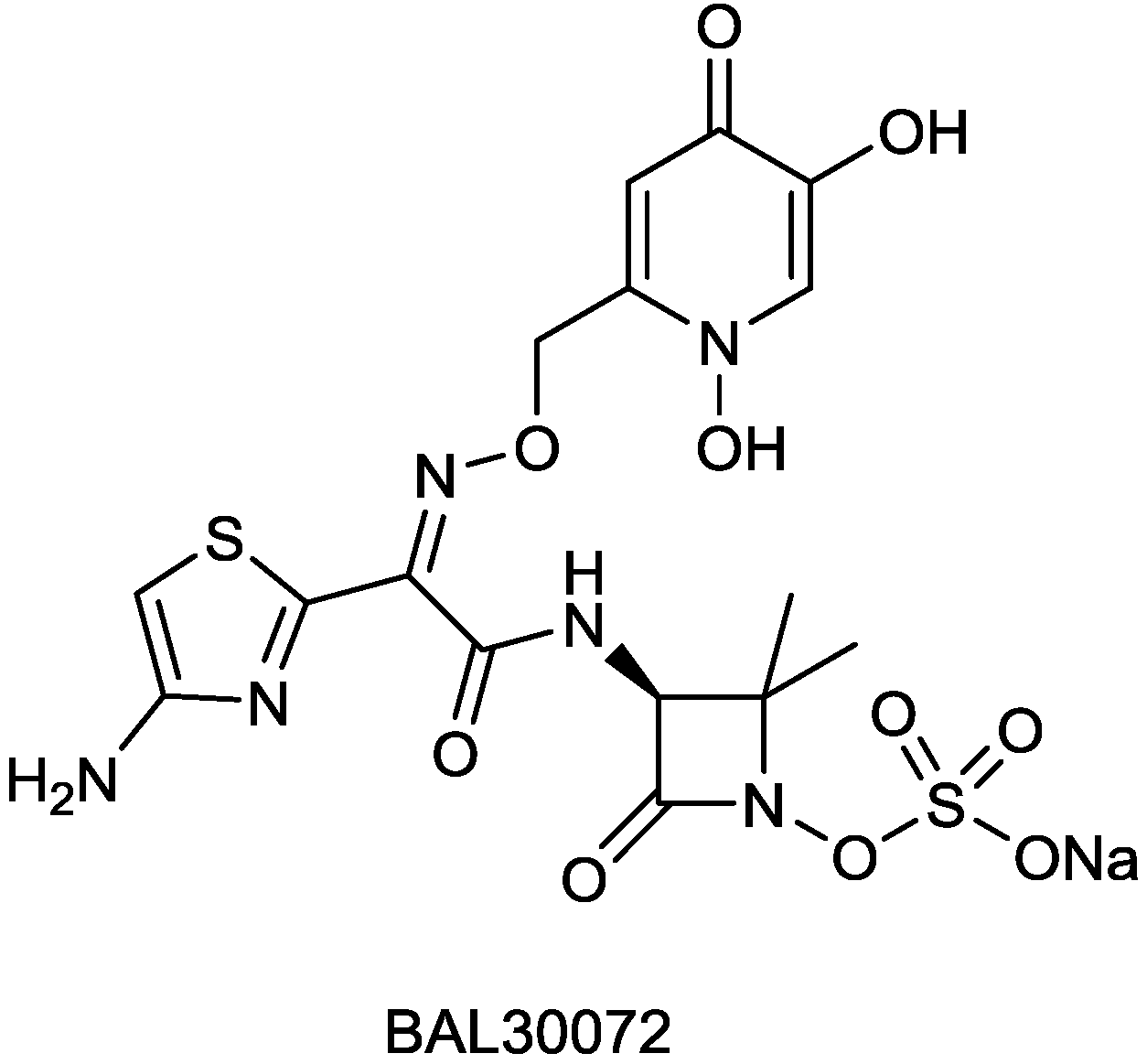
2.1.6. S-649266 (GSK-2696266)
2.1.7. ATM-AVI
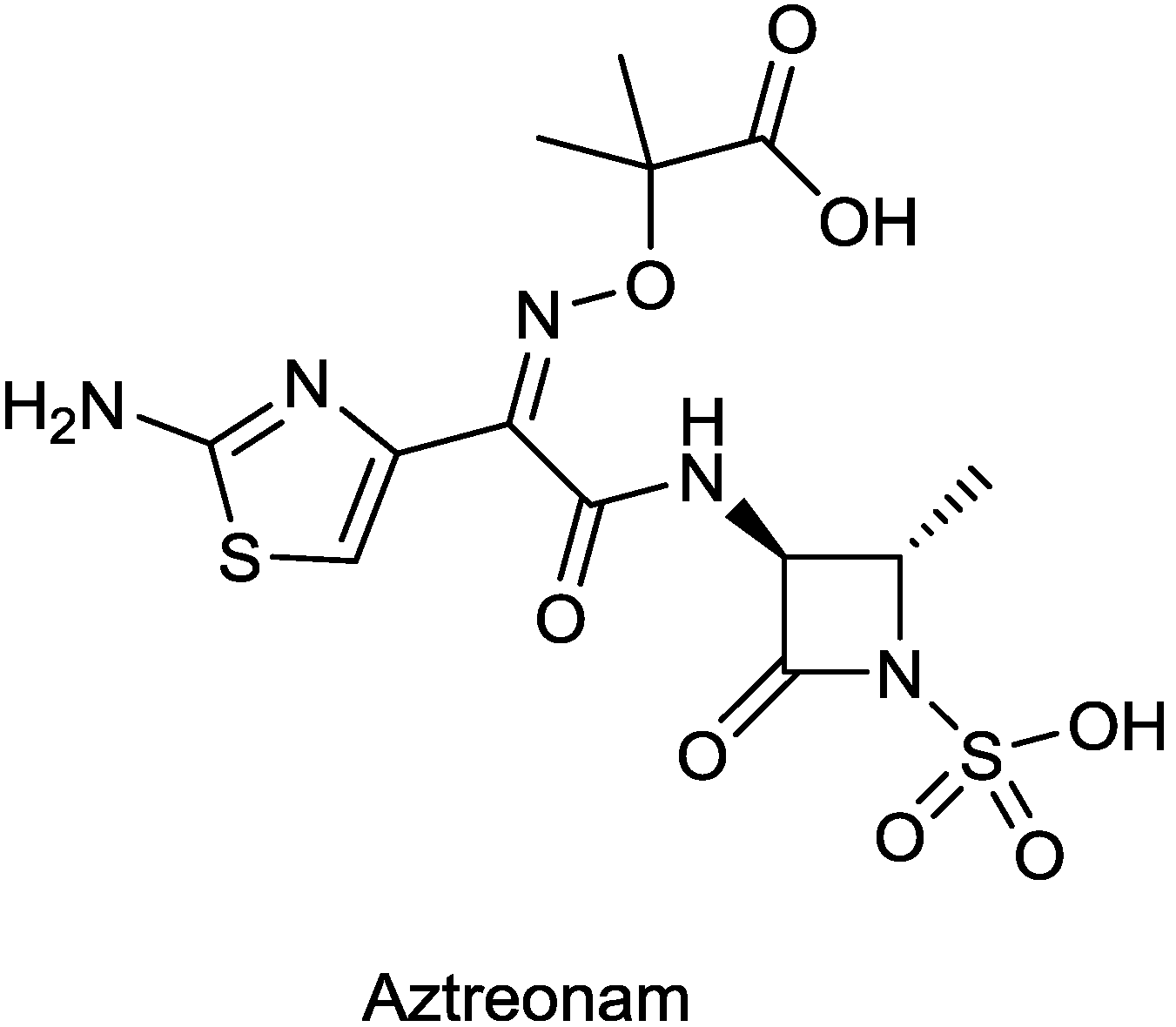
2.1.8. Biapenem/RPX7009
2.1.9. TD-1792
2.1.10. FPI-1465
2.1.11. Sulfonamido Boronic Acids
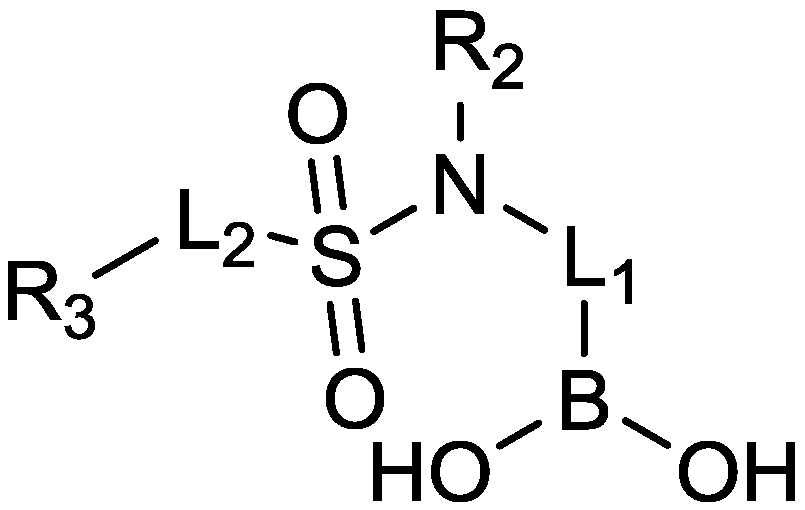
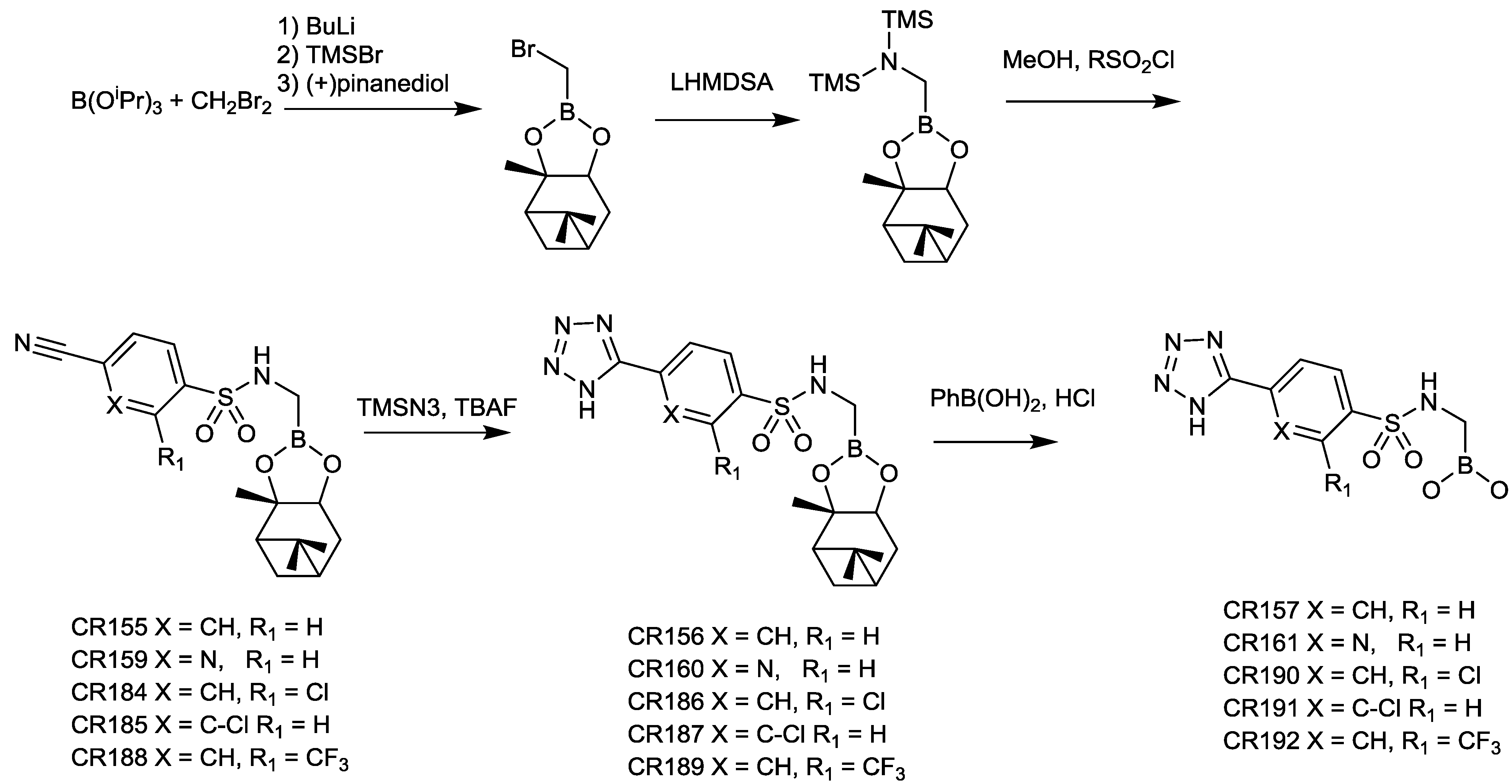
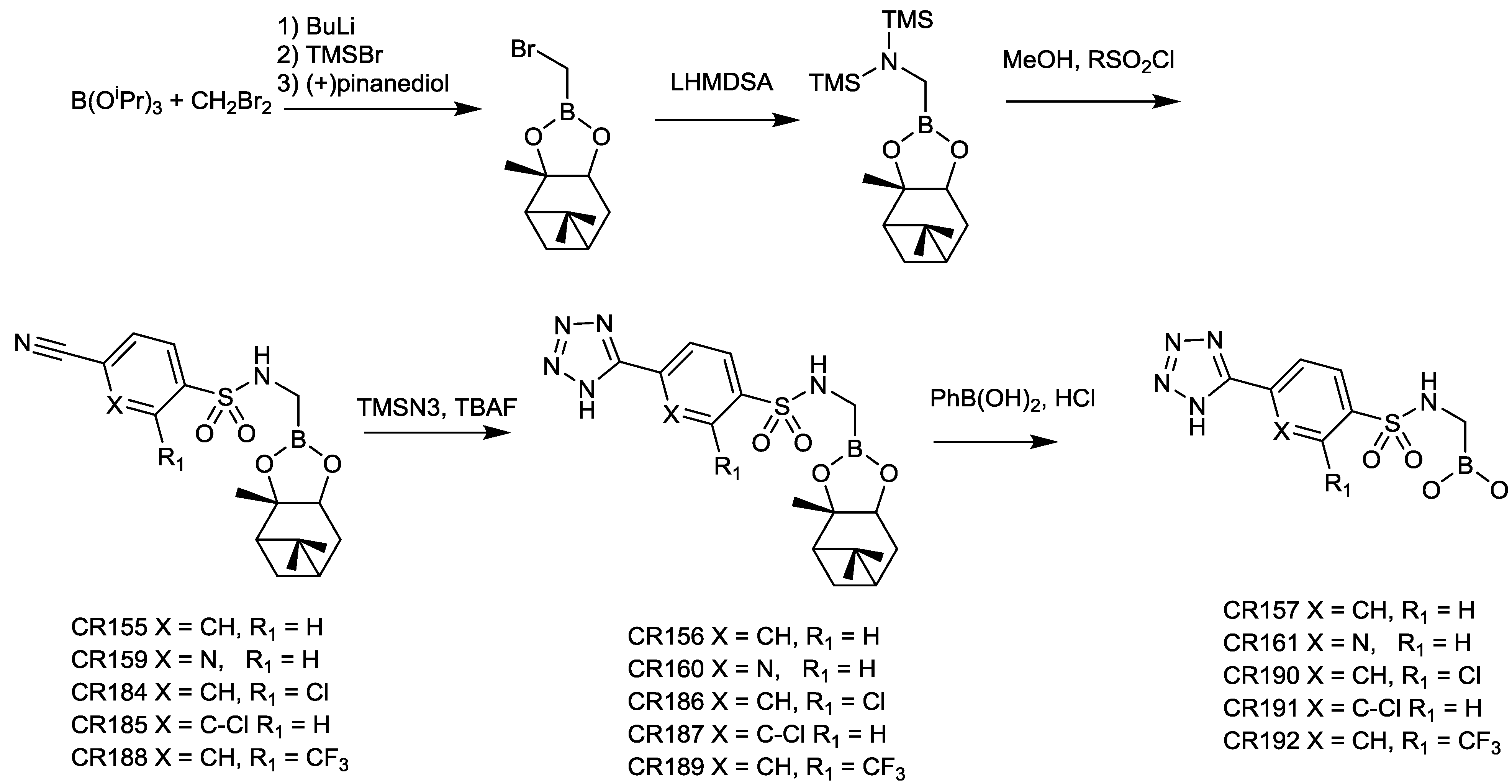
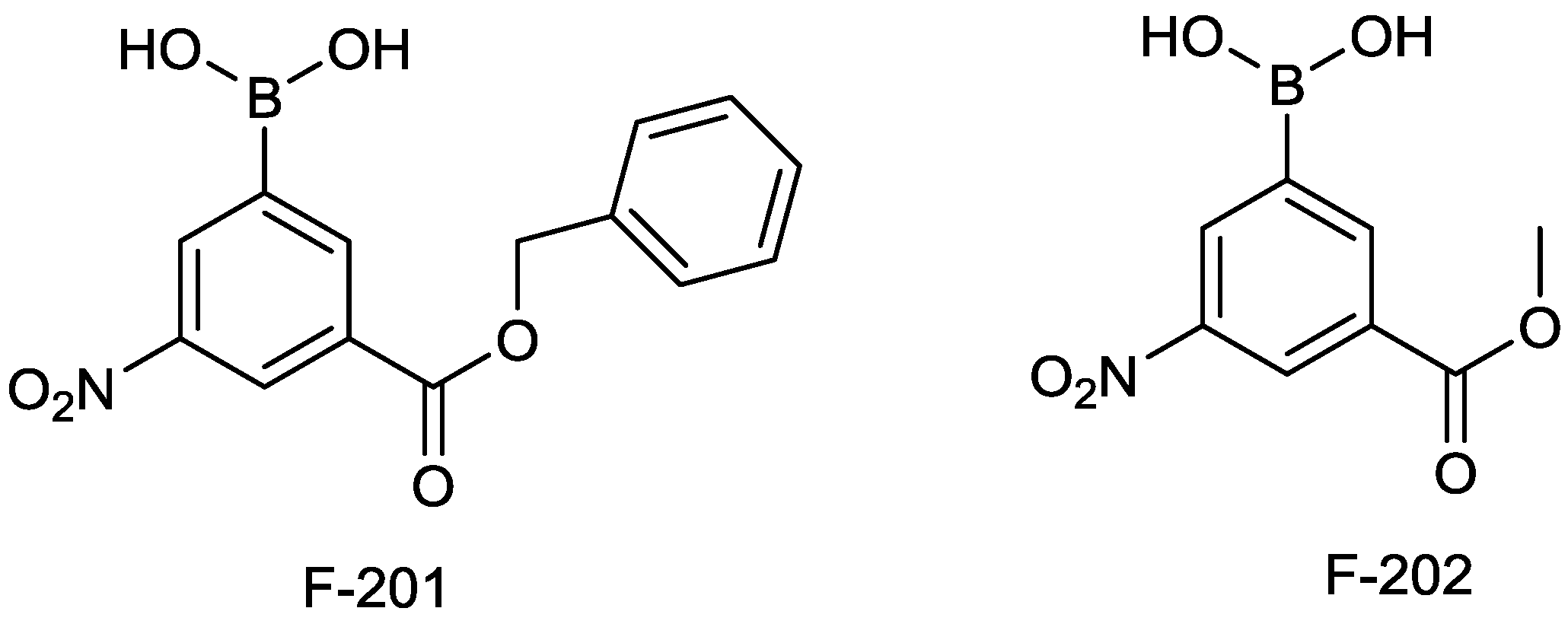
2.1.12. Arylboronic Acids
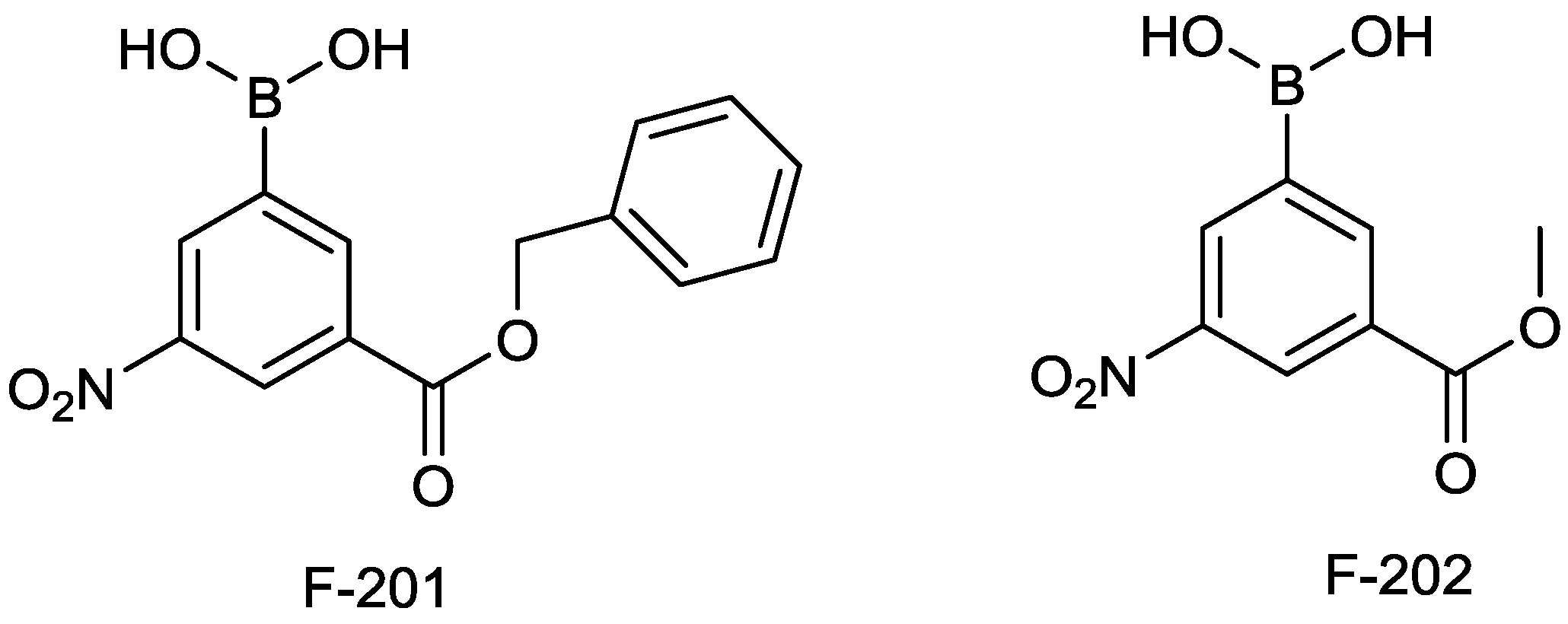
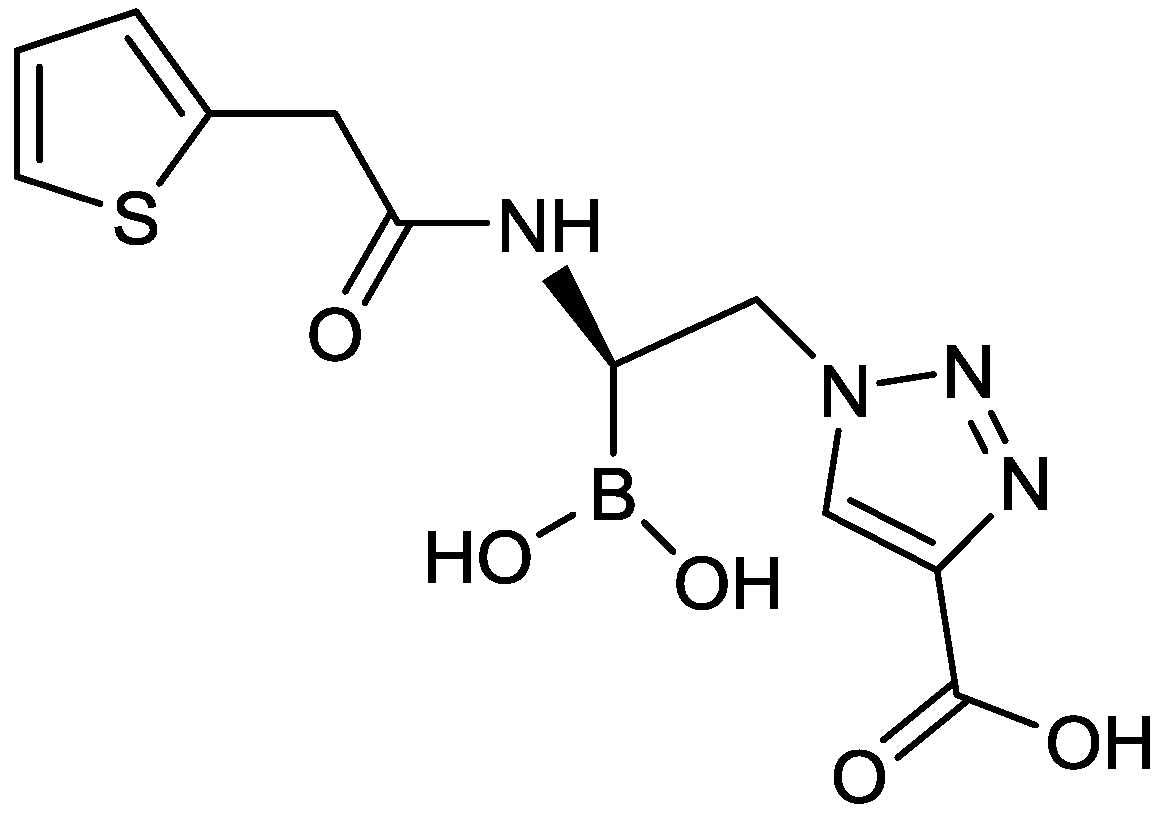
2.1.13. Triazole-Substituted Boronic Acids
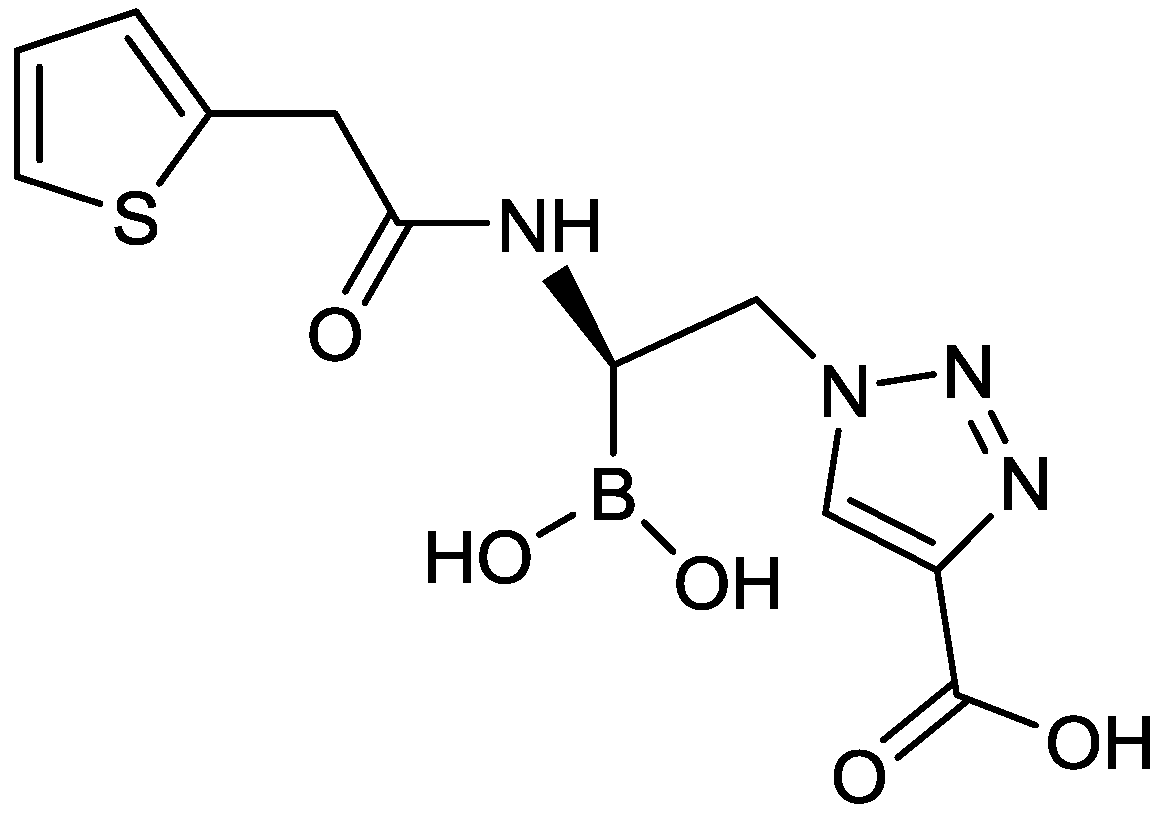
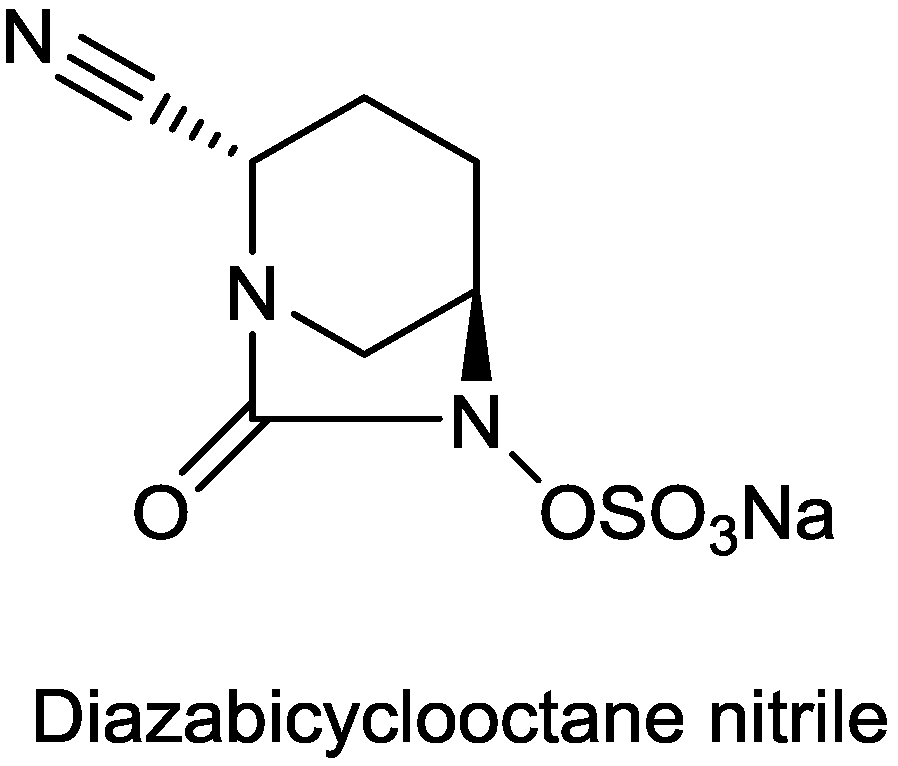
2.1.14. Diazabicyclooctane Nitrile
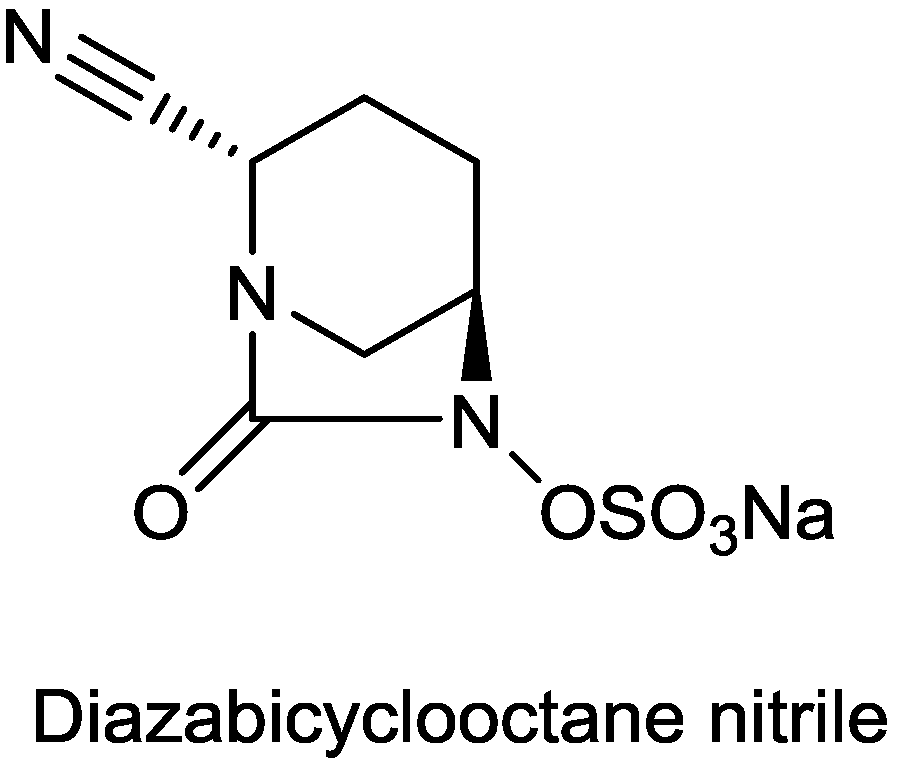
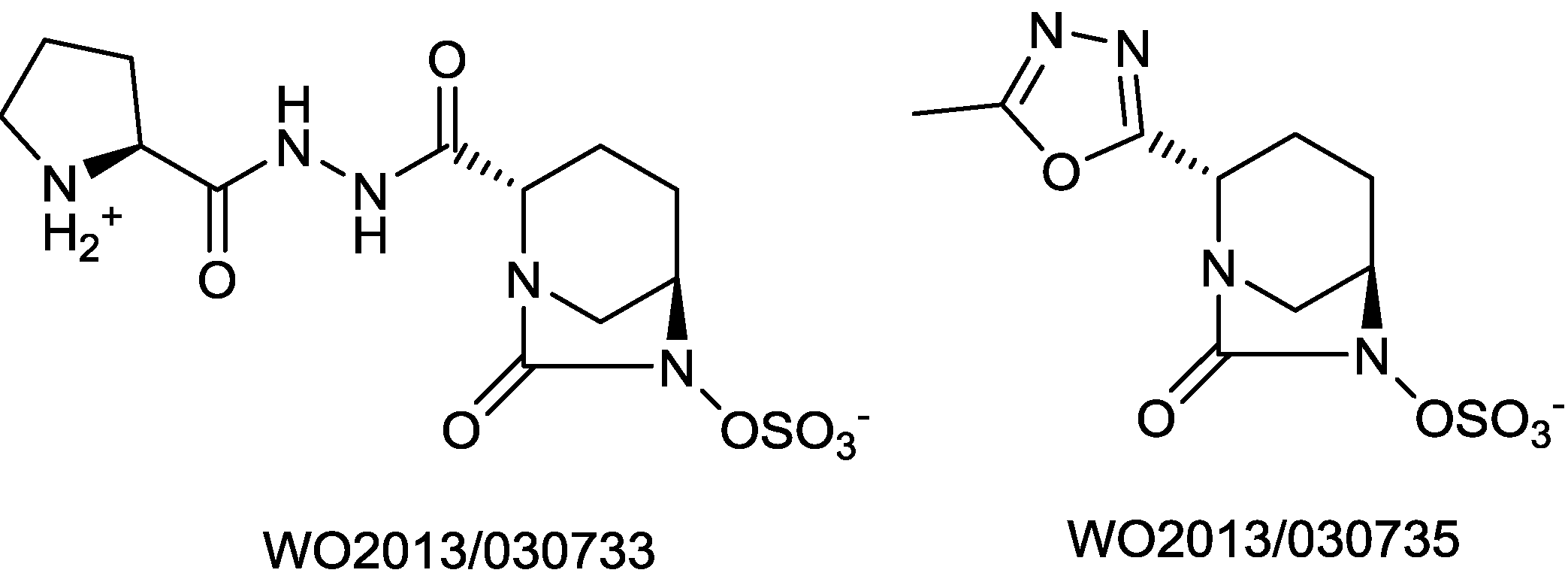
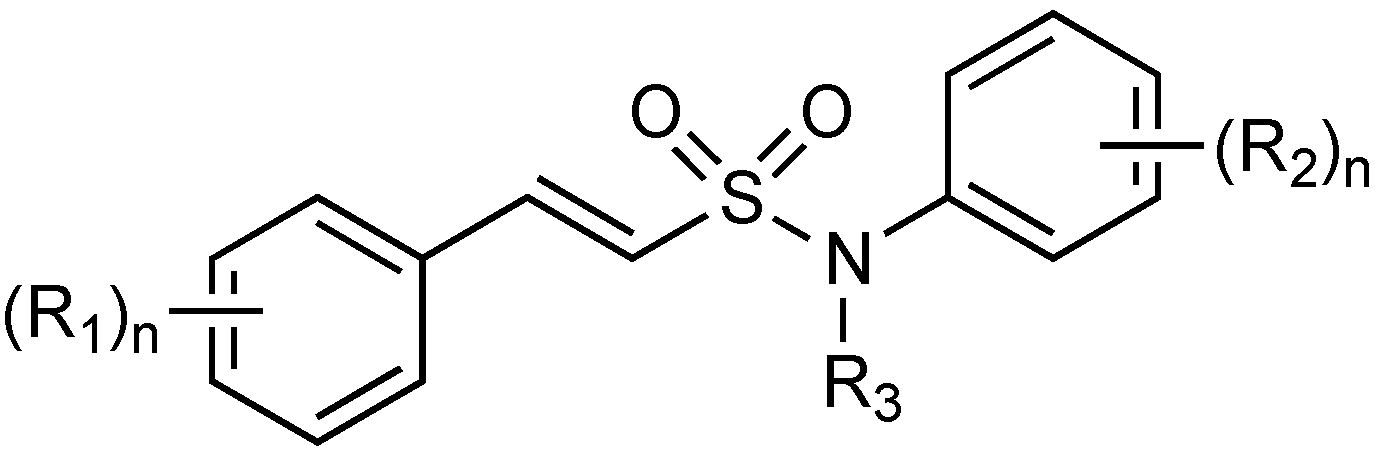
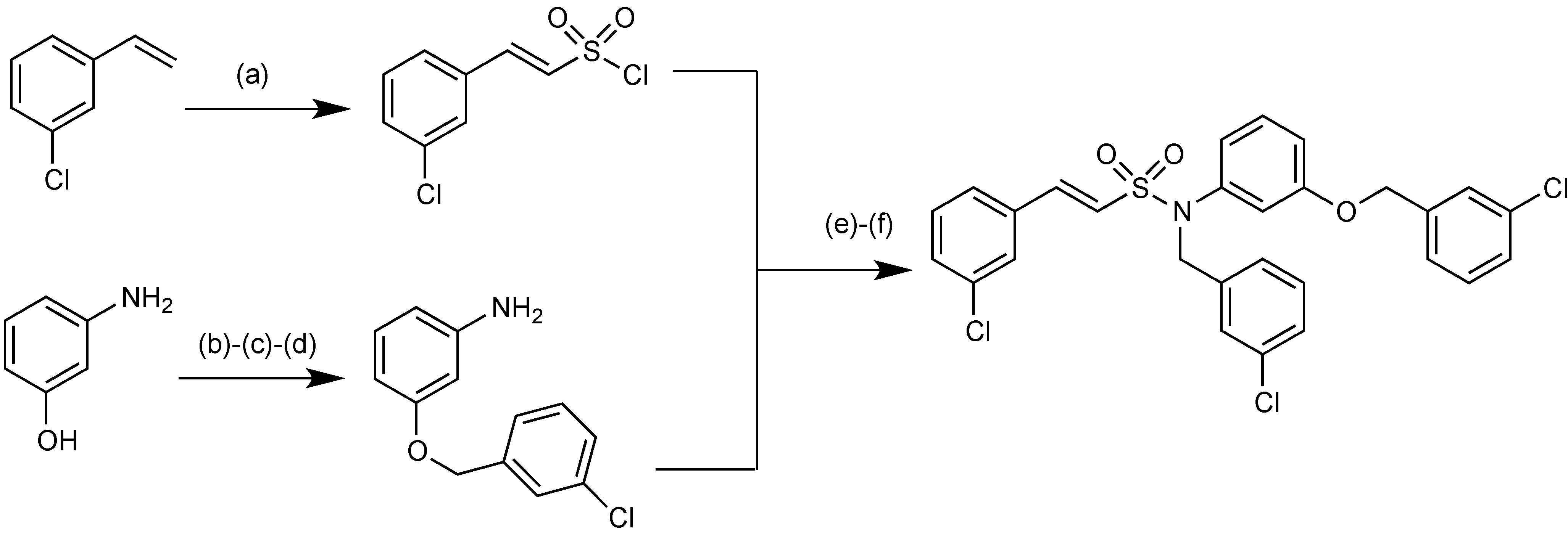
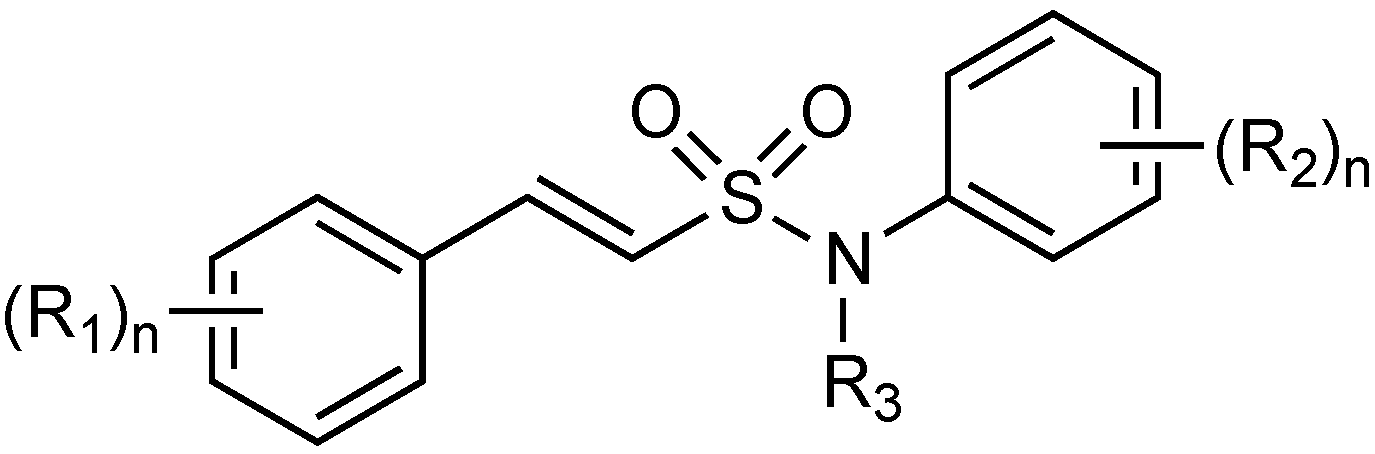
2.1.15. Sulfonamido β-lactamase Inhibitors: John Hopkins University
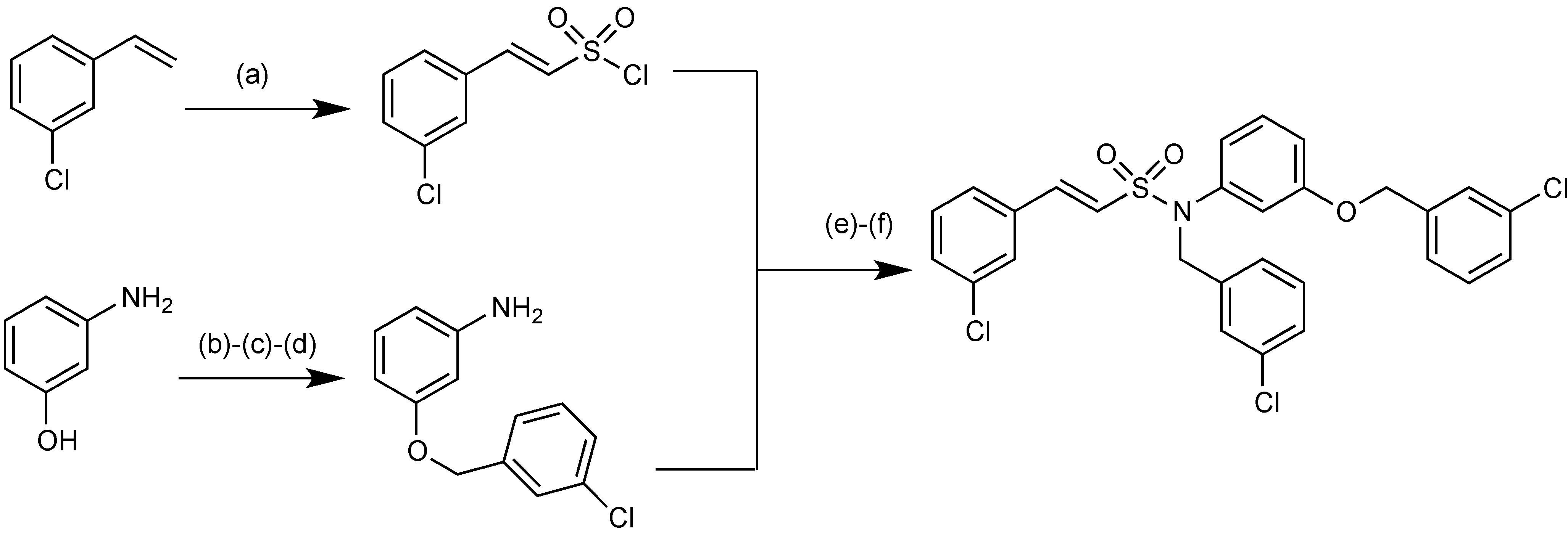
2.1.16. MG96077
2.1.17. CB-027
2.1.18. FSI-1671
2.1.19. Antibacterial Diazabicyclooctanes
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shlaes, D.M. Antibiotics: The Perfect Storm; Springer Dordrec: Heilderberg, London, NY, USA, 2010. [Google Scholar]
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the eskape pathogens. Expert Rev. Anti-Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef]
- Kallen, A.J.; Srinivasan, A. Current epidemiology of multidrug-resistant gram-negative bacilli in the United States. Infect. Control. Hosp. Epidemiol. 2010, 31, S51–S54. [Google Scholar] [CrossRef]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: No eskape! An update from the infectious diseases society of america. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef]
