
Forced Degradation Studies and Development and Validation of HPLC-UV Method for the Analysis of Velpatasvir Copovidone Solid Dispersion

 ,
, .jpg) , and
, and
Abstract
:1. Introduction
2. Experimental
2.1. Materials and Chemicals
2.2. Instrumentation
2.3. Reference and Sample Stock Solutions
2.4. Forced Degradation Studies
2.5. Optimization of Chromatographic Conditions
3. Results and Discussion
3.1. Method Validation
3.2. Recovery Studies
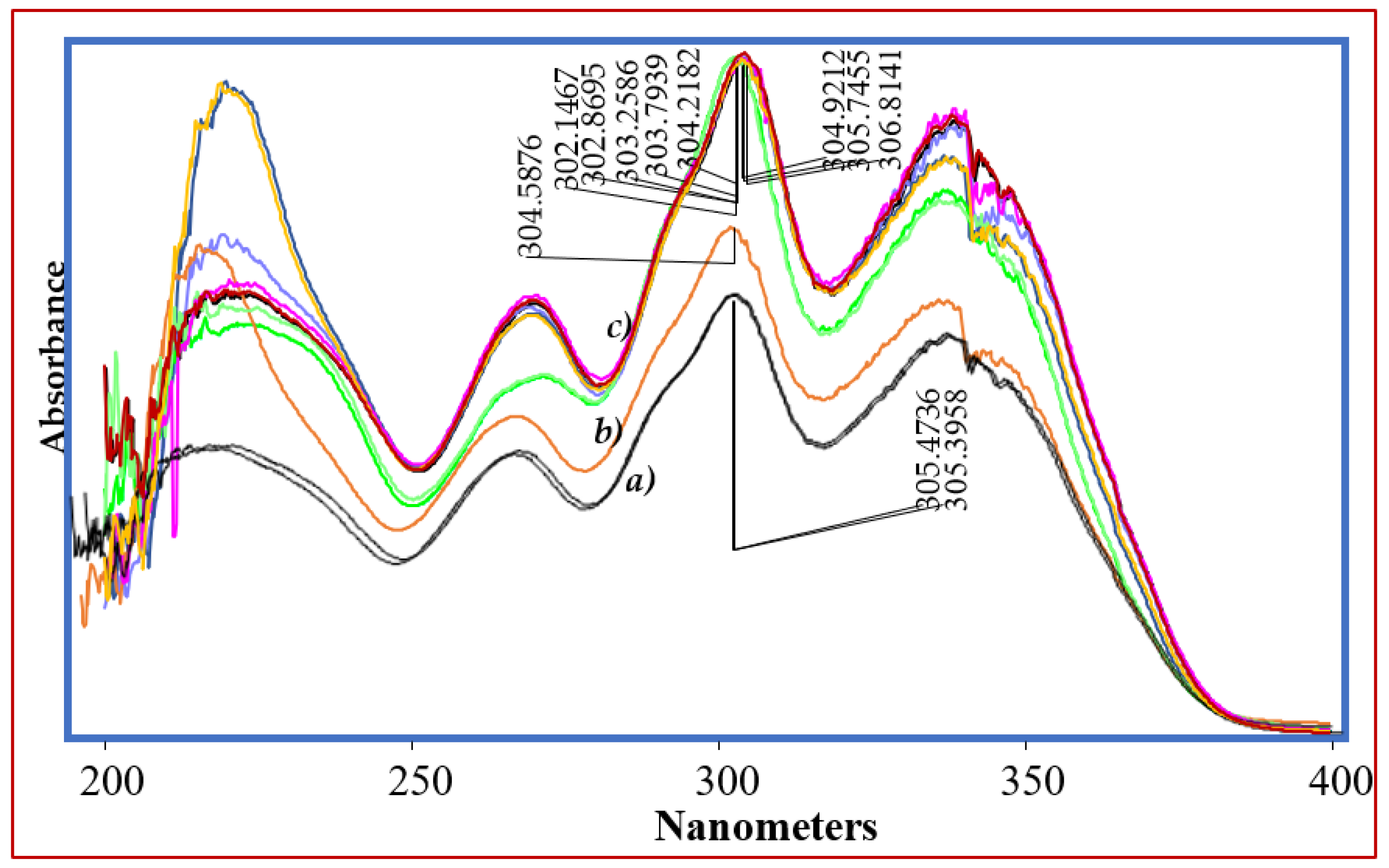
3.3. Specificity
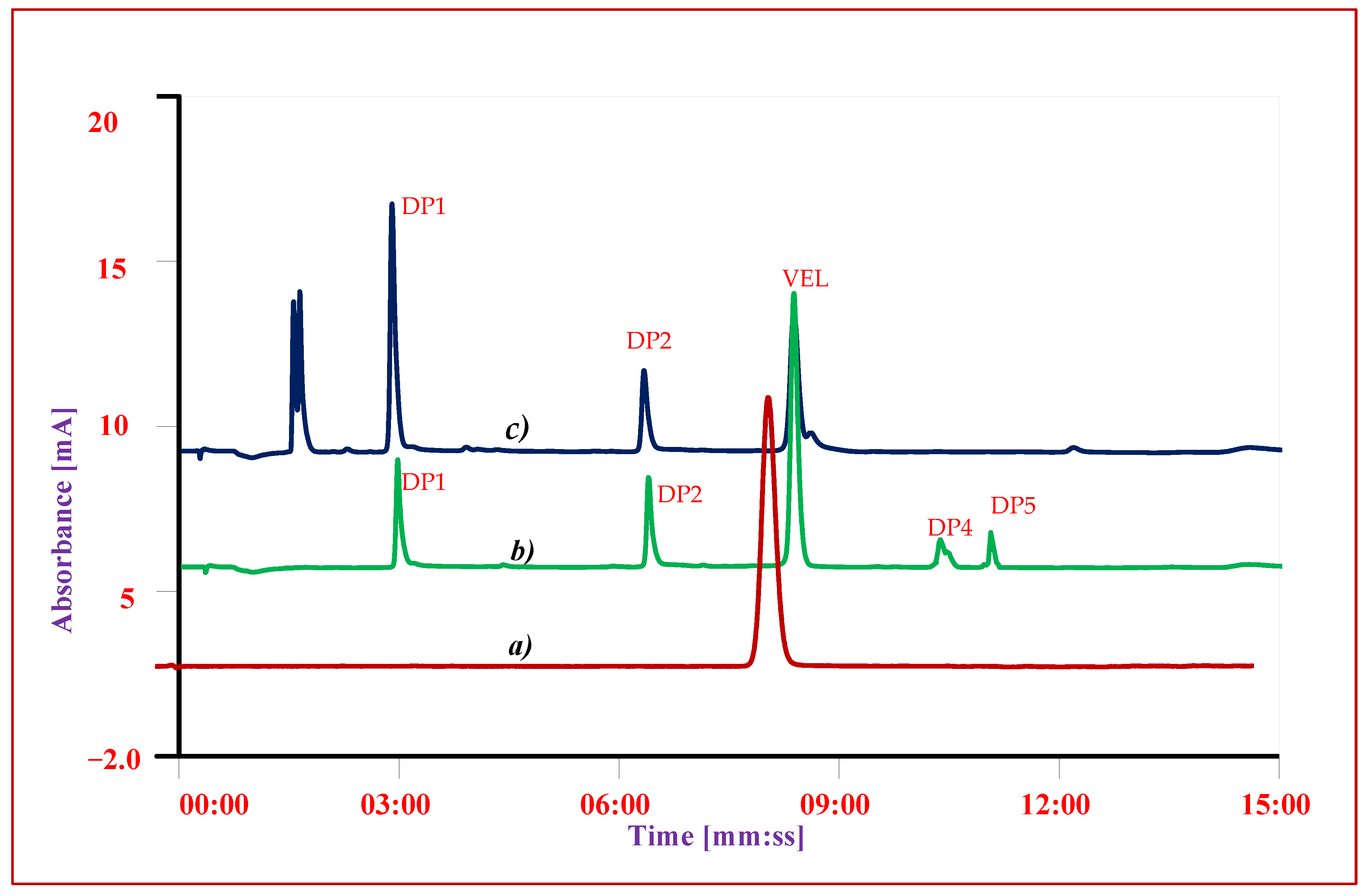
3.4. Precision
3.5. Robustness
