Impact of Parenteral Ceftiofur on Developmental Dynamics of Early Life Fecal Microbiota and Antibiotic Resistome in Neonatal Lambs

 ,
,  ,
,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Impact of CCFA on the Lamb’s Body Weight
2.2. Whole Genome Shotgun Sequencing Summary
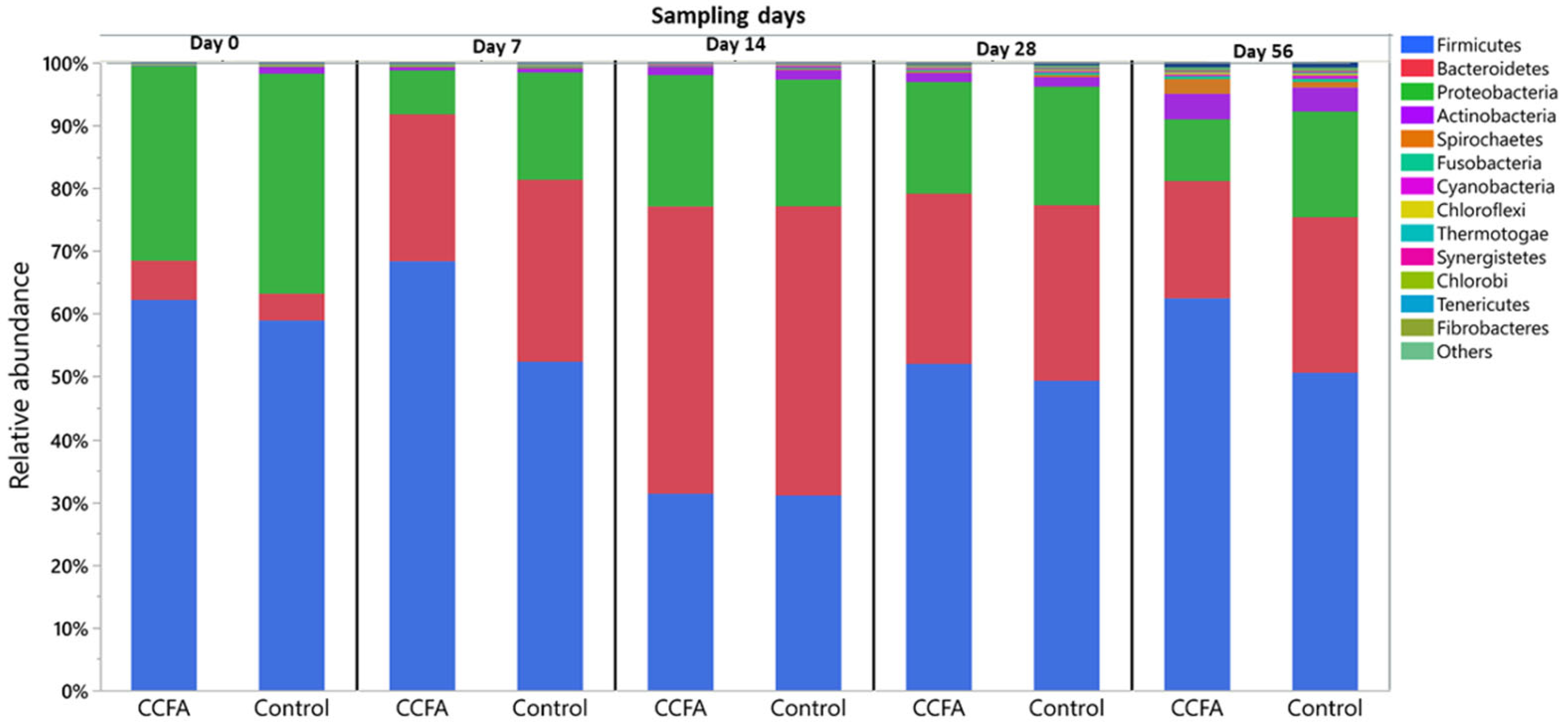
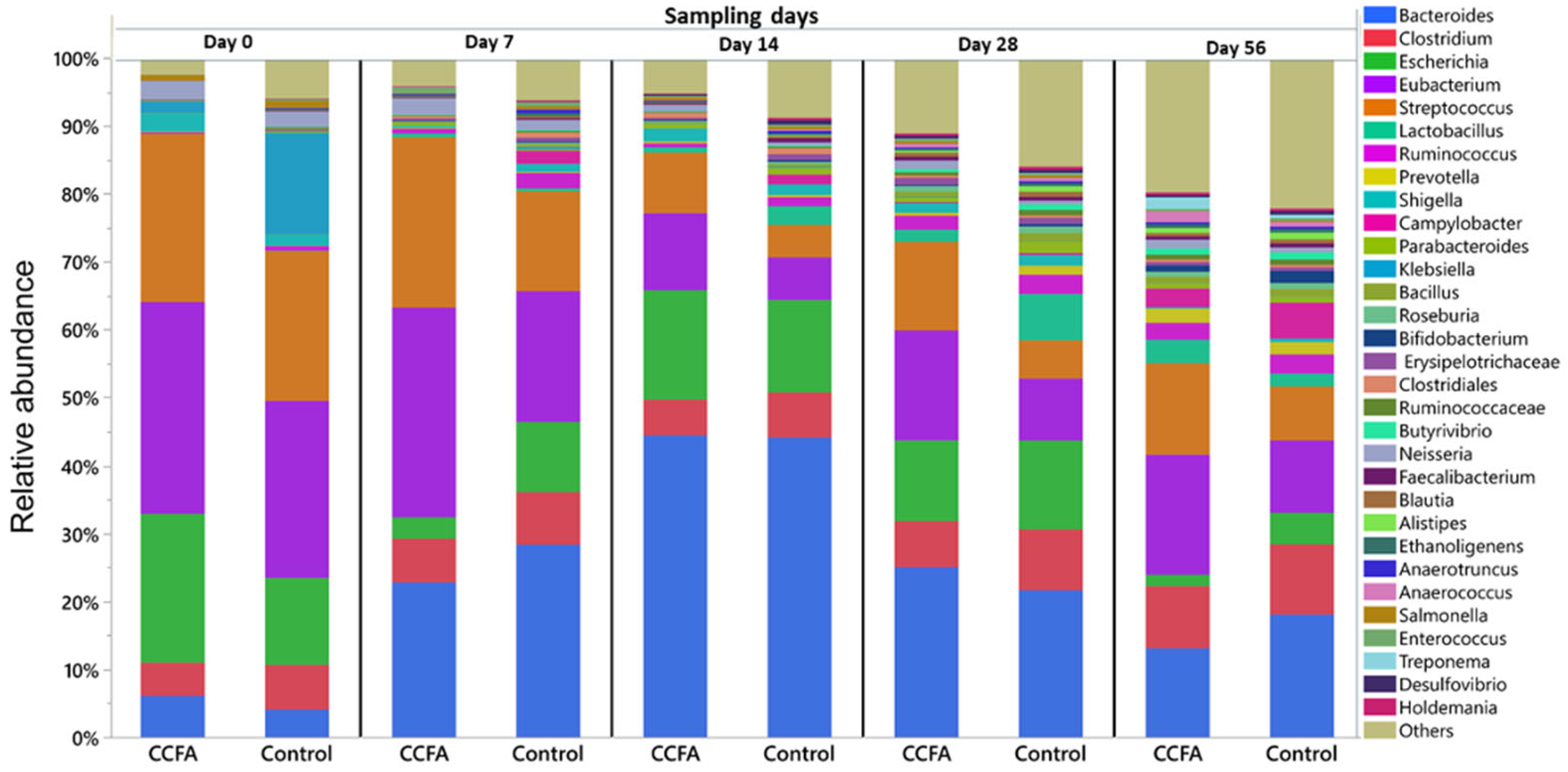
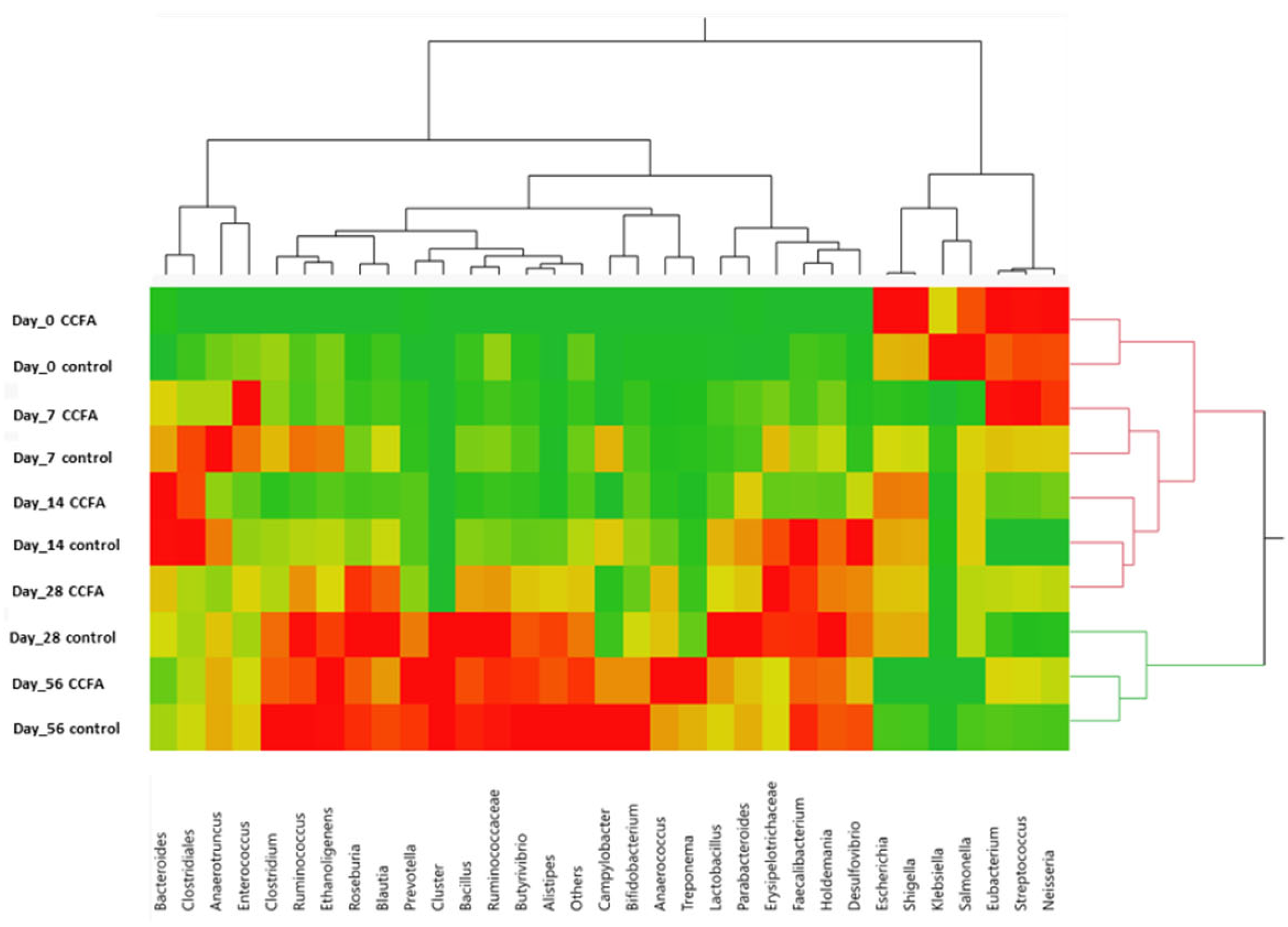
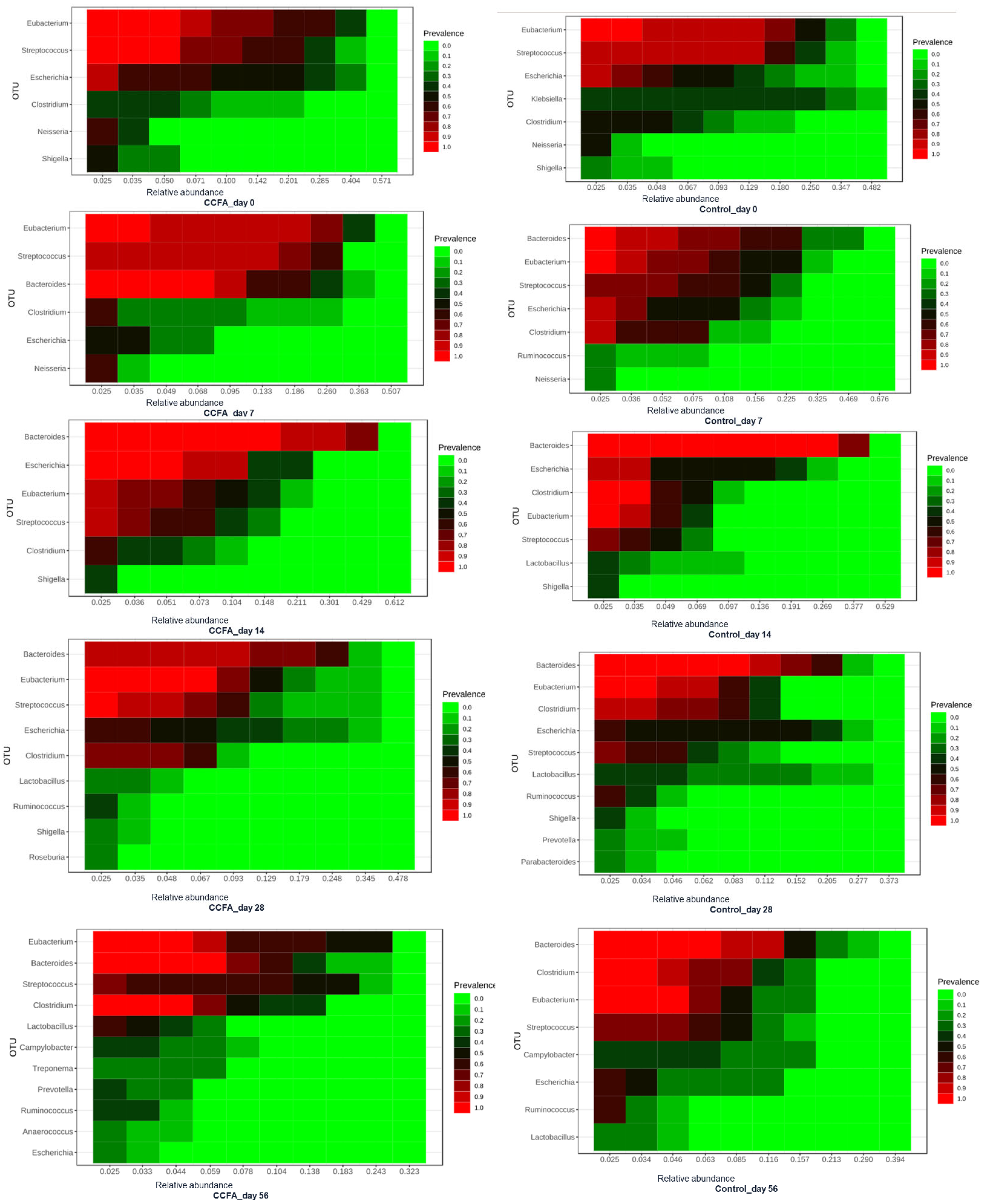
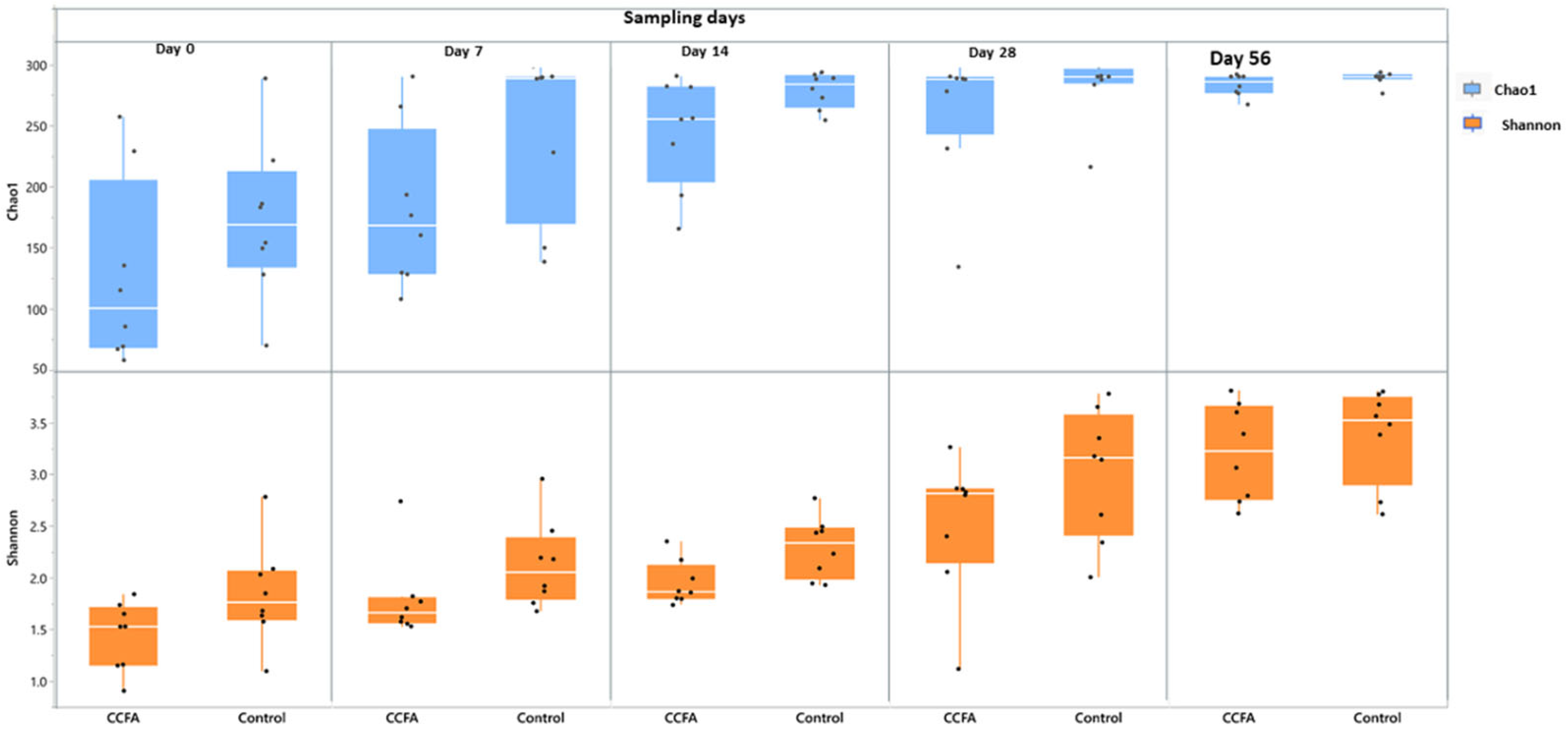
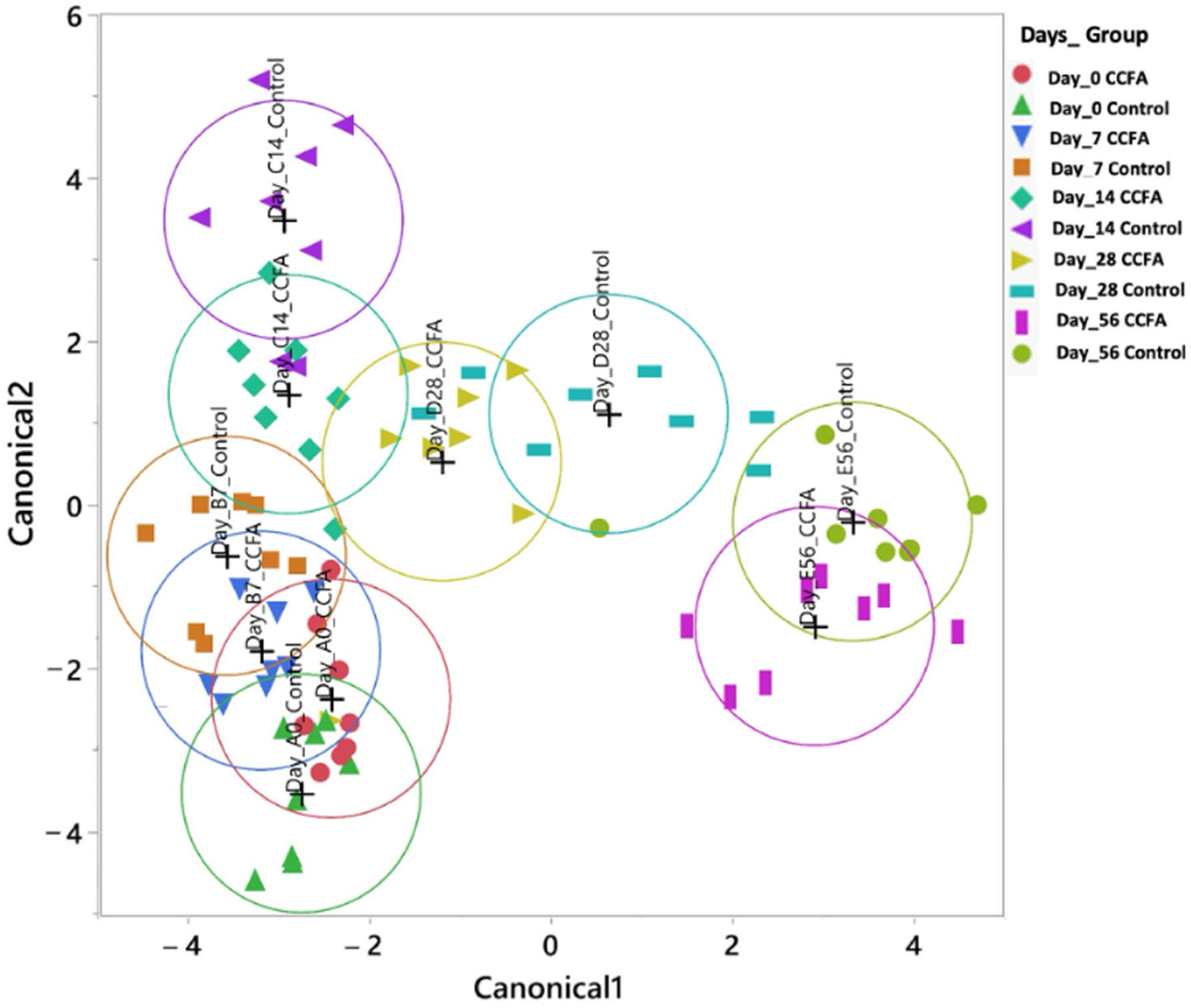
2.3. Effects of the Early Ceftiofur Treatment on the Lamb Fecal Microbiome Composition and Diversity
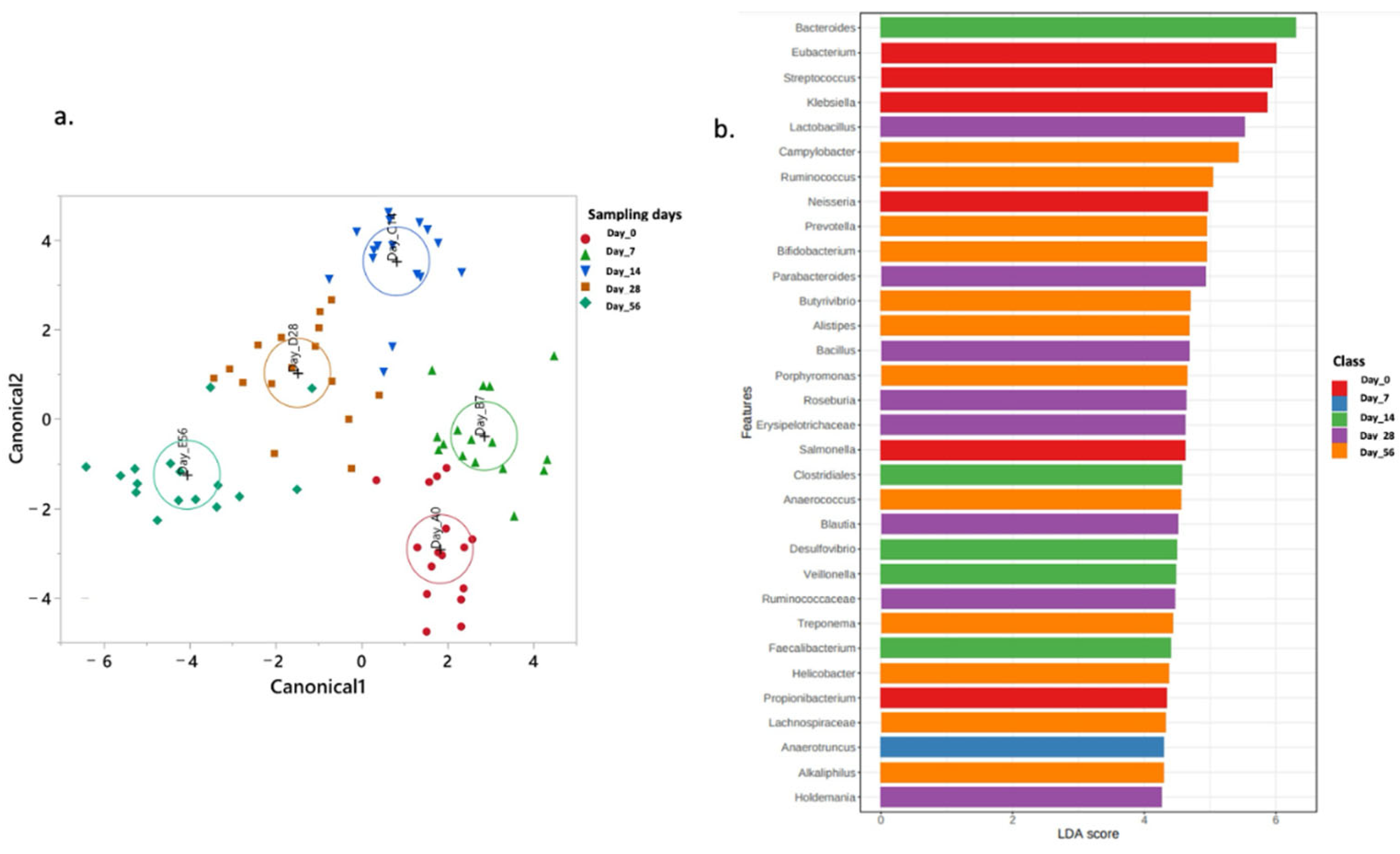
2.4. Normal Fecal Microbiota Development
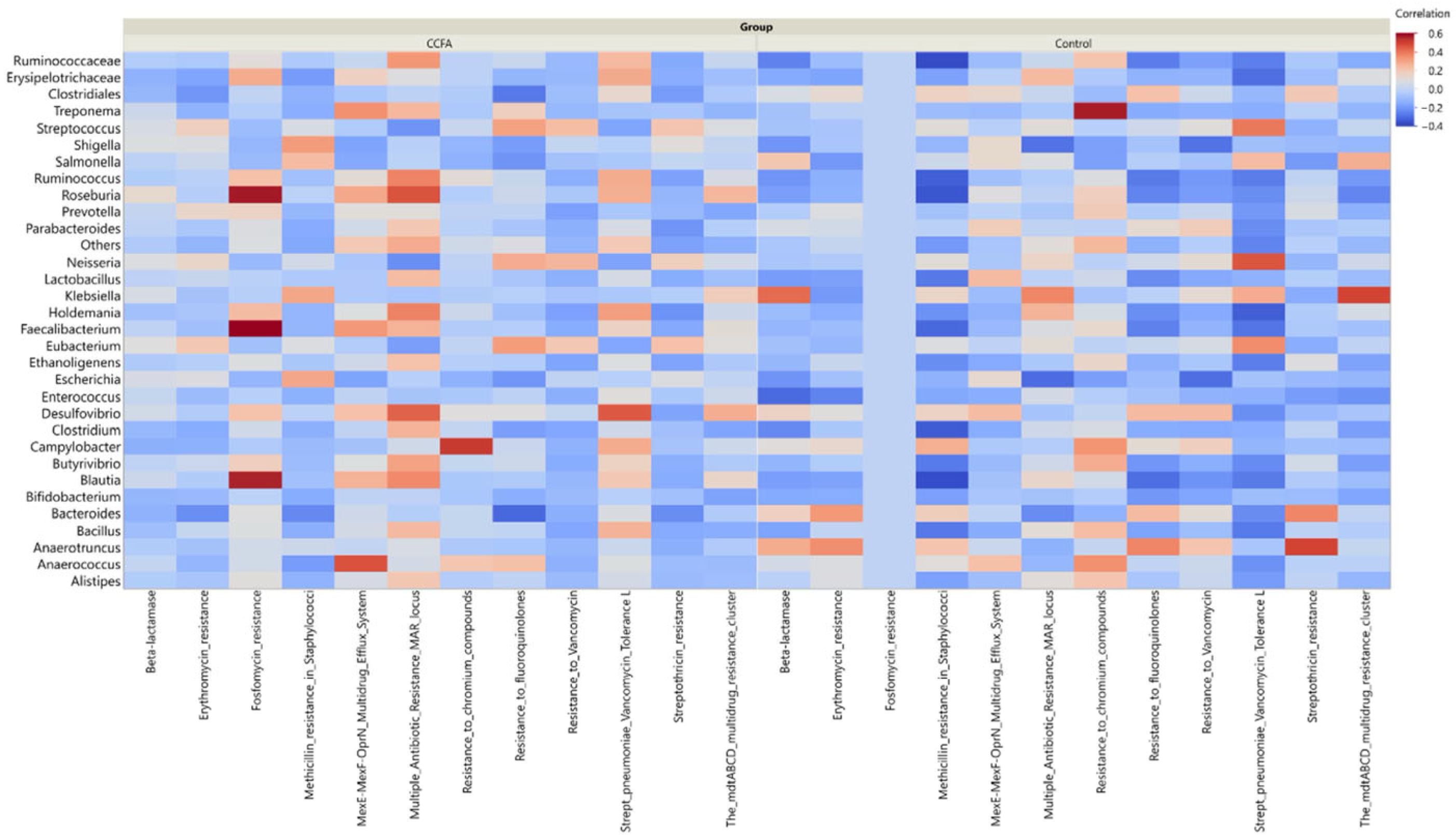
2.5. Effect of Ceftiofur on the Microbial Functional Resistance Genes
3. Discussion
4. Materials and Methods
4.1. Experimental Design and Sample Collection
4.1.1. Animals
4.1.2. Challenge
4.1.3. Samples
4.1.4. Ethics Statement of Animal Use
4.2. DNA Extraction
4.3. Whole Genome Shotgun DNA Sequencing (WGSS)
4.4. Bioinformatic Analysis
4.4.1. Sequence Reads Processing
4.4.2. Operational Taxonomic Units (OTUs) Analysis
4.4.3. Beta-Lactam Resistome Identification
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CCFA | Ceftiofur Crystalline Free Acid |
| ARG(s) | Antibiotic Resistance Gene(s) |
| WGSS | Whole Genome Shotgun Sequencing |
| DNA | Deoxyribonucleic Acid |
| OTUs | Operational Taxonomic Units |
| MG-RAST | Metagenomics Rapid Annotation using Subsystem Technology |
| RefSeq | Reference Sequence Database |
| PCA | Principal Component Analysis |
| CDA | Canonical Discriminant Analysis |
| LDA | Linear Discriminant Analysis |
| LEfSe | Linear Discriminant Analysis Effect Size |
| PCR | Polymerase Chain Reaction |
References
- Alonso, V.R.; Guarner, F. Linking the gut microbiota to human health. Br. J. Nutr. 2013, 109, S21–S26. [Google Scholar] [CrossRef] [PubMed]
- Malmuthuge, N.; Griebel, P.J.; Guan, L.L. The gut microbiome and its potential role in the development and function of newborn calf gastrointestinal tract. Front. Vet. Sci. 2015, 2, 36. [Google Scholar] [CrossRef]
- Van Den Broek, M.F.; De Boeck, I.; Kiekens, F.; Boudewyns, A.; Vanderveken, O.M.; Lebeer, S. Translating recent microbiome insights in otitis media into probiotic strategies. Clin. Microbiol. Rev. 2019, 32, e00010–e00018. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.; Batool, A.; Arif, S. Healthy Cattle Microbiome and Dysbiosis in Diseased Phenotypes. Ruminants 2022, 2, 134–156. [Google Scholar] [CrossRef]
