3. Discussion
The balance between AMR and fitness/virulence in MRSA is highly complex [
24]. Various factors, including genomic backgrounds, responses to antimicrobial exposure, mechanisms of AMR resistance, and the nature of infections (acute or chronic), can influence this interplay. AMR often comes at the cost of overall bacterial fitness, affecting growth ability, competitiveness, and virulence, influencing the microorganism’s transmission and spread in drug-free environments [
13,
25].
The FC molecular basis remains poorly characterized and understood. AMR can reduce biological fitness as antimicrobials target essential bacterial functions such as cell wall biosynthesis, chromosome supercoiling, RNA transcription, and protein synthesis. The core-genome and, thus, its essential genes, control these critical pathways necessary for survival and growth. This means that mutations or changes in their expression can significantly impact fitness [
6]. Similarly, virulence is closely linked to pathogen fitness [
24], and, hence, can be subject to selection. For directly transmitted pathogens, higher growth-rates within the host are linked to higher transmission-rates and better microorganism fitness [
26].
Based on these observations, for the first time, our study tried to close the knowledge gap regarding the interconnections among XDR MOA-related AMR-mechanisms, genomic proneness, the FC omic basis, and compensatory mutational and transcriptional adaptations to acquire and trade off XDR in high-risk HA-, LA-, and CA-MRSA.
This research investigated three isogenic high-risk HA-MRSA, LA-MRSA, and CA-MRSA clones with different genomic backgrounds, expressing DAP/DAL-GLY resistance, paying increasing AMR FCs to explore the FC omic molecular basis, MOA-related AMR-mechanisms, the “FC-compensatory” adaptations, and genomic proneness in this extreme drug resistance (XDR).
Our integrated omic data underpinned the molecular role of the EG and VG gene pools as strategic ‘hot spots’ of compensatory mutational and transcriptional adaptations in the transition from susceptibility to mono- or cross-resistance to second-line DAP/DAL and first-line GLY antimicrobials in high-risk HA-MRSA, LA-MRSA and CA-MRSA acquiring a low or high FC burden.
Firstly, it was mandatory to characterize the MOA-related resistance mechanisms, responsible for the mono-DAP-R in ST-5 N315 HA-GSSA, cross-DAP-R/hGISA in ST398 LA-MRSA, and cross-DAP-R/DAL-R/GISA in ST-1 MW2 CA-MRSA, to outline their impact on vital cellular functions and their association with the omic FC burden in a specific genomic background.
In ST-5 N315 HA-MRSA, mono-DAP-R appeared to be associated with a dual drug-repulsion resistance-mechanism based on “target-enrichment” and “positive-charge enrichment” features. This was related to a concomitant over-expression of tarL/F (target-enrichment) and dltA/C (positive-charge enrichment), implicated in WTA biosynthesis and modification, coupled to an over-expression of the phospholipid biosynthesis-related gene pgsA (target-enrichment); however, these were not co-expressed with mprF (positive-charge enrichment). These changes can putatively result in increases to the WTA abundance and their D-alanylation