- Llarrull, L.I.; Testero, S.A.; Fisher, J.F.; Mobashery, S. The future of the β-lactams. Curr. Opin. Microbiol. 2010, 13, 551–557. [Google Scholar] [CrossRef]
- Worthington, R.J.; Melander, C. Overcoming resistance to β-lactam antibiotics. J. Org. Chem. 2013, 78, 4207–4213. [Google Scholar] [CrossRef]
- Shlaes, D.M. New β-lactam–β-lactamase inhibitor combinations in clinical development. Ann. N. Y. Acad. Sci. 2013, 1277, 105–114. [Google Scholar] [CrossRef]
- Drawz, S.M.; Papp-Wallace, K.M.; Bonomo, R.A. New β-lactamase inhibitors: A therapeutic renaissance in an MDR World. Antimicrob. Agents Chemother. 2014, 58, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S.; Blaskovich, M.A.; Cooper, M.A. Antibiotics in the clinical pipeline in 2013. J. Antibiot. 2013, 66, 571–591. [Google Scholar] [CrossRef]
- Cooper, M.A.; Shlaes, D. Fix the antibiotics pipeline. Nature 2011, 472, 32. [Google Scholar] [CrossRef]
- Laxminarayan, R.; Powers, J.H. Antibacterial R&D incentives. Nat. Rev. Drug Discov. 2011, 10, 727–728. [Google Scholar] [CrossRef]
- Spellberg, B.; Sharma, P.; Rex, J.H. The critical impact of time discounting on economic incentives to overcome the antibiotic market failure. Nat. Rev. Drug Discov. 2012, 11, 168. [Google Scholar] [CrossRef]
- So, A.; Gupta, N.; Brahmachari, S.; Chopra, I.; Munos, B.; Nathan, C.; Outterson, K.; Paccaud, J.; Payne, D.; Peeling, R. Towards new business models for R&D for novel antibiotics. Drug Resist. Updates 2011, 14, 88–94. [Google Scholar] [CrossRef]
- Boucher, H.W. The 10 × '20 initiative: Pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clin. Infect. Dis. 2010, 50, 1081–1083. [Google Scholar] [CrossRef]