- Kraimi, N.; Dawkins, M.; Gebhardt-Henrich, S.G.; Velge, P.; Rychlik, I.; Volf, J.; Creach, P.; Smith, A.; Colles, F.; Leterrier, C. Influence of the microbiota-gut-brain axis on behavior and welfare in farm animals: A review. Physiol. Behav. 2019, 210, 112658. [Google Scholar] [CrossRef]
- Guo, C.Y.; Ji, S.K.; Yan, H.; Wang, Y.J.; Liu, J.J.; Cao, Z.J.; Yang, H.J.; Zhang, W.J.; Li, S.L. Dynamic change of the gastrointestinal bacterial ecology in cows from birth to adulthood. MicrobiologyOpen 2020, 9, e1119. [Google Scholar] [CrossRef]
- Li, R.W.; Connor, E.E.; Li, C.; Baldwin, V.; Ransom, L.; Sparks, M.E. Characterization of the rumen microbiota of pre-ruminant calves using metagenomic tools. Environ. Microbiol. 2012, 14, 129–139. [Google Scholar] [CrossRef]
- Oikonomou, G.; Teixeira, A.G.V.; Foditsch, C.; Bicalho, M.L.; Machado, V.S.; Bicalho, R.C. Fecal microbial diversity in pre-weaned dairy calves as described by pyrosequencing of metagenomic 16S rDNA. Associations of Faecalibacterium species with health and growth. PLoS ONE 2013, 8, e63157. [Google Scholar] [CrossRef] [PubMed]
- Malmuthuge, N.; Griebel, P.J.; Guan, L.L. Taxonomic identification of commensal bacteria associated with the mucosa and digesta throughout the gastrointestinal tracts of preweaned calves. Appl. Environ. Microbiol. 2014, 80, 2021–2028. [Google Scholar] [CrossRef]
- Malmuthuge, N.; Li, M.; Chen, Y.; Fries, P.; Griebel, P.J.; Baurhoo, B.; Zhao, X.; Guan, L.L. Distinct commensal bacteria associated with ingesta and mucosal epithelium in the gastrointestinal tracts of calves and chickens. FEMS Microbiol. Ecol. 2012, 79, 337–347. [Google Scholar] [CrossRef]
- Sommer, F.; Bäckhed, F. The gut microbiota—Masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef]
- Henderson, G.; Cox, F.; Ganesh, S.; Jonker, A.; Young, W.; Janssen, P.H. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Rep. 2015, 5, 14567. [Google Scholar] [CrossRef]
- Weimer, P.; Stevenson, D.; Mantovani, H.; Man, S. Host specificity of the ruminal bacterial community in the dairy cow following near-total exchange of ruminal contents. J. Dairy Sci. 2010, 93, 5902–5912. [Google Scholar] [CrossRef]