rate, as well as an exclusive putative increase in the membrane-bound phosphatidylglycerol (PG) amount without an increase in its L-lysinylation rate. This differential expression profile supports a putative overall increase in cell-wall and cell-membrane positive net-charge, even though by different mechanisms. Despite our analysis identifying an MI-nsSNP T345I in MprF, its role remained unclear. This mutation is predicted to decrease the protein stability and functionality (−0.86 ΔΔG), supporting a possible decline in the L-lysinylation rate of membrane-bound PG. This could reduce the positive net-charge of the cell-envelope. These findings aligned with previous studies that question the direct functional impact of MprF SNPs on resistance mechanisms [
27].
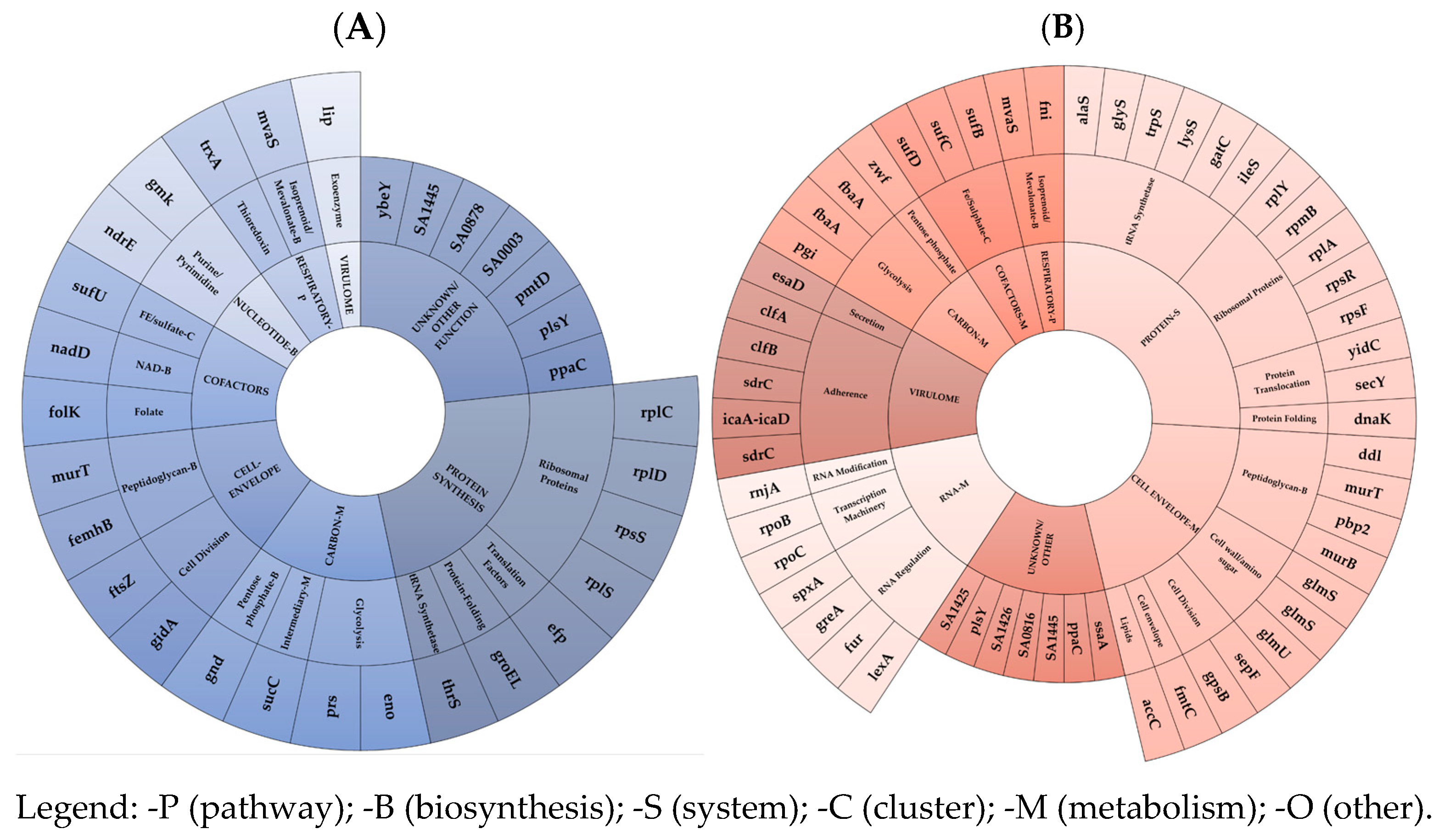
The acquisition of mono-DAP-R in the ST-5 N315 HA-MRSA genomic background resulted in very low FCs, with minimal reductions in growth ability, competitiveness, and virulence (both in vitro and in vivo models) [
13]. This correlated with the reported extra-small-scale mutation- and transcription-driven FC burden. Mutation-driven FC burden was only in charge of two MI-nsSNPs in genes regulating the cell-envelope charge (
mprF) and peptidoglycan metabolism (
fmhB). The transcription-driven FC burden was related to an under-expression of an extra-small pool of EG and VG mRNAs and asRNAs. mRNA under-expression was observed, in decreasing order, in the protein synthesis - (mainly in the ribosomal protein EG subset), unknown/other, followed by the cell-envelope (mainly in the peptidoglycan along with cell division EG subsets), and carbon metabolism in the (mainly in glycolysis EG subset) EG clusters. Interestingly, the lowest under-expression trend was found in the VG cluster. Consequently, in ST-5 N315 HA-MRSA, it can be speculated that there was a minor amount of different coding mRNAs, including the ribosomal protein coding targets, i.e., RpsS (small ribosomal subunit protein uS19), involved in a complex with S13 that binds strongly to the 16S ribosomal RNA, RplS (large ribosomal subunit protein bL19), involved in the structure and function of the aminoacyl-tRNA binding site, and RplD (large ribosomal subunit protein uL4) and RplC (large ribosomal subunit protein uL3), implicated in the assembly of the ribosomal 50S subunit; the Unknown targets, i.e., ppaC, plsY, pmtD, ybeY, SA0003, SA0878, SA1445; the peptidoglycan biosynthesis targets, i.e., MurT (Lipid-II isoglutaminyl-synthase complex) catalyzing the formation of alpha-D-isoglutamine in the cell wall lipid II stem peptide and femX (Lipid II–glycine glycyltransferase), responsible for the incorporation of the first glycine of the pentaglycine interpeptide bridge; the cell-division-related genes, i.e., FtsZ (an essential protein that forms a contractile ring structure (Z ring) at the cell division site) and gidA (NAD-binding protein), which forms tRNA-cmnm5s2U34; the glycolysis-related targets, i.e., Prs (ribose-phosphate pyrophosphokinase), involved in the biosynthesis of the central metabolite phospho-alpha-D-ribosyl-1-pyrophosphate (PRPP); and Eno (Enolase), catalyzing the reversible conversion of 2-phosphoglycerate into phosphoenolpyruvate.