- Talbot, G.H. B-Lactam antimicrobials: What have you done for me lately? Ann. N. Y. Acad. Sci. 2013, 1277, 76–83. [Google Scholar] [CrossRef]
- Jernigan, M.G.; Press, E.G.; Nguyen, M.H.; Clancy, C.J.; Shields, R.K. The combination of doripenem and colistin is bactericidal and synergistic against colistin-resistant, carbapenemase-producing Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2012, 56, 3395–3398. [Google Scholar] [CrossRef]
- Lampri, N.; Galani, I.; Poulakou, G.; Katsarolis, I.; Petrikkos, G.; Giamarellou, H.; Souli, M. Mecillinam/clavulanate combination: A possible option for the treatment of community-acquired uncomplicated urinary tract infections caused by extended-spectrum β-lactamase-producing Escherichia coli. J. Antimicrob. Chemother. 2012, 67, 2424–2428. [Google Scholar] [CrossRef]
- Pallett, A.; Hand, K. Complicated urinary tract infections: Practical solutions for the treatment of multiresistant gram-negative bacteria. J. Antimicrob. Chemother. 2010, 65, iii25–iii33. [Google Scholar] [PubMed]
- Ge, Y.; Whitehouse, M.; Friedland, I.; Talbot, G.H. Pharmacokinetics and safety of CXA-101, a new antipseudomonal cephalosporin, in healthy adult male and female subjects receiving single-and multiple-dose intravenous infusions. Antimicrob. Agents Chemother. 2010, 54, 3427–3431. [Google Scholar] [CrossRef]
- Toda, A.; Ohki, H.; Yamanaka, T.; Murano, K.; Okuda, S.; Kawabata, K.; Hatano, K.; Matsuda, K.; Misumi, K.; Itoh, K. Synthesis and SAR of novel parenteral anti-pseudomonal cephalosporins: Discovery of fr 264205. Bioorg. Med. Chem. Lett. 2008, 18, 4849–4852. [Google Scholar] [CrossRef]