- Benson, A.K.; Kelly, S.A.; Legge, R.; Ma, F.; Low, S.J.; Kim, J.; Zhang, M.; Oh, P.L.; Nehrenberg, D.; Hua, K. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. USA 2010, 107, 18933–18938. [Google Scholar] [CrossRef]
- Yeoman, C.J.; Ishaq, S.L.; Bichi, E.; Olivo, S.K.; Lowe, J.; Aldridge, B.M. Biogeographical differences in the influence of maternal microbial sources on the early successional development of the bovine neonatal gastrointestinal tract. Sci. Rep. 2018, 8, 3197. [Google Scholar] [CrossRef]
- Cammack, K.M.; Austin, K.J.; Lamberson, W.R.; Conant, G.C.; Cunningham, H.C. Ruminnat nutrition symposium: Tiny but mighty: The role of the rumen microbes in livestock production. J. Anim. Sci. 2018, 96, 752–770. [Google Scholar] [CrossRef]
- Wang, J.; Fan, H.; Han, Y.; Zhao, J.; Zhou, Z. Characterization of the microbial communities along the gastrointestinal tract of sheep by 454 pyrosequencing analysis. Asian-Australas. J. Anim. Sci. 2016, 30, 100. [Google Scholar] [CrossRef]
- Li, K.; Shi, B.; Na, R. The colonization of rumen microbiota and intervention in pre-weaned ruminants. Animals 2023, 13, 994. [Google Scholar] [CrossRef]
- Xiao, H.; Yan, H.; Tian, P.; Ji, S.; Zhao, W.; Lu, C.; Zhang, Y.; Liu, Y. The effect of early colonized gut microbiota on the growth performance of suckling lambs. Front. Microbiol. 2023, 14, 1273444. [Google Scholar] [CrossRef]
- Blaser, M.J. Antibiotic use and its consequences for the normal microbiome. Science 2016, 352, 544–545. [Google Scholar] [CrossRef]
- Batey, R.; Nilon, P.; Page, S.; Browning, G.; Norris, J. Antimicrobial prescribing guidelines for sheep. Aust. Vet. J. 2024, 102, 103. [Google Scholar] [CrossRef]
- Foster, D.M.; Jacob, M.E.; Farmer, K.A.; Callahan, B.J.; Theriot, C.M.; Kathariou, S.; Cernicchiaro, N.; Prange, T.; Papich, M.G. Ceftiofur formulation differentially affects the intestinal drug concentration, resistance of fecal Escherichia coli, and the microbiome of steers. PLoS ONE 2019, 14, e0223378. [Google Scholar] [CrossRef]
- Rivera-Garcia, S.; Angelos, J.A.; Rowe, J.D.; Byrne, B.A.; Wetzlich, S.E.; Van Liew, D.B.; Tell, L.A. Pharmacokinetics of ceftiofur crystalline-free acid following subcutaneous administration of a single dose to sheep. Am. J. Vet. Res. 2014, 75, 290–295. [Google Scholar] [CrossRef]
- Sawant, A.; Sordillo, L.; Jayarao, B. A survey on antibiotic usage in dairy herds in Pennsylvania. J. Dairy Sci. 2005, 88, 2991–2999. [Google Scholar] [CrossRef]