Concurrently, dysregulations via asRNAs were found to be prevalent in the following clusters and subsets: in the protein synthesis EG cluster, mainly in the tRNA synthesis subset, via the under-expression of ileS (isoleucine–tRNA ligase), gatC (aspartyl/glutamyl-tRNA(Asn/Gln) amidotransferase subunit C), and the over-expression of lysS (Lysine–tRNA ligase), trpS (tryptophan–tRNA ligase), alaS (alanine–tRNA ligase), glyS (Glycine–tRNA ligase), and in the ribosomal proteins, via the over-expression of rpsF, rpsR, rplA, rpmB, rplY; in the cell-envelope metabolism EG cluster, mainly in peptidoglycan biosynthesis, via the over-expression of pbp2 (peptidoglycan carboxypeptidase), murT (Lipid II isoglutaminyl synthase), murB (UDP-N-acetylenolpyruvoylglucosamine reductase), and ddl (D-alanine–D-alanine ligase); as well as in the teichoic acid modification subset, via the over-expression of tagH (teichoic acids export ATP-binding protein), llm (lipophilic protein), dltA (D-alanine–D-alanyl carrier protein ligase), and cell-wall amino sugar subsets via the under-expression of glmS (glutamine–fructose-6-phosphate) and the over-expression of glmS, glmU (bifunctional protein GlmU), catalyzing the last two sequential reactions in the de novo biosynthetic pathway for the UDP-N-acetylglucosamine (UDP-GlcNAc); in the unknown/other EG cluster; in the RNA-metabolism EG cluster, mainly in the RNA-regulation subset, via the under-expression of greA, spxA and over-expression of fur (ferric uptake regulation protein), lexA (LexA repressor), repressing the genes involved in the response to DNA damage (SOS response), as well as in basic transcription machinery, via the over-expression of rpoB (DNA-directed RNA polymerase subunit beta) and rpoC (DNA-directed RNA polymerase subunit beta’); and in the virulome cluster, mainly in the adhesion subset, via the under-expression and over-expression of two different loci in sdrC, and the over-expression of clfB, clfA, and icaA–icaD.
These transcriptomic data demonstrated the smallest decrease in the transcriptional rate of the EG and VG clusters associated with the mono-DAP-R acquisition in ST-5 N315 HA-MRSA.
This relatively minimal mutation- and transcription-driven omic FC burden provided significant molecular evidence supporting and agreeing with the very low in vivo FC burden (very low reduction in growth performance, competitiveness, and virulence) as previously outlined in ST5 N315 DAP-R HA-MRSA [
13].
Regarding ST398 DAP-R LA-hGISA, cross-DAP-R/hGISA could seem putatively associated with a GLY-trapping within a thicker cell wall and an alternative “target-enrichment” related to a DAP charge-repulsion mechanism. hGISA was caused by increased peptidoglycan monomers, leading to a thickened cell wall, determining a putative GLY entrapment. This occurred via the over-expression of genes involved in the cell-envelope metabolism, particularly those associated with peptidoglycan biosynthesis, such as murI (glutamate racemase) and murA (UDP-N-acetylglucosamine 1-carboxyvinyltransferase 1), which plays a central role in the core steps of biosynthesis.
Similarly, DAP-R seems determined by a “target-enrichment” mechanism leading to a more copious amount of WTAs, despite not being associated with their increased D-alanylation rate. Similarly, an increased positive cell wall net-charge seems determined by an increased target abundance. This occurred through the over-expression of WTA modification and biosynthesis EG tarL (CDP-glycerol glycerophosphotransferase family protein), not co-expressed with the dlt-operon.
On the contrary, no charge repulsion mechanism due to mprF differential expression was demonstrated, and the role of MprF S295L remains dubious, as commented upon in previous publications [
27].
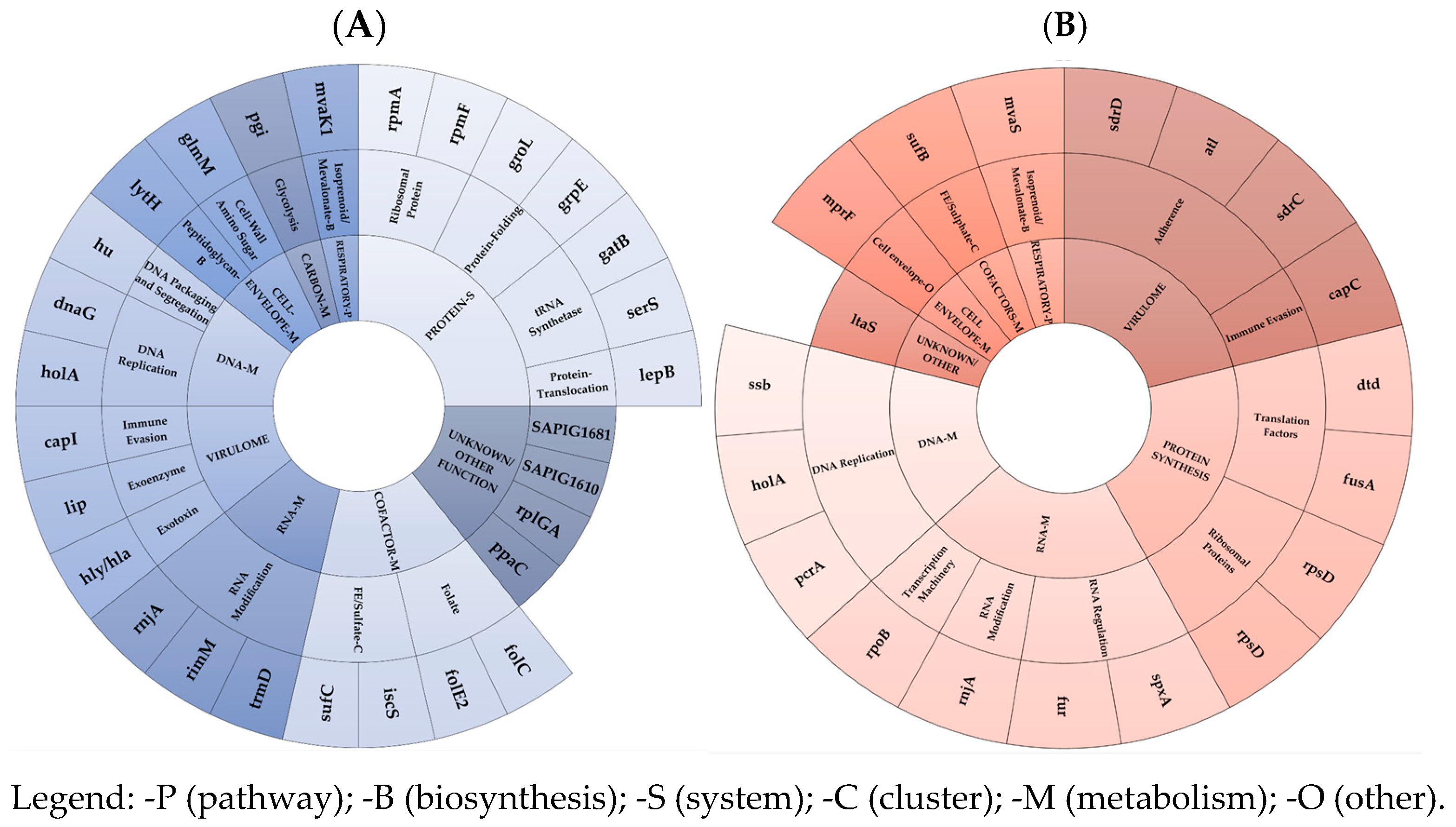
The acquisition of cross-DAP-R/hGISA in ST398 LA-MRSA conferred low in vivo FCs, with a medium reduction in growth ability, a low decrease in competitiveness, and no effects on virulence (both in vitro and in vivo model) [