- Ohki, H.; Okuda, S.; Yamanaka, T.; Ohgaki, M.; Toda, A.; Kawabata, K.; Inoue, S.; Misumi, K.; Itoh, K.; Satoh, K. Cephem compounds. WO/2004/039814, 2004. [Google Scholar]
- Zamorano, L.; Juan, C.; Fernández-Olmos, A.; Ge, Y.; Cantón, R.; Oliver, A. Activity of the new cephalosporin CXA-101 (FR264205) against Pseudomonas aeruginosa isolates fromchronically—Infected cystic fibrosis patients. Clin. Microbiol. Infect. 2010, 16, 1482–1487. [Google Scholar] [CrossRef]
- Moyá, B.; Zamorano, L.; Juan, C.; Ge, Y.; Oliver, A. Affinity of the new cephalosporin CXA-101 to penicillin-binding proteins of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2010, 54, 3933–3937. [Google Scholar] [CrossRef]
- Livermore, D.M.; Mushtaq, S.; Ge, Y.; Warner, M. Activity of cephalosporin CXA-101 (FR264205) against Pseudomonas aeruginosa and Burkholderia cepacia group strains and isolates. Int. J. Antimicrob. Agents 2009, 34, 402–406. [Google Scholar] [CrossRef]
- Juan, C.; Zamorano, L.; Pérez, J.L.; Ge, Y.; Oliver, A. Activity of a new antipseudomonal cephalosporin, CXA-101 (FR264205), against carbapenem-resistant and multidrug-resistant Pseudomonas aeruginosa clinical strains. Antimicrob. Agents Chemother. 2010, 54, 846–851. [Google Scholar] [CrossRef]
- Takeda, S.; Ishii, Y.; Hatano, K.; Tateda, K.; Yamaguchi, K. Stability of fr 264205 against ampc β-lactamase of Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 2007, 30, 443–445. [Google Scholar] [CrossRef]
- Takeda, S.; Nakai, T.; Wakai, Y.; Ikeda, F.; Hatano, K. In vitro and in vivo activities of a new cephalosporin, fr264205, against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2007, 51, 826–830. [Google Scholar] [CrossRef]