3.6. Linearity and Range
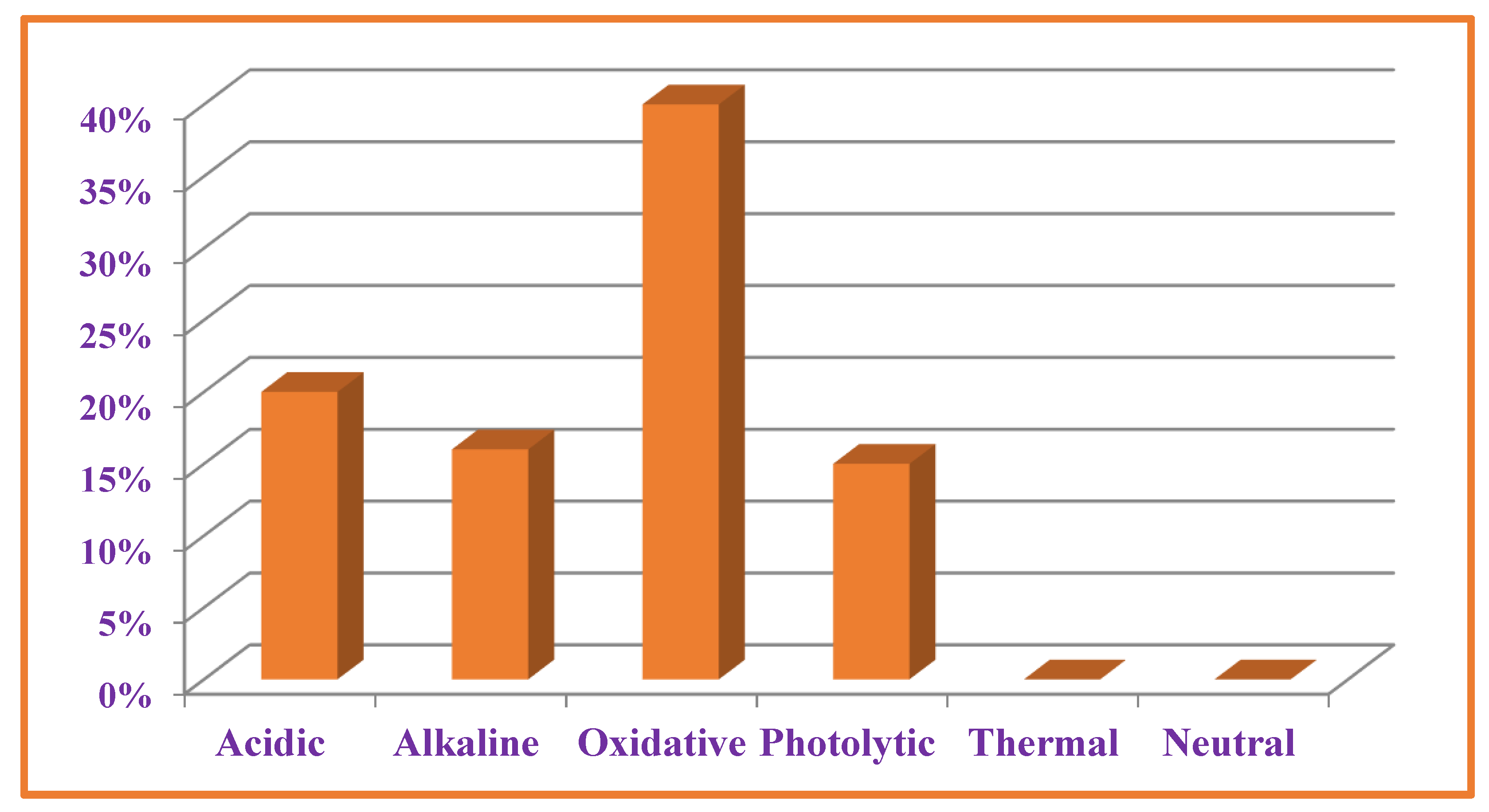
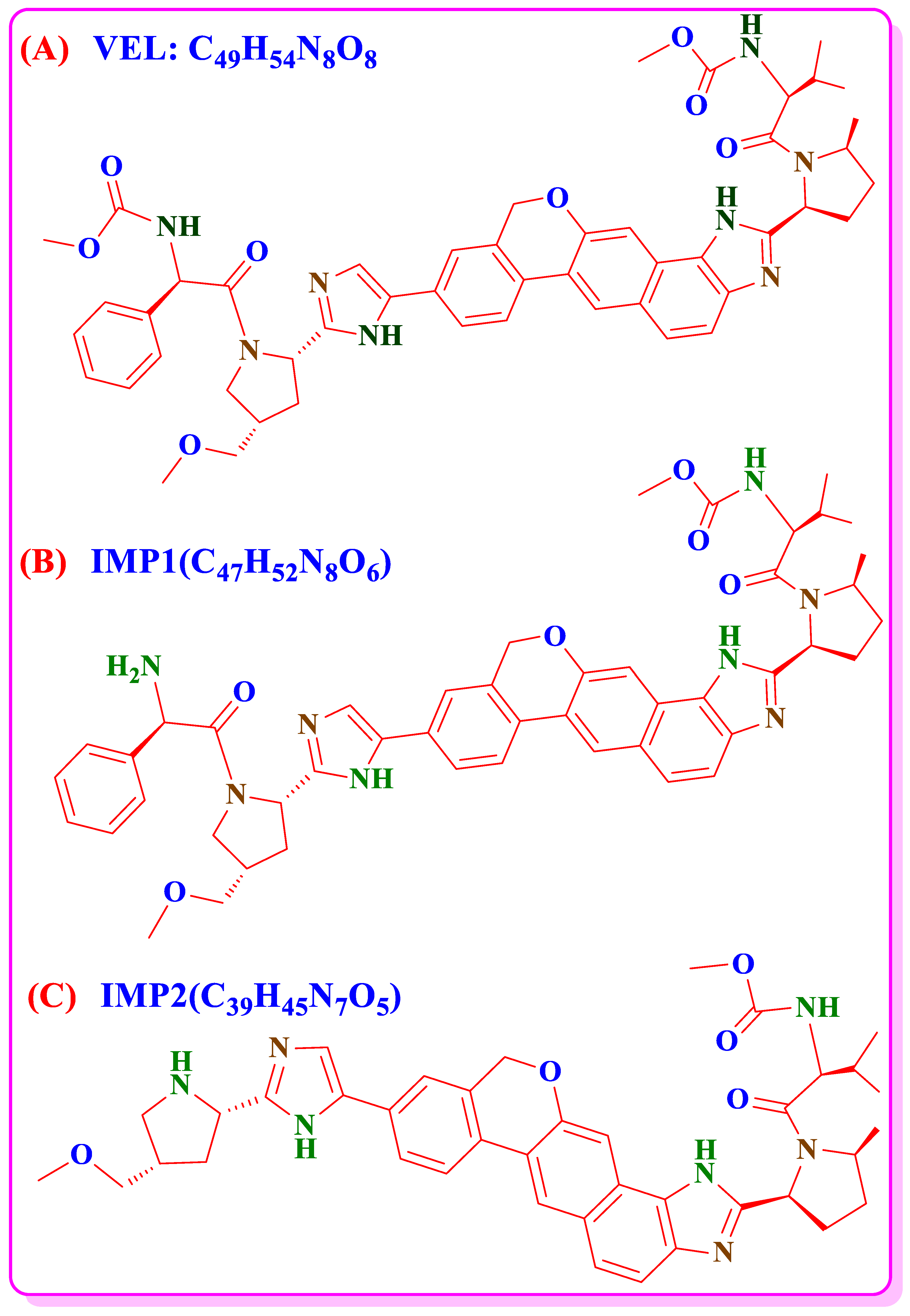