13]. This corresponded to a small mutation- and transcription-driven FC burden, marked by the under-expression of a medium-sized set of EG and VG targets. In detail, the mutation-driven FC burden was linked to four EG MI-SNPs, including a gene regulating the cell-envelope charge and an unknown EG exported protein (SAPIG1881). Interestingly, the two remaining nsSNPs were found as compensatory mutations in the basic transcription machinery-EGs, rpoB (DNA-directed RNA polymerase subunit beta), and rpoC (DNA-directed RNA polymerase subunit beta’). One VG mutation was, uniquely, in the cell-wall-associated fibronectin-binding protein coding ebh. The transcription-driven FC-burden was mainly due to a small-size under-expression of EG and VG mRNAs as well as asRNAs. This was observed, in decreasing order, in the protein synthesis (mainly in the tRNA synthetase, ribosomal protein, and protein-folding EG subsets), unknown, cofactor-metabolism (mainly in FE sulphate and folate EG subsets), and DNA-metabolism EG cluster (mainly in the DNA-replication EG subset). Interestingly, a very-small-scale differential expression was defined in the VG cluster, with only three under-expressed virulence factors in the immune evasion, exoenzyme, and exotoxin VG subsets, in agreement with the lack of FCs in the in vitro and in vivo virulence [
13], and the RNA metabolism EG cluster (mainly in the RNA-modification subset). In ST398 DAP-R LA-hGISA, lower coding mRNA can be reported among in the following: in tRNA-synthetases, i.e., serS (serine-tRNA synthetase) and gatC (asparaginyl-tRNA or glutaminyl-tRNA synthetase); in ribosomal proteins, i.e., rpmF (a large ribosomal subunit protein bL32 and rpmA (large ribosomal subunit protein bL27); in protein-folding targets, i.e., GrpE related to the hyperosmotic and heat shock by preventing the aggregation of stress-denatured proteins and GroEL; in unknown, i.e., ppaC, rplGA, SAPIG1610, SAPIG1681; in Fe-sulfate cofactor targets, i.e., SufC (Fe-S cluster assembly ATPase) and IscS (probable tRNA sulfurtransferase) catalyzing the ATP-dependent transfer of a sulfur to tRNA to produce 4-thiouridine in position 8 of tRNAs, which functions as a near-UV photosensor; in the folate-cluster target FolE2 (GTP cyclohydrolase) and folC (tetrahydrofolate synthase); in the DNA-replication targets, i.e., dnaG (DNA primase), an RNA polymerase, catalyzes the synthesis of short RNA molecules used as primers for DNA polymerase during DNA replication, such as holA (DNA polymerase III subunit delta), and DNA packaging and segregation hu (DNA-binding protein HU); in virulome, i.e., immune evasion SAPIG0171 capsular polysaccharide biosynthesis protein, exoenzyme SAPIG2721 GehA lipase, and exotoxin SAPIG1158 alpha-hemolysin, as well as in the RNA metabolism, mainly in the RNA modification rnjA (ribonuclease J 2), rimM (ribosome maturation factor RimM), and trmD (tRNA (guanine-N(1)-)-methyltransferase).
Concurrently, dysregulations via asRNAs were demonstrated in the RNA-metabolism EG cluster, mainly in the RNA-modification subset, via the under-expression of rnjA (Ribonuclease J 2), involved in the maturation of rRNA, and in some organisms, mRNA maturation and/or decay, as well as in the RNA-regulation subset, via the under-expression of fur (ferric uptake regulation protein) and the over-expression of spxA (global transcriptional regulator Spx), which were also demonstrated to be involved in the global transcriptional regulator—which plays a key role in the stress response. Dysregulations via asRNAs were also demonstrated in the following subsets: in the basic transcription machinery subset, via the under-expression of rpoB (DNA-directed RNA polymerase subunit beta), catalyzing the transcription of DNA into RNA using the four ribonucleoside triphosphates as substrates; in the protein synthesis subset, via the dysregulation in ribosomal protein rpsD and the over-expression of the translation factor dtd (D-aminoacyl-tRNA deacylase) and fusA (Elongation factor G); in DNA-metabolism EG clusters, i.e., the DNA-replication subset, via the under-expression of ssb (single-stranded DNA-binding protein), holA (DNA polymerase III subunit delta), and pcrA Elicase (ATP-dependent DNA helicase PcrA); in the virulome cluster, via the under-expression of sdrC, as well as in the over-expression of atl, sdrD, adhesins, and in the immune evasion of capC.