- Livermore, D.M.; Mushtaq, S.; Ge, Y. Chequerboard titration of cephalosporin CXA-101 (FR264205) and tazobactam versus β-lactamase-producing Enterobacteriaceae. J. Antimicrob. Chemother. 2010, 65, 1972–1974. [Google Scholar] [CrossRef] [PubMed]
- Chandorkar, G.; Huntington, J.; Parsons, T.; Umeh, O. Methods for treating intrapulmonary infections. WO 2,013,036,783, 2013. [Google Scholar]
- Richards, D.M.; Brogden, R. Ceftazidime. Drugs 1985, 29, 105–161. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M.; Blaser, M.; Carrs, O.; Cassell, G.; Fishman, N.; Guidos, R.; Levy, S.; Powers, J.; Norrby, R.; Tillotson, G. Discovery research: The scientific challenge of finding new antibiotics. J. Antimicrob. Chemother. 2011, 66, 1941–1944. [Google Scholar] [CrossRef] [PubMed]
- Drawz, S.M.; Bonomo, R.A. Three decades of β-lactamase inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201. [Google Scholar] [CrossRef]
- Lagacé-Wiens, P.; Walkty, A.; Karlowsky, J.A. Ceftazidime-avibactam: An evidence-based review of its pharmacology and potential use in the treatment of gram-negative bacterial infections. Core Evid. 2014, 9, 13–25. [Google Scholar] [PubMed]
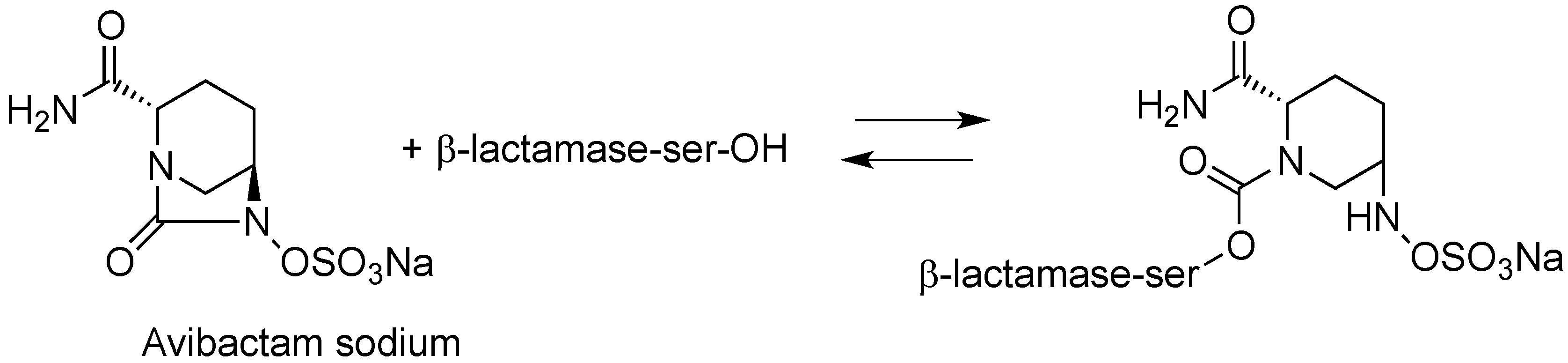
- Ehmann, D.E.; Jahić, H.; Ross, P.L.; Gu, R.-F.; Hu, J.; Kern, G.; Walkup, G.K.; Fisher, S.L. Avibactam is a covalent, reversible, non–β-lactam β-lactamase inhibitor. Proc. Natl. Acad. Sci. USA 2012, 109, 11663–11668. [Google Scholar] [CrossRef] [PubMed]
- Ehmann, D.E.; Jahic, H.; Ross, P.L.; Gu, R.-F.; Hu, J.; Durand-Réville, T.F.; Lahiri, S.; Thresher, J.; Livchak, S.; Gao, N.; et al. Kinetics of avibactam inhibition against class A, C, and D β-lactamases. J. Biol. Chem. 2013, 288, 27960–27961. [Google Scholar] [CrossRef]