- Wang, C.; Wei, S.; Chen, N.; Xiang, Y.; Wang, Y.; Jin, M. Characteristics of gut microbiota in pigs with different breeds, growth periods and genders. Microb. Biotechnol. 2022, 15, 793–804. [Google Scholar] [CrossRef]
- Kobayashi, R.; Nagaoka, K.; Nishimura, N.; Koike, S.; Takahashi, E.; Niimi, K.; Murase, H.; Kinjo, T.; Tsukahara, T.; Inoue, R. Comparison of the fecal microbiota of two monogastric herbivorous and five omnivorous mammals. Anim. Sci. J. 2020, 91, e13366. [Google Scholar] [CrossRef]
- Esteban-Blanco, C.; Gutiérrez-Gil, B.; Marina, H.; Pelayo, R.; Suárez-Vega, A.; Acedo, A.; Arranz, J.-J. The milk microbiota of the spanish churra sheep breed: New insights into the complexity of the milk microbiome of dairy species. Animals 2020, 10, 1463. [Google Scholar] [CrossRef]
- Pantoja-Feliciano, I.G.; Clemente, J.C.; Costello, E.K.; Perez, M.E.; Blaser, M.J.; Knight, R.; Dominguez-Bello, M.G. Biphasic assembly of the murine intestinal microbiota during early development. ISME J. 2013, 7, 1112–1115. [Google Scholar] [CrossRef]
- Minato, H.; Otsuka, M.; Shirasaka, S.; Itabashi, H.; Mitsumori, M. Colonization of microorganisms in the rumen of young calves. J. Gen. Appl. Microbiol. 1992, 38, 447–456. [Google Scholar] [CrossRef]
- Jami, E.; Israel, A.; Kotser, A.; Mizrahi, I. Exploring the bovine rumen bacterial community from birth to adulthood. ISME J. 2013, 7, 1069–1079. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef]
- Arshad, M.A.; Hassan, F.-U.; Rehman, M.S.; Huws, S.A.; Cheng, Y.; Din, A.U. Gut microbiome colonization and development in neonatal ruminants: Strategies, prospects, and opportunities. Anim. Nutr. 2021, 7, 883–895. [Google Scholar] [CrossRef]
- O’Mahony, L.; McCarthy, J.; Kelly, P.; Hurley, G.; Luo, F.; Chen, K.; O’Sullivan, G.C.; Kiely, B.; Collins, J.K.; Shanahan, F. Lactobacillus and bifidobacterium in irritable bowel syndrome: Symptom responses and relationship to cytokine profiles. Gastroenterology 2005, 128, 541–551. [Google Scholar] [CrossRef]
- Kim, Y.-H.; Nagata, R.; Ohtani, N.; Ichijo, T.; Ikuta, K.; Sato, S. Effects of dietary forage and calf starter diet on ruminal pH and bacteria in Holstein calves during weaning transition. Front. Microbiol. 2016, 7, 1575. [Google Scholar] [CrossRef]
- Kim, E.-T.; Lee, S.-J.; Kim, T.-Y.; Lee, H.-G.; Atikur, R.M.; Gu, B.-H.; Kim, D.-H.; Park, B.-Y.; Son, J.-K.; Kim, M.-H. Dynamic changes in fecal microbial communities of neonatal dairy calves by aging and diarrhea. Animals 2021, 11, 1113. [Google Scholar] [CrossRef]