These transcriptomic data evidenced a small-scale reduction in the EG and VG expression profile related to the cross-DAP-R/hGISA in ST398 LA-MRSA.
This low mutation- and transcription-driven omic FC-burden provides coherent molecular evidence supporting and agreeing with the low in vivo FC burden characterizing ST398 DAP-R LA-hGISA [
13].
Concerning ST-1 MW2 CA-GISA, the cross-DAP-R/DAL-R/GISA in this genomic background seems to be associated with the transcription-driven MOA-related AMR-mechanisms. DAP-R is related to a dual mechanism of “positive-charge enrichment” by a co-overexpression of dltA/B/D and mprF. This speculatively increased the cell wall and cell-membrane positive net-charge, resulting in a resistance mechanism based on charge-repulsion for a “target-modification enrichment” mechanism. Although our data showed an MI-nsSNP, determining T345A in MprF, its precise role is, similarly, unclear. This mutation resulted in a greater decrease in protein stability (−1.62 DDG change), suggesting a potential reduction in MprF functionality. The putative reduced L-lysinylation rate of membrane-bound PG, caused by the T345A in MprF, could lower the cell’s positive net charge, weakening the charge-repulsion resistance mechanism against positively charged compounds such as GLYs or DAP. However, mprF co-overexpression might compensate for the decreased protein functionality, enabling it to still play a key role in maintaining resistance to DAP. In addition, the low availability of the D-Ala-D-Ala terminus precursor, due to the under-expression of alanine racemase (alr) and D-Ala-D-Ala ligase (ddl), could result in a reduced teichoic acid D-alanylation rate, weakening the charge-repulsion resistance mechanism. Consequently, it can be hypothesized that this deficiency was compensated by the co-overexpression of dltA/B/D and mprF, which may help retain resistance. DAL-R/GISA was previously related to a key MOA-related target differential expression, which includes the under-expression of the alanine racemase (alr) and D-Ala-D-Ala ligase (ddl) involved in peptidoglycan biosynthesis. These genes are essential for the D-Ala-D-Ala terminus synthesis in the peptidoglycan monomer, which plays a critical role in transpeptidation. This expression profile indicates a reduced availability of D-Ala precursors and decreased D-Ala-D-Ala binding, which are targets of DAL and GLY antimicrobials. Interestingly, this was associated with numerous accessory dysregulated pathways supporting the key one, as previously published [
23].
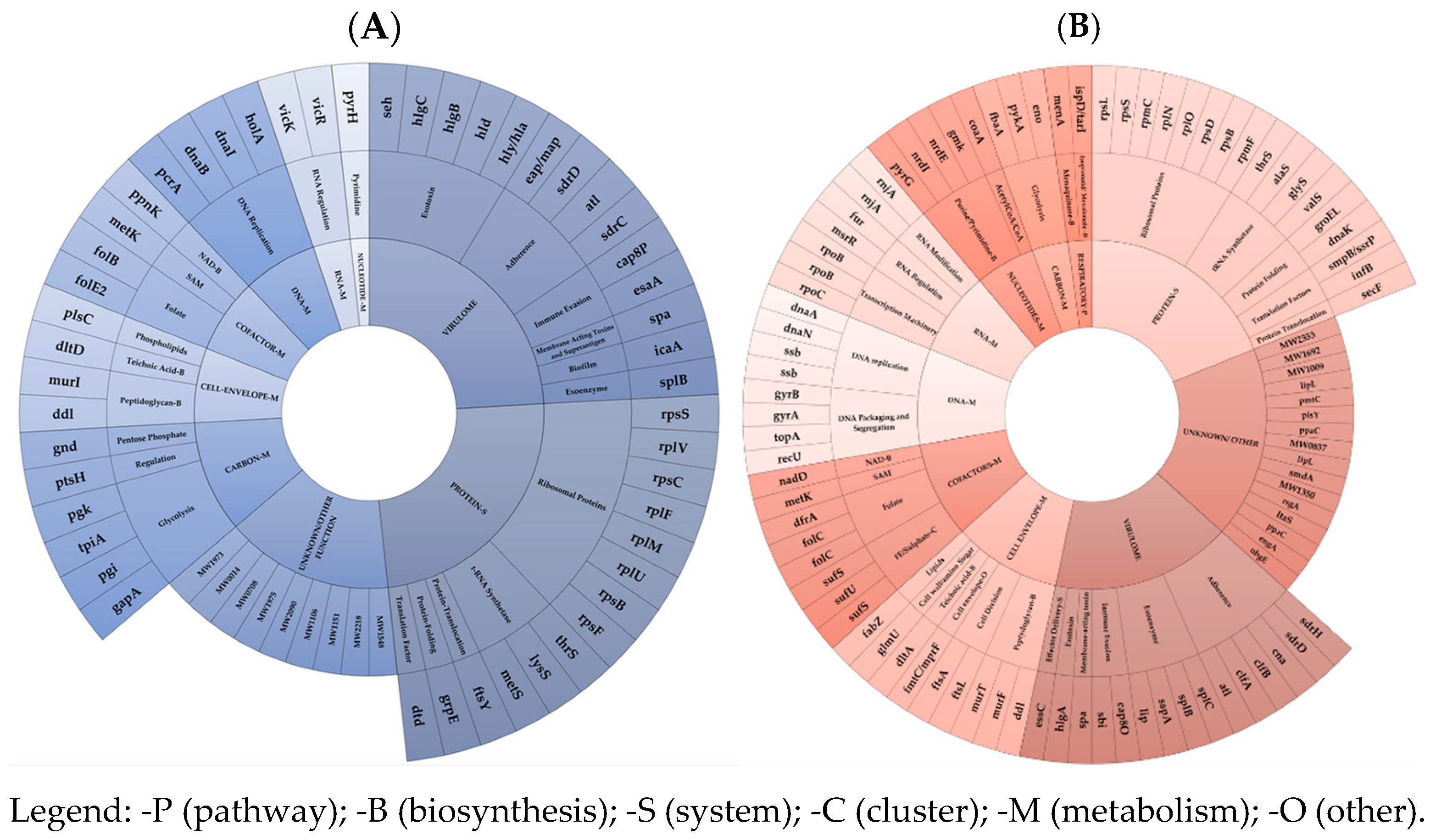
In ST-1 MW2 CA-GISA, the acquisition of cross-DAP-R/DAL-R/GISA, resulting in high in vivo FCs (severely impaired growth performance, reduced competitiveness, and decreased in vitro and in vivo virulence) [