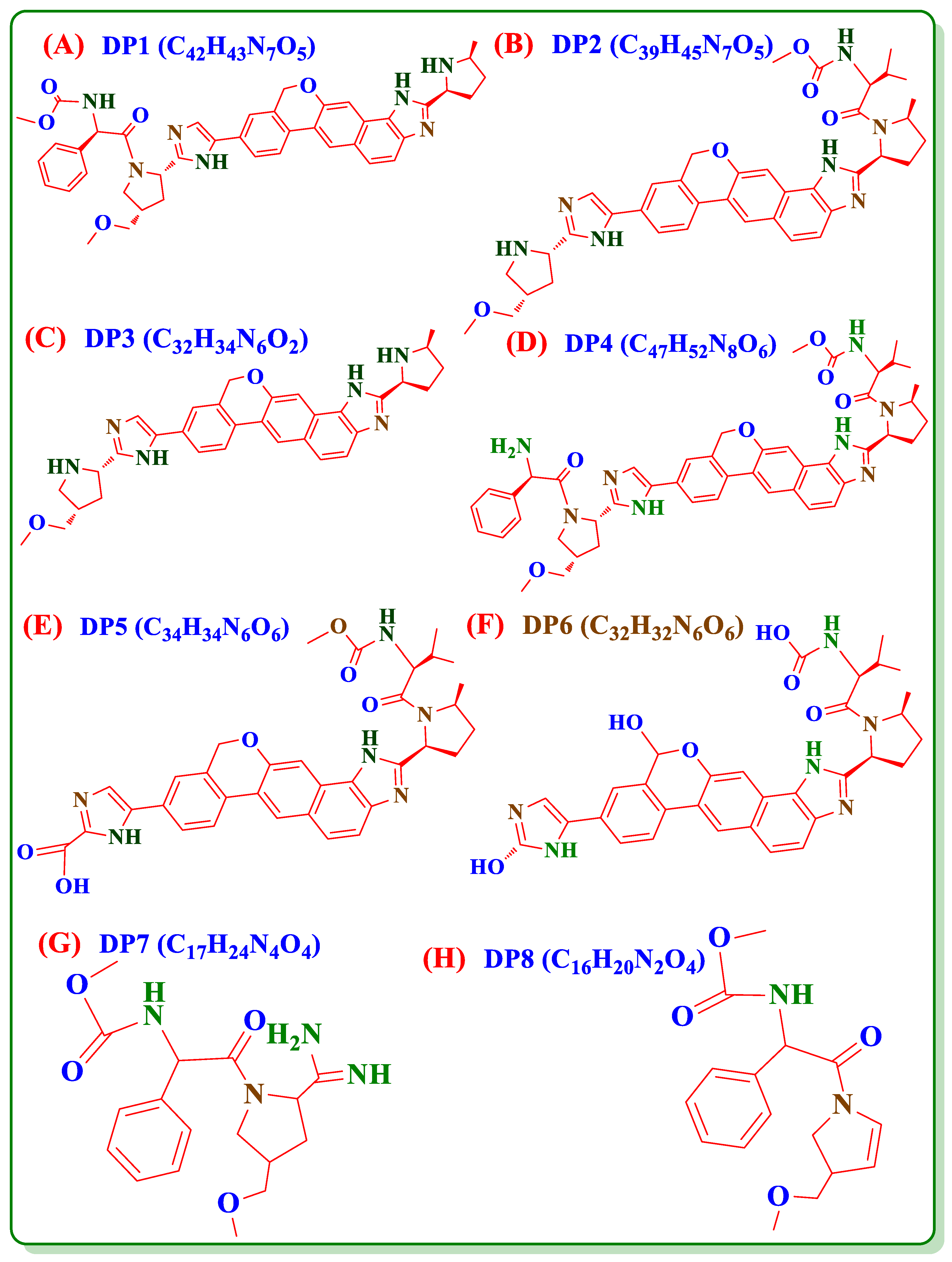
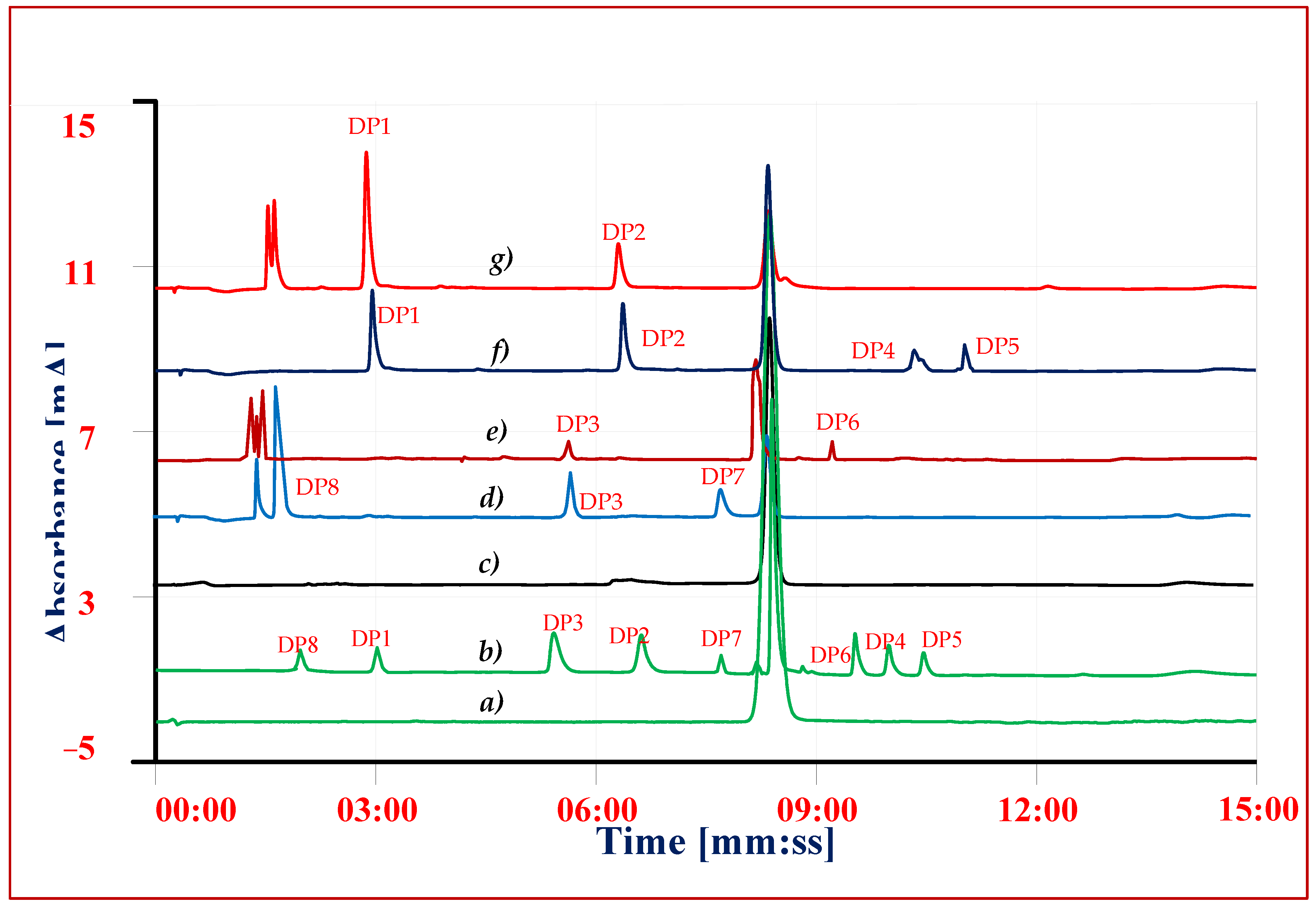
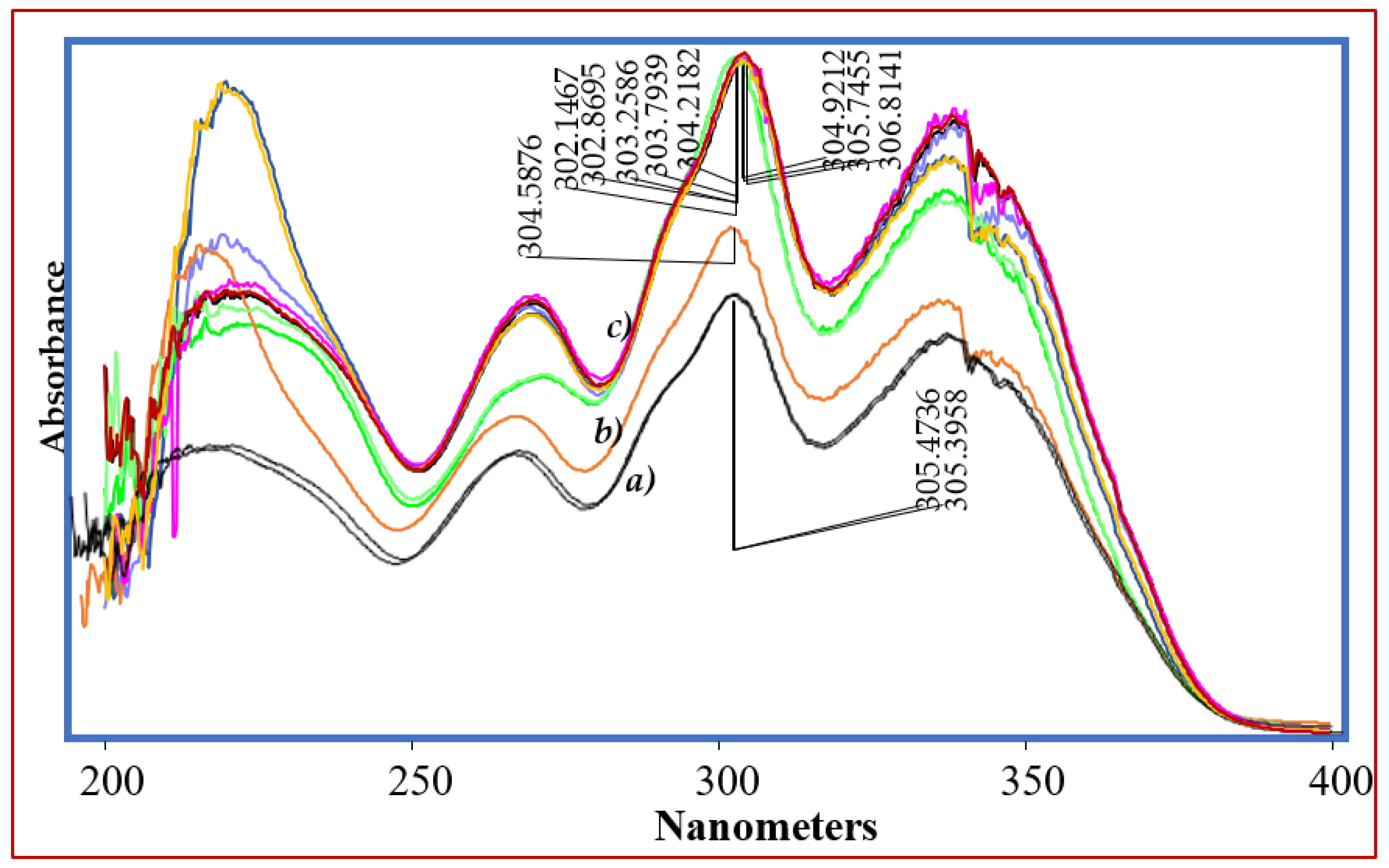