- Singer, R.S.; Patterson, S.K.; Wallace, R.L. Effects of therapeutic ceftiofur administration to dairy cattle on Escherichia coli dynamics in the intestinal tract. Appl. Environ. Microbiol. 2008, 74, 6956–6962. [Google Scholar] [CrossRef]
- Hornish, R.E.; Katarski, S. Cephalosporins in veterinary medicine-ceftiofur use in food animals. Curr. Top. Med. Chem. 2002, 2, 717–731. [Google Scholar] [CrossRef]
- Wren, C.M.; Cowper, J.; Greer, N.; Goldin, L.; Perry, A. Effect of reduced fluoroquinolone use on cephalosporin use, susceptibilities and Clostridioides difficile infections. Antibiotics 2022, 11, 1312. [Google Scholar] [CrossRef]
- Beyer, A.; Baumann, S.; Scherz, G.; Stahl, J.; von Bergen, M.; Friese, A.; Roesler, U.; Kietzmann, M.; Honscha, W. Effects of ceftiofur treatment on the susceptibility of commensal porcine E. coli–comparison between treated and untreated animals housed in the same stable. BMC Vet. Res. 2015, 11, 265. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, F.; Fischer-Tlustos, A.; Neves, A.; He, Z.; Steele, M.; Guan, L. Metagenomic analysis revealed the individualized shift in ileal microbiome of neonatal calves in response to delaying the first colostrum feeding. J. Dairy Sci. 2021, 104, 8783–8797. [Google Scholar] [CrossRef]
- Ruczizka, U.; Metzler-Zebeli, B.; Unterweger, C.; Mann, E.; Schwarz, L.; Knecht, C.; Hennig-Pauka, I. Early parenteral administration of ceftiofur has gender-specific short-and long-term effects on the fecal microbiota and growth in pigs from the suckling to growing phase. Animals 2019, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Iwiński, H.; Wódz, K.; Chodkowska, K.; Nowak, T.; Różański, H. In vitro evaluation of antimicrobial effect of phytobiotics mixture on Salmonella spp. isolated from chicken broiler. Antibiotics 2022, 11, 868. [Google Scholar] [CrossRef]
- VT Nair, D.; Venkitanarayanan, K.; Kollanoor Johny, A. Antibiotic-resistant Salmonella in the food supply and the potential role of antibiotic alternatives for control. Foods 2018, 7, 167. [Google Scholar] [CrossRef]
- Deusch, S.; Tilocca, B.; Camarinha-Silva, A.; Seifert, J. News in livestock research—Use of Omics-technologies to study the microbiota in the gastrointestinal tract of farm animals. Comput. Struct. Biotechnol. J. 2015, 13, 55–63. [Google Scholar] [CrossRef]