13], was associated with a large-scale mutation-driven and transcription-driven FC burden by large-scale under-expression of the EG and VG targets.
The mutation-driven FC burden was in charge of MI-nsSNPs in the cell-envelope charge and peptidoglycan biosynthesis EG subsets (similar to DAP-R HA-GSSA) including the T345A in MprF (DAP/GLY-R), I121N in MurG (indirectly supporting GLY-RS), F74L in TagH (teichoic acid biosynthesis), H481Y in RpoB (RIF-R and transcription), and in an unknown/other EG cluster, i.e., I186M in GdpG (β-lactam resistance and GLY-cross-resistance). Furthermore, wild-type-revertant mutations included V329G in GlnA (glutamine biosynthesis, carbon/intermediary metabolism) and N177K in RecU (DNA metabolism/DNA packaging).
An additional mutation-driven burden was found in the VG cluster. This included Y130H in Cap8H and V120G in Cap8K (capsular polysaccharide synthesis enzymes), T1313S SdrD (serine-aspartate repeat adhesion protein), V1768D in Ebh (cell-wall-associated fibronectin-binding protein), and only one wild-type-revertant MI-nsSNP D635E in the ClfB wild type (adhesion clumping factor B).
The transcription-driven FC burden was predominantly characterized by massive under-expression across all EG clusters and VG clusters. EG mRNA under-expression, in decreasing order, was most notable in the protein synthesis- (predominantly in the ribosomal protein EG subset), VG (mainly in the exotoxin, immune evasion subsets and equally adhesins), and in unknown/other clusters. A smaller under-expression was observed in the carbon metabolism, DNA-, cofactor metabolism, and cell-envelope EG clusters. In ST-1 MW2 CA-GISA, a lower coding mRNA amount was mainly found for the gene-pool including rpsF, rpsB, rpsS, rplU, rplM, rplF, rpsC, and rplV (ribosomal proteins); metS, lysS, and thrS (t-RNA synthetase); in virulence genes, i.e., hla, hld, hlgB, hlgC (hemolysin), seh (enterotoxins), in sdrC, sdrD, eap/map, atl (Adhesins), and in esaA, cap8P (immune evasion); in unknown targets, i.e., MW1973, MW0014, MW0708, MW1975, MW2090, MW1106, MW1151, MW2218, and MW1548; in glycolysis-related genes, i.e., pgk (phosphoglycerate kinase), tpiA (triosephosphate isomerase), pgi (glucose-6-phosphate isomerase), and gap (glyceraldehyde-3-phosphate dehydrogenase 1); in DNA replication genes, i.e., holA (DNA polymerase III subunit delta), dnaI (primosomal protein), dnaB (replicative DNA helicase), and pcrA (ATP-dependent DNA helicase PcrA); in folate-cofactor targets, i.e., folB (dihydroneopterin aldolase) and folE2 (GTP cyclohydrolase FolE2); peptidoglycan biosynthesis genes, i.e., murI (glutamate racemase) and ddl (D-alanine–D-alanine ligase).