- Lahiri, S.D.; Mangani, S.; Durand-Reville, T.; Benvenuti, M.; de Luca, F.; Sanyal, G.; Docquier, J.-D. Structural insight into potent broad-spectrum inhibition with reversible recyclization mechanism: Avibactam in complex with CTX-M-15 and Pseudomonas aeruginosa Ampc β-lactamases. Antimicrob. Agents Chemother. 2013, 57, 2496–2505. [Google Scholar] [CrossRef] [PubMed]
- Walkty, A.; DeCorby, M.; Lagacé-Wiens, P.; Karlowsky, J.; Hoban, D.; Zhanel, G. In vitro activity of ceftazidime combined with nxl104 versus Pseudomonas aeruginosa isolates obtained from patients in canadian hospitals (canward 2009 study). Antimicrob. Agents Chemother. 2011, 55, 2992–2994. [Google Scholar] [CrossRef]
- Levasseur, P.; Girard, A.-M.; Claudon, M.; Goossens, H.; Black, M.T.; Coleman, K.; Miossec, C. In vitro antibacterial activity of the ceftazidime-avibactam (nxl104) combination against Pseudomonas aeruginosa clinical isolates. Antimicrob. Agents Chemother. 2012, 56, 1606–1608. [Google Scholar] [CrossRef]
- Mushtaq, S.; Warner, M.; Livermore, D.M. In vitro activity of ceftazidime+nxl104 against Pseudomonas aeruginosa and other non-fermenters. J. Antimicrob Chemother. 2010, 65, 2376–2381. [Google Scholar] [CrossRef]
- Boyd, J.A.; Cherryman, J.H.; Golden, M.; Kalyan, Y.B.; Lawton, G.R.; Milne, D.; Phillips, A.J.; Racha, S.; Ronsheim, M.S.; Telford, A. Process for preparing heterocyclic compounds including trans-7-oxo-6-(sulphooxy)-1,6-diazabicyclo[3,2,1]octane-2-carboxamide and salts thereof. WO 2,012,172,368, 2012. [Google Scholar]
- Dedhiya, M.G.; Bhattacharya, S.; Ducandas, V.; Giuliani, A.; Ravaux, V.; Bonnet, A.; Priour, A.; Spargo, P.L. Novel crystalline forms of trans-7-oxo-6-(sulphooxy)-1,6-diazabicyclo[3,2,1]octane-2-carboxamide sodium salt. US20130267480 A1.