- Nowland, T.L.; Torok, V.A.; Low, W.Y.; Barton, M.D.; Plush, K.J.; Kirkwood, R.N. Faecal microbiota analysis of piglets during lactation. Animals 2020, 10, 762. [Google Scholar] [CrossRef] [PubMed]
- Rutjens, S.; Vereecke, N.; De Spiegelaere, W.; Croubels, S.; Devreese, M. Intestinal Exposure to Ceftiofur and Cefquinome after Intramuscular Treatment and the Impact of Ceftiofur on the Pig Fecal Microbiome and Resistome. Antibiotics 2022, 11, 342. [Google Scholar] [CrossRef]
- Candon, S.; Perez-Arroyo, A.; Marquet, C.; Valette, F.; Foray, A.-P.; Pelletier, B.; Milani, C.; Ventura, M.; Bach, J.-F.; Chatenoud, L. Antibiotics in early life alter the gut microbiome and increase disease incidence in a spontaneous mouse model of autoimmune insulin-dependent diabetes. PLoS ONE 2015, 10, e0125448. [Google Scholar] [CrossRef]
- Huda, M.N.; Salvador, A.C.; Barrington, W.T.; Gacasan, C.A.; D’Souza, E.M.; Ramirez, L.D.; Threadgill, D.W.; Bennett, B.J. Gut microbiota and host genetics modulate the effect of diverse diet patterns on metabolic health. Front. Nutr. 2022, 9, 896348. [Google Scholar] [CrossRef]
- Thompson, C.L.; Wang, B.; Holmes, A.J. The immediate environment during postnatal development has long-term impact on gut community structure in pigs. ISME J. 2008, 2, 739–748. [Google Scholar] [CrossRef]
- Schwarzer, M.; Strigini, M.; Leulier, F. Gut microbiota and host juvenile growth. Calcif. Tissue Int. 2018, 102, 387–405. [Google Scholar] [CrossRef]
- Liu, L.; Kirst, M.E.; Zhao, L.; Li, E.; Wang, G.P. Microbiome Resilience despite a Profound Loss of Minority Microbiota following Clindamycin Challenge in Humanized Gnotobiotic Mice. Microbiol. Spectr. 2022, 10, e01960-e01921. [Google Scholar] [CrossRef]
- Bengtsson-Palme, J.; Kristiansson, E.; Larsson, D.J. Environmental factors influencing the development and spread of antibiotic resistance. FEMS Microbiol. Rev. 2018, 42, fux053. [Google Scholar] [CrossRef] [PubMed]
- Berglund, B. Environmental dissemination of antibiotic resistance genes and correlation to anthropogenic contamination with antibiotics. Infect. Ecol. Epidemiol. 2015, 5, 28564. [Google Scholar] [CrossRef] [PubMed]
- Gasparrini, A.J.; Wang, B.; Sun, X.; Kennedy, E.A.; Hernandez-Leyva, A.; Ndao, I.M.; Tarr, P.I.; Warner, B.B.; Dantas, G. Persistent metagenomic signatures of early-life hospitalization and antibiotic treatment in the infant gut microbiota and resistome. Nat. Microbiol. 2019, 4, 2285–2297. [Google Scholar] [CrossRef]