Additionally, putative key transcriptional dysregulations via asRNAs were progressively found in the protein synthesis, mainly in the following subsets: in the ribosomal protein EG subset via the under-expressions in rpmF, rpsD, rplO, rplN, rpmC, rpsS, rpsB, and the over-expression of rpsL; in the t-RNA synthetase subset, via the under-expressions in glyS, alaS, thrS, and the over-expression of valS; in unknown/other, i.e., obgE, engA, MW0544, MW0837, MW1104, MW1350, MW1727, MW1860, MW0681, and OE in MW1860, MW1240, MW0544, MW1009, MW1692, MW2353, and MW1872; in the VG cluster, mainly in the adhesin subset, via the under-expressions in sdrD, clfA, clfB, atl, can, and the over-expression of sdrH; in the exoenzyme subset, via the under-expression of lip, sspA, splB, and the over-expression of splC; in the folate subset, via under the expression of dfrA, and the dysregulation in folC; in the Fe/sulphate subset, via the under-expression of sufS (cysteine desulfurase), sufU (iron–sulfur cluster assembly scaffold protein IscU), and the over-expression of sufS; in the cell-envelope EG cluster, mainly in the peptidoglycan biosynthesis subset, via the under-expression of murF (UDP-N-acetylmuramoyl-L-alanyl-D-glutamate–L-lysine ligase), murT (Lipid II isoglutaminyl synthase), and ddl (D-alanine–D-alanine ligase); in the DNA-metabolism EG cluster, mainly in the DNA-replication subset, via the under-expression of dnaA (chromosomal replication initiator protein DnaA), dnaN (Holliday junction branch migration complex subunit RuvB), and dysregulation in ssb (single-stranded DNA-binding protein); and in the DNA packaging and segregation subset, via the under-expression of gyrA (DNA gyrase subunit A), gyrB (DNA gyrase subunit B), topA (DNA topoisomerase 1), and the over-expression of recU (Holliday junction resolvase RecU). A smaller dysregulation occurred in the RNA-metabolism EG cluster, mainly in the following subsets: in the basic transcription machinery subset, via the under-expression of rpoB (DNA-directed RNA polymerase subunit beta) and the over-expression of two diverse rpoB and rpoC (DNA-directed RNA polymerase subunit beta’); in the nucleotide metabolism EG, mainly in the purine and pyrimidine biosynthesis subset, via the under-expression of gmk (guanylate kinase), nrdE (Ribonucleoside-diphosphate reductase), and the over-expression of nrdI (Protein NrdI) and pyrG (CTP synthase); and in the carbon-metabolism EG cluster, mainly in the glycolysis-subset, via the under-expression of pykA (pyruvate kinase), fbaA (fructose-bisphosphate aldolase), and Eno (Enolase).
Transcriptomics demonstrates that cross-DAP-R/DAL-R/GISA required a drastic decrease in the overall transcription trend of essential and virulence genes, in contrast to the mono-DAP-R and cross-DAP/hGISA acquisition.
This maximal mutation- and transcription-driven omic FC burden provides robust molecular-level supporting evidence, in agreement with the high in vivo FC-burden (huge/high decrease in growth-performance, competitiveness, and virulence) previously reported in ST-1 MW2 CA-GISA [
13].
Noteworthy considerations need to be addressed about the molecular basis of genomic proneness and its “compensatory-skills” to acquire and maintain the omic adaptations necessary for XDR.
First, the FC compensatory adaptations are not random but, on the contrary, they systematically appear in key genomic regions—particularly those involved in protein synthesis, cell-envelope metabolism (a hub for lipopeptide, β-lactam, and glycopeptide resistance), and the virulome—revealing a harmonized evolutionary response to antibiotic pressure.
In the core-genome EG clusters, we observed coordinated transcription-shifts and allelic diversification, suggesting an evolved genomic plasticity that supports adaptability in variable-scale prone genomes, particularly those undergoing genome reduction, rearrangement, or horizontal acquisition events. These compensatory mechanisms are amplified in high-risk clones from hospital-acquired (HA), livestock-associated (LA), and community-acquired (CA) MRSA lineages, each presenting distinct environmental and host-imposed selective landscapes.
In LA-MRSA, HA-MRSA, and CA-GISA, the EG clusters related to protein synthesis, cell envelope, and virulence play a key role in the omic basis of FCs.
From a molecular point of view, it remains to be considered that the differential expression can lead to a major or minor amount of the mRNA target genes, whilst the differential expression of regulatory asRNAs can lead to the up- or down-regulation of its regulated targets or pathways via an increase or decrease in transcriptional or translational rate. Furthermore, asRNAs can regulate gene expression by several mechanisms. One is a transcriptional interference, i.e., RNA polymerase that transcribes the asRNA physically blocks the transcription of the sense-strand. Post-transcriptionally, asRNAs can alter the structure of the sense RNA, affecting its translation or promoting degradation by RNases. RNase III, which cleaves double-stranded RNA, plays a key role in this process [
28,
29,
30,
31,
32,
33].
Looking only at the mostly involved clusters, the protein synthesis of the EG cluster involves pathways such as ribosomal proteins, t-RNA synthetase, translocation factors, protein folding, and protein translocation. Adaptations in these pathways can disrupt normal protein synthesis, reducing translation efficiency and causing defective protein production, misfolded proteins, and impairing translation. These changes may require additional energy, negatively affecting fitness.
The cell-envelope metabolism EG cluster, enriched with seven essential pathways such as peptidoglycan biosynthesis and cell-envelope modification, can confer mono or cross DAP and DAL/GLY-R resistance. These adaptations alter cell morphology, impair envelope integrity, reduce stress resistance, and compromise growth, all linked to adaptive lipoglycopeptide (DAL)/glycopeptide (GLY) resistance mechanisms.
The VG cluster involves adaptations in exotoxin, immune evasion enzymes, adhesins, exoenzymes, biofilm, superantigens, toxins, and secretion systems, which can drastically change virulence and pathogenicity.