- Shlaes, D.; Levasseur, P. Use of (1R,2S,5R)1,6-diazabicyclo[3,2,1]octane-2-carboxamide,7-oxo-6-(sulfooxy)-, monosodium salt as a diagnostic reagent for detecting serine beta-lactamases. EP 2,135,959, 2009. [Google Scholar]
- Abe, T.; Okue, M.; Sakamaki, Y. Optically-active diazabicyclooctane derivative and method for manufacturing same. WO/2012/086241, 2012. [Google Scholar]
- Goodman, J.J.; Martin, S.I. Critical appraisal of ceftaroline in the management of community-acquired bacterial pneumonia and skin infections. Ther. Clin. Risk Manag. 2012, 8, 149–156. [Google Scholar] [PubMed]
- Bassetti, M.; Ginocchio, F.; Mikulska, M.; Taramasso, L.; Giacobbe, D.R. Will new antimicrobials overcome resistance among gram-negatives? Expert Rev. Anti-Infect. Ther. 2011, 9, 909–922. [Google Scholar] [CrossRef]
- Corey, G.R.; Wilcox, M.; Talbot, G.H.; Friedland, H.D.; Baculik, T.; Witherell, G.W.; Critchley, I.; Das, A.F.; Thye, D. Integrated analysis of canvas 1 and 2: Phase 3, multicenter, randomized, double-blind studies to evaluate the safety and efficacy of ceftaroline versus vancomycin plus aztreonam in complicated skin and skin-structure infection. Clin. Infect. Dis. 2010, 51, 641–650. [Google Scholar] [CrossRef]
- Goldstein, E.J.C.; Citron, D.M.; Merriam, C.V.; Tyrrell, K.L. Comparative in vitro activity of ceftaroline, ceftaroline-avibactam, and other antimicrobial agents against aerobic and anaerobic bacteria cultured from infected diabetic foot wounds. Diagn. Microbiol. Infect. Dis. 2013, 76, 347–351. [Google Scholar] [CrossRef]
- Eleftheriadou, I.; Tentolouris, N.; Argiana, V.; Jude, E.; Boulton, A.J. Methicillin-resistant Staphylococcus aureus in diabetic foot infections. Drugs 2010, 70, 1785–1797. [Google Scholar] [CrossRef] [PubMed]
- Castanheira, M.; Sader, H.S.; Farrell, D.J.; Mendes, R.E.; Jones, R.N. Activity of ceftaroline-avibactam tested against gram-negative organism populations, including strains expressing one or more β-lactamases and methicillin-resistant Staphylococcus aureus carrying various staphylococcal cassette chromosome mec types. Antimicrob. Agents Chemother. 2012, 56, 4779–4785. [Google Scholar] [CrossRef]
- Louie, A.; Castanheira, M.; Liu, W.; Grasso, C.; Jones, R.N.; Williams, G.; Critchley, I.; Thye, D.; Brown, D.; vanScoy, B. Pharmacodynamics of β-lactamase inhibition by nxl104 in combination with ceftaroline: Examining organisms with multiple types of β-lactamases. Antimicrob. Agents Chemother. 2012, 56, 258–270. [Google Scholar] [CrossRef]
- Mangion, I.K.; Ruck, R.T.; Rivera, N.; Huffman, M.A.; Shevlin, M. A concise synthesis of a β-lactamase inhibitor. Org. Lett. 2011, 13, 5480–5483. [Google Scholar] [CrossRef]
- Rodloff, A.; Goldstein, E.; Torres, A. Two decades of imipenem therapy. J. Antimicrob. Chemother. 2006, 58, 916–929. [Google Scholar] [CrossRef]
- Hirsch, E.B.; Ledesma, K.R.; Chang, K.-T.; Schwartz, M.S.; Motyl, M.R.; Tam, V.H. In vitro activity of mk-7655, a novel β-lactamase inhibitor, in combination with imipenem against carbapenem-resistant gram-negative bacteria. Antimicrob. Agents Chemother. 2012, 56, 3753–3757. [Google Scholar] [CrossRef]
- Young, K.; Hackel, M.; Lascols, C. Response to imipenem plus mk-7655,a novel β-lactamase inhibitor, in a surveillance study population of Pseudomonas aeruginosa from smart 2009, C2–724. In Proceedings of the 52nd Interscience Conference on Antimicrobial Agents Chemotherapy (ICAAC), San Francisco, CA, USA, 13 September 2012; pp. 9–12.
- Miller, S.P.; Zhong, Y.-L.; Liu, Z.; Simeone, M.; Yasuda, N.; Limanto, J.; Chen, Z.; Lynch, J.; Capodanno, V. Practical and cost-effective manufacturing route for the synthesis of a β-lactamase inhibitor. Org. Lett. 2014, 16, 174–177. [Google Scholar] [CrossRef]
- Sun, W.-W.; Cao, P.; Mei, R.-Q.; Li, Y.; Ma, Y.-L.; Wu, B. Palladium-catalyzed unactivated c(sp3)–h bond activation and intramolecular amination of carboxamides: A new approach to β-lactams. Org. Lett. 2013, 16, 480–483. [Google Scholar] [PubMed]
- Klein, C.; Hüttel, W. A simple procedure for selective hydroxylation of L-proline and L-pipecolic acid with recombinantly expressed proline hydroxylases. Adv. Synth. Catal. 2011, 353, 1375–1383. [Google Scholar] [CrossRef]
- Schmitt-Hoffmann, A.; Roos, B.; Maares, J. Pharmacokinetics and safety of the novel sulfactam antibiotic bal30072 after single ascending dose infusions in healthy volunteers, A2–572. In Proceedings of the 51st Interscience Conference on Antimicrobial Agents and Chemother (ICAAC), Chicago, IL, USA, 17–20 September 2011; pp. 17–20.
- Livermore, D.M.; Mushtaq, S. Ptx 2416, a dihydropyridone monobactam vs. Pseudomonas aeruginosa strains with characterized resistances. In Proceedings of the 43rd Interscience Conference Antimicrobial Agents Chemotherapyan American Society for Microbiology (ASM), Washington, DC, USA, 2003. Poster F-554.
- Van Delden, C.; Page, M.G.; Köhler, T. Involvement of Fe uptake systems and AmpC β-lactamase in susceptibility to the siderophore monosulfactam bal30072 in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2013, 57, 2095–2102. [Google Scholar] [CrossRef]
- Page, M.G.; Dantier, C.; Desarbre, E. In vitro properties of bal30072, a novel siderophore sulfactam with activity against multiresistant gram-negative bacilli. Antimicrob. Agents Chemother. 2010, 54, 2291–2302. [Google Scholar] [CrossRef]
- Hofer, B.; Dantier, C.; Gebhardt, K.; Desarbre, E.; Schmitt-Hoffmann, A.; Page, M.G. Combined effects of the siderophore monosulfactam bal30072 and carbapenems on multidrug-resistant gram-negative bacilli. J. Antimicrob. Chemother. 2013, 68, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Rhomberg, P.; Flamm, R.; Jones, R.; Sader, H. Antimicrobial activity of bal30072, alone and in combination with meropenem tested against gram-negative bacteria causing serious infections in hospitals from China, India, Latin America and Southeast Asia-Pacific. In Proceedings of the 52nd Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC), San Francisco, CA, USA, 9–12 September 2012; p. E-193.