- Sheedy, D.B.; Okello, E.; Williams, D.R.; Precht, K.; Cella, E.; Lehenbauer, T.W.; Aly, S.S. Effect of antimicrobial treatment on the dynamics of ceftiofur resistance in Enterobacteriaceae from adult California dairy cows. Microorganisms 2021, 9, 828. [Google Scholar] [CrossRef]
- Jiang, X.; Yang, H.; Dettman, B.; Doyle, M.P. Analysis of fecal microbial flora for antibiotic resistance in ceftiofur-treated calves. Foodbourne Pathog. Dis. 2006, 3, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Weinroth, M.D.; Scott, H.M.; Norby, B.; Loneragan, G.H.; Noyes, N.R.; Rovira, P.; Doster, E.; Yang, X.; Woerner, D.R.; Morley, P.S. Effects of ceftiofur and chlortetracycline on the resistomes of feedlot cattle. Appl. Environ. Microbiol. 2018, 84, e00610–e00618. [Google Scholar] [CrossRef]
- Dong, L.; Meng, L.; Liu, H.; Wu, H.; Schroyen, M.; Zheng, N.; Wang, J. Effect of Cephalosporin Treatment on the Microbiota and Antibiotic Resistance Genes in Feces of Dairy Cows with Clinical Mastitis. Antibiotics 2022, 11, 117. [Google Scholar] [CrossRef]
- Gibson, M.K.; Crofts, T.S.; Dantas, G. Antibiotics and the developing infant gut microbiota and resistome. Curr. Opin. Microbiol. 2015, 27, 51–56. [Google Scholar] [CrossRef]
- Martin, K.L.; Clapham, M.O.; Davis, J.L.; Baynes, R.E.; Lin, Z.; Vickroy, T.W.; Riviere, J.E.; Tell, L.A. Extralabel drug use in small ruminants. J. Am. Vet. Med. Assoc. 2018, 253, 1001–1009. [Google Scholar] [CrossRef]
- McMahon, T.; Abdelmesih, M.; Gill, A. Evaluation of DNA extraction methods for the detection of Shiga toxin producing Escherichia coli in food by polymerase chain reaction. Int. J. Food Microbiol. 2023, 404, 110317. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. Babraham Bioinformatics-FastQC a Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 22 April 2025).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A. The metagenomics RAST server–a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [PubMed]
- Lindgreen, S.; Adair, K.L.; Gardner, P.P. An evaluation of the accuracy and speed of metagenome analysis tools. Sci. Rep. 2016, 6, 19233. [Google Scholar] [CrossRef] [PubMed]