Successively, it is mandatory to point out the key role of the occurrence of the mitigating molecular factors, i.e., compensatory mutations or “transcription-shifts”. In ST-1 MW2 CA-GISA, the high-impact nsSNP in agrA (Arg170*), quorum sensing master regulator of the staphylococcal virulence, the MI-nsSNP in RNA polymerase beta subunit coding gene, rpoB, together with the large-size transcription-shift confer to this genome high-proneness and compensatory skills to balance FCs due to XDR. ST-1 MW2 CA-MRSA results in a highly prone genome able to support and balance the massive XDR omic burden, despite its intrinsic nature to carry a lower AMR rate, sufficient for spreading in low antimicrobial pressure community settings. Based on actual knowledge, the agrA and rpoB compensatory mutations drastically reduced virulence and transcription rate, representing compensatory features allowing the chance to use cell resources to do other things, i.e., shifting the metabolism towards the new XDR-related pathways [
12]. The acquisition of cross-DAP-R/DAL-R/GISA required complex and massive mutational and transcriptional rearrangements. These support the XDR mechanism, significantly reducing microorganisms’ fitness in free-drug conditions. The adaptations leading to FCs by high growth retardation, poor competitiveness, and low virulence impose an energetic and metabolic burden, compromising vital functions and virulence. To express cross-DAP/DAL/GLY MOA-related AMR mechanisms, CA-GISA requires additional efforts to synthesize the new enzymes necessary to activate the complex network of metabolic resistance pathways and to reallocate the available resources. This represents a critical factor in microbial competition [
34]. At the phenotypic level, this matches the observed slowing of growth and poor competitiveness [
13], whilst at the omic level, it can align with transcriptional compensatory adaptations in essential genes involved in vital pathways, such as protein synthesis, cell-envelope metabolism, cofactor biosynthesis, DNA–RNA processes, carbon and nucleotide metabolism, and the respiratory pathway, all influencing growth performance.
In the ST398 mono-DAP-R LA-MRSA genomic background, the MI-nsSNPs in genes coding the beta and beta’ subunits of the RNA polymerase, rpoB and rpoC, as well as the small-size transcription-shifts confers to this genome a “FC-compensatory” moderate-proneness and, thus, compensatory skills to balance the costs for the XDR acquisition. The ST398 LA-MRSA genomic background indeed acquires a lower resistance-rate, i.e., a cross-DAP-R/hGISA, requiring a smaller-scale mutation- and transcription-driven burden. The cross-DAP-R/hGISA mechanism is due to a different resistance mechanism caused by GLY-trapping and a “target-enrichment” DAP-alternative repulsion mechanism. The former arises from the differential expression of two genes; however, whilst this affects a high-interconnected pathway such as the peptidoglycan biosynthesis, contrarily, the latter results from a single-gene differential expression (tarL), implicated in a more tailored pathway such as the WTA modification and biosynthesis.
In the ST-5 N315 HA-MRSA genomic background, no compensatory mutations were observed; thus, the extra-small-size compensatory transcriptional shifts allow the genome a low proneness to balance the costs for XDR. The ST-5 N315 HA-MRSA genomic background can face the acquisition of mono-DAP-R, requiring an extra-small-scale and transcriptional burden. The DAP-R mechanism is, in fact, due to the over-expression of tarF/L and dltA/C, both exclusively tailored to the cell-envelope’s positive net-charge modifications.
Trying to close the gap between omic FC and their clinical and molecular implications, our findings underscore the significance of the functional compensatory landscape (“FC-compensatory” mechanisms) within essential genomic modules—specifically those involved in protein synthesis, cell-envelope metabolism, and the virulome—in shaping the evolution of extreme drug resistance (XDR) in MRSA. The existence of FC compensatory adaptations across these genomic compartments facilitates MRSA in achieving a balance between resistance and fitness, thereby aiming to optimize persistence, virulence, and transmissibility.
These compensatory landscapes appear to be a critical evolutionary response to the selective pressures exerted by antimicrobials targeting the multi-faceted mechanisms of action (MOAs) of frontline antibiotics. This supports a model in which “FC compensatory” features are not merely casual, but integral to the success of XDR MRSA in diverse ecological contexts. Adapting critical cellular functions impacted by resistance mechanisms, these compensatory elements aim to optimize the persistence and transmissibility of high-risk MRSA lineages, thereby posing an ongoing threat to patient outcomes, and thus, to human health.
In the clinical context, there is an urgent need for therapeutic strategies that anticipate or disrupt such compensatory adaptations, potentially targeting these molecular trade-offs to reinstate susceptibility and to limit the spread of XDR MRSA. This significantly undermines treatment efficacy, increases the burden of care, and leads to poorer outcomes for patients across all exposure contexts—hospital, livestock-associated, and community-acquired.
A comprehensive understanding of the compensatory landscape has the potential to enhance risk stratification, guide therapeutic choices, and most significantly, inform the development of treatments that target these adaptations. This could affect the pathogen's ability to sustain resistance without sacrificing virulence or metabolic integrity. Furthermore, such treatments could reverse the rising tide of treatment failure and improve patient outcomes in the face of an evolving and deeply adaptive pathogen. The integration of compensatory dynamics into our understanding of resistance evolution offers a refined framework for predicting MRSA behavior across different epidemiological niches. Furthermore, it provides a foundation for the rational design of next-generation antimicrobials and diagnostics aimed at intercepting adaptive resilience in pathogenic bacteria.
In conclusion, our data first demonstrated the role of compensatory features appearing in protein synthesis, cell-envelope metabolism, EG clusters, and virulome as key mediators of “FC compensatory” adaptations related to the XDR MOA-related resistance in variable-scale prone genomes of high-risk HA-, LA-, and CA-MRSA.


 ,
, 

{kind=link}
{kind=link}
{kind=link}