- De Souza Mendes, C.; de Souza Antunes, A. Pipeline of known chemical classes of antibiotics. Antibiotics 2013, 2, 500–534. [Google Scholar] [CrossRef]
- Crandon, J.L.; Nicolau, D.P. Human simulated studies of aztreonam and aztreonam-avibactam to evaluate activity against challenging gram-negative organisms, including metallo-β-lactamase producers. Antimicrob. Agents Chemother. 2013, 57, 3299–3306. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M.; Mushtaq, S. Activity of biapenem (rpx2003) combined with the boronate β-lactamase inhibitor rpx7009 against carbapenem-resistant enterobacteriaceae. J. Antimicrob. Chemother. 2013, 68, 1825–1831. [Google Scholar] [CrossRef]
- Hecker, S.J.; Reddy, K.R.; Totrov, M.; Hirst, G.C.; Lomovskaya, O.; Griffith, D.C.; King, P.; Tsivkovski, R.; Sun, D.; Sabet, M.; et al. Discovery of rpx 7009, a broad-spectrum β-lactamase inhibitor with utility vs. class a serine carbapenemases. In 52nd Interscience Conference on Antimicrobial Agents Chemotherapy (ICAAC), San Francisco, IL, USA, 9–12 September 2012; p. F-848.
- Lomovskaya, O.; King, P.; Sun, D.; Griffith, D.C.; Hecker, S.J.; Dudley, M.N. Microbiological characterization of beta-lactamase inhibitor rpx7009. In 52nd Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC), San Francisco, CA, USA, 9–12 September 2012; p. F-850.
- Blais, J.; Lewis, S.R.; Krause, K.M.; Benton, B.M. Antistaphylococcal activity of TD-1792, a multivalent glycopeptide-cephalosporin antibiotic. Antimicrob. Agents Chemother. 2012, 56, 1584–1587. [Google Scholar] [CrossRef]
- Fedora pharmaceuticals demonstrates that FPI-1465 increases activity of certain antibiotics against drug-resistant bacteri. Available online: http://www.fedorapharma.com/instantedit/files/Fedora _ICAAC_data_091213.pdf (accessed on 12 September 2013).
- Mendes, R.; Rhomberg, P.; Becker, H.; Jones, R. Activity of β-lactam agents tested in combination with novel β-lactamase inhibitor compounds against enterobacteriaceae producing extended-spectrum β-lactamases. In Proceedings of the 53nd International Interscience Conference on Antimicrobial Agents Chemotherapy, Denver, CO, USA, 10–13 September 2013; p. F-1188.
- Maiti, S.N.; Nguyen, D.; Ling, R.; Ha, C.M.; Ganguly, B.; Ou, L.; Shan, R.; Singh, R.; Kully, M.; Khan, J.; et al. Design, synthesis and structure activity relationship of novel substituted amides containing diaza bicyclic heterocyclic compounds as broad-spectrum β-lactamase inhibitors. In 53nd International Interscience Conference on Antimicrobial Agents Chemotherapy, Denver, CO, USA, 11 September 2013; p. F-1190.
- Shoichet, B.K.; Prati, F.; Caselli, E.; Romagnoli, C.; Eidam, O. Beta-lactamase inhibitors. WO 2013/056163, 2013. [Google Scholar]
- Eidam, O.; Romagnoli, C.; Dalmasso, G.; Barelier, S.; Caselli, E.; Bonnet, R.; Shoichet, B.K.; Prati, F. Fragment-guided design of subnanomolar β-lactamase inhibitors active in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 17448–17453. [Google Scholar] [CrossRef]
- Prati, F.; Caselli, E. Boronic acid inhibitors of beta-lactamases. WO 2013/053372, 2013. [Google Scholar]
- Patil, V.T.; Tadiparthi, R.; Birajdar, S.; Bhagwat, S. Preparation of trans-7-oxo-6(sulfoxy)-1,6-diazabicyclo[3.2.1]octane-2carbonitrile salts for the treatment of bacterial infections. WO 2013/038330, 2013. [Google Scholar]
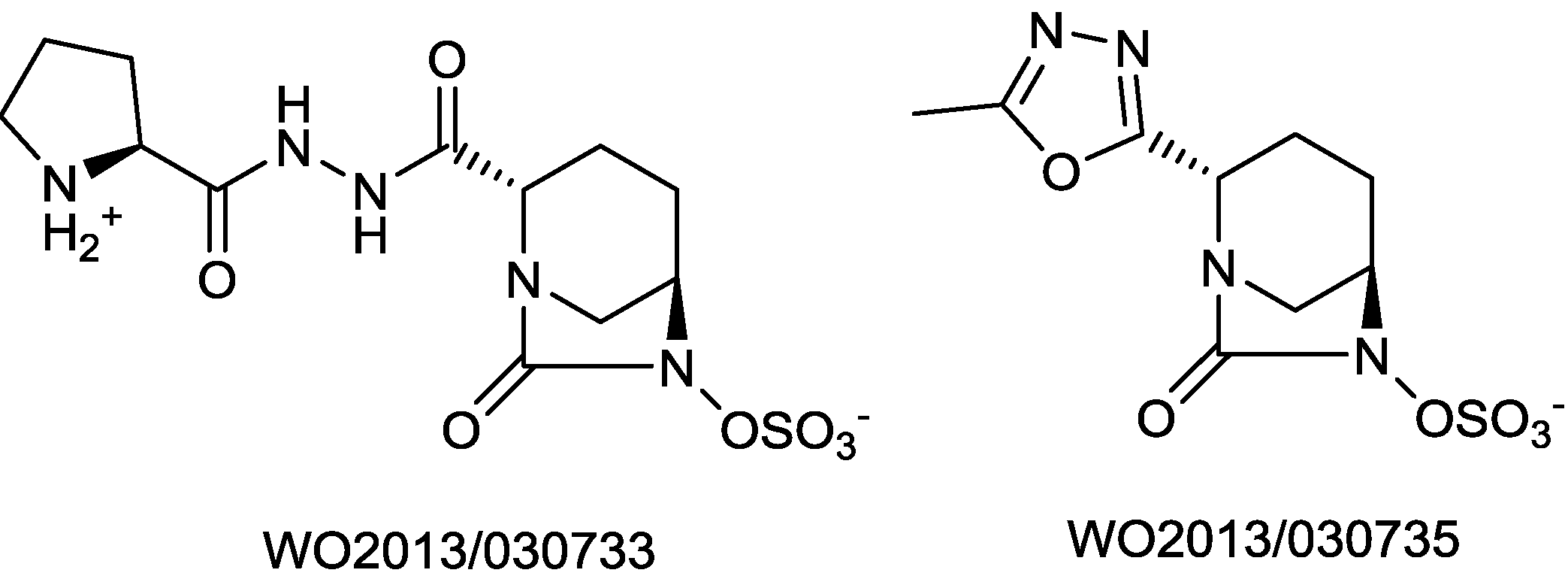
- Patel, M.V.; Deshpande, P.K.; Bhawsar, S.; Bhagwat, S.; Jafri, M.A.; Mishra, A.; Pavase, L.; Gupta, S.; Kale, R.; Joshi, S. 1,6-diazabicyclo[3,2,1]octan-7-one derivatives and their use in the treatment of bacterial infections. WO 2013/030733, 2013. [Google Scholar]
- Bhagwat, S.; Deshpande, P.K.; Bhawsar, S.; Patil, V.J.; Tadiparthi, R.; Pawar, S.S.; Jadhav, S.B.; Dabhade, S.K.; Deshmukh, V.V.D.B; Birajdar, S.; et al. 1,6-diazabicyclo[3,2,1]octan-7-one derivatives and their use in the treatment of bacterial infections. WO 2013/030735, 2013. [Google Scholar]
- Freire, E.; Siles, R.; Ross, P.C. High affinity beta lactamase inhibitors. WO 2013/056079, 2013. [Google Scholar]
- Zhang, S.; Chuong, L.I.M.C.; Khang, I.C.; Alsup, A.; Li, T.; Arya, A.; He, Y.; Yin, N.; Rock, J.; Abel, C.; et al. In vivo efficacy of cb-027 against methicillin-resistant Staphylococcus aureus, and ceftazidime-resistant Pseudomonas aeruginosa and Klebsiella pneumoniae infections in mice. In Proceedings of the 52nd International Interscience Conference on Antimicrobial Agents and Chemotherapy, San Francisco, CA, USA, 10 September 2012; p. F-846.
- Joo, H.; Choi, W.-B.; Kim, D.-I.; Kowalik, E.; Hager, M.W.; Mao, S.; Li, Y.; Liu, S. In Fsi-1671, a novel anti-acinetobacter carbapenem; in vivo efficacy against carbapenem-resistance gram-negative bacterial infection. In Proceedings of the 53rd International Interscience Conference on Antimicrobial Agents and Chemotherapy, Denver, CO, USA, 11 September 2013; p. F-1201.
- Biondi, S.; Long, S.; Panunzio, M.; Qin, W.L. Current Trends in β-Lactam Based β-Lactamases Inhibitors. Curr. Med. Chem. 2011, 18, 4223–4236. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Qin, W.; Panunzio, M.; Biondi, S. β-Lactam Antibiotics Renaissance. Antibiotics 2014, 3, 193-215. https://doi.org/10.3390/antibiotics3020193
Qin W, Panunzio M, Biondi S. β-Lactam Antibiotics Renaissance. Antibiotics. 2014; 3(2):193-215. https://doi.org/10.3390/antibiotics3020193
Chicago/Turabian StyleQin, Wenling, Mauro Panunzio, and Stefano Biondi. 2014. "β-Lactam Antibiotics Renaissance" Antibiotics 3, no. 2: 193-215. https://doi.org/10.3390/antibiotics3020193
APA StyleQin, W., Panunzio, M., & Biondi, S. (2014). β-Lactam Antibiotics Renaissance. Antibiotics, 3(2), 193-215. https://doi.org/10.3390/antibiotics3020193