- Keegan, K.P.; Glass, E.M.; Meyer, F. MG-RAST, a metagenomics service for analysis of microbial community structure and function. In Microbial Environmental Genomics (MEG); Springer: Berlin/Heidelberg, Germany, 2016; pp. 207–233. [Google Scholar]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Zeineldin, M.; Megahed, A.; Burton, B.; Blair, B.; Aldridge, B.; Lowe, J.F. Effect of single dose of antimicrobial administration at birth on fecal microbiota development and prevalence of antimicrobial resistance genes in piglets. Front. Microbiol. 2019, 10, 1414. [Google Scholar] [CrossRef]
- Collado, N.; Buttiglieri, G.; Marti, E.; Ferrando-Climent, L.; Rodriguez-Mozaz, S.; Barceló, D.; Comas, J.; Rodriguez-Roda, I. Effects on activated sludge bacterial community exposed to sulfamethoxazole. Chemosphere 2013, 93, 99–106. [Google Scholar] [CrossRef]
- Dutil, L.; Irwin, R.; Finley, R.; Ng, L.K.; Avery, B.; Boerlin, P.; Bourgault, A.M.; Cole, L.; Daignault, D.; Desruisseau, A.; et al. Ceftiofur resistance in Salmonella enterica serovar Heidelberg from chicken meat and humans, Canada. Emerg. Infect. Dis. 2010, 16, 48. [Google Scholar] [CrossRef]
- Rafii, F.; Williams, A.J.; Park, M.; Sims, L.M.; Heinze, T.M.; Cerniglia, C.E.; Sutherland, J.B. Isolation of bacterial strains from bovine fecal microflora capable of degradation of ceftiofur. Vet. Microbiol. 2009, 139, 89–96. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donia, M.; Aref, N.-E.; Zeineldin, M.; Megahed, A.; Blair, B.; Lowe, J.; Aldridge, B. Impact of Parenteral Ceftiofur on Developmental Dynamics of Early Life Fecal Microbiota and Antibiotic Resistome in Neonatal Lambs. Antibiotics 2025, 14, 434. https://doi.org/10.3390/antibiotics14050434
Donia M, Aref N-E, Zeineldin M, Megahed A, Blair B, Lowe J, Aldridge B. Impact of Parenteral Ceftiofur on Developmental Dynamics of Early Life Fecal Microbiota and Antibiotic Resistome in Neonatal Lambs. Antibiotics. 2025; 14(5):434. https://doi.org/10.3390/antibiotics14050434
Chicago/Turabian StyleDonia, Mohamed, Nasr-Eldin Aref, Mohamed Zeineldin, Ameer Megahed, Benjamin Blair, James Lowe, and Brian Aldridge. 2025. "Impact of Parenteral Ceftiofur on Developmental Dynamics of Early Life Fecal Microbiota and Antibiotic Resistome in Neonatal Lambs" Antibiotics 14, no. 5: 434. https://doi.org/10.3390/antibiotics14050434
APA StyleDonia, M., Aref, N.-E., Zeineldin, M., Megahed, A., Blair, B., Lowe, J., & Aldridge, B. (2025). Impact of Parenteral Ceftiofur on Developmental Dynamics of Early Life Fecal Microbiota and Antibiotic Resistome in Neonatal Lambs. Antibiotics, 14(5), 434. https://doi.org/10.3390/antibiotics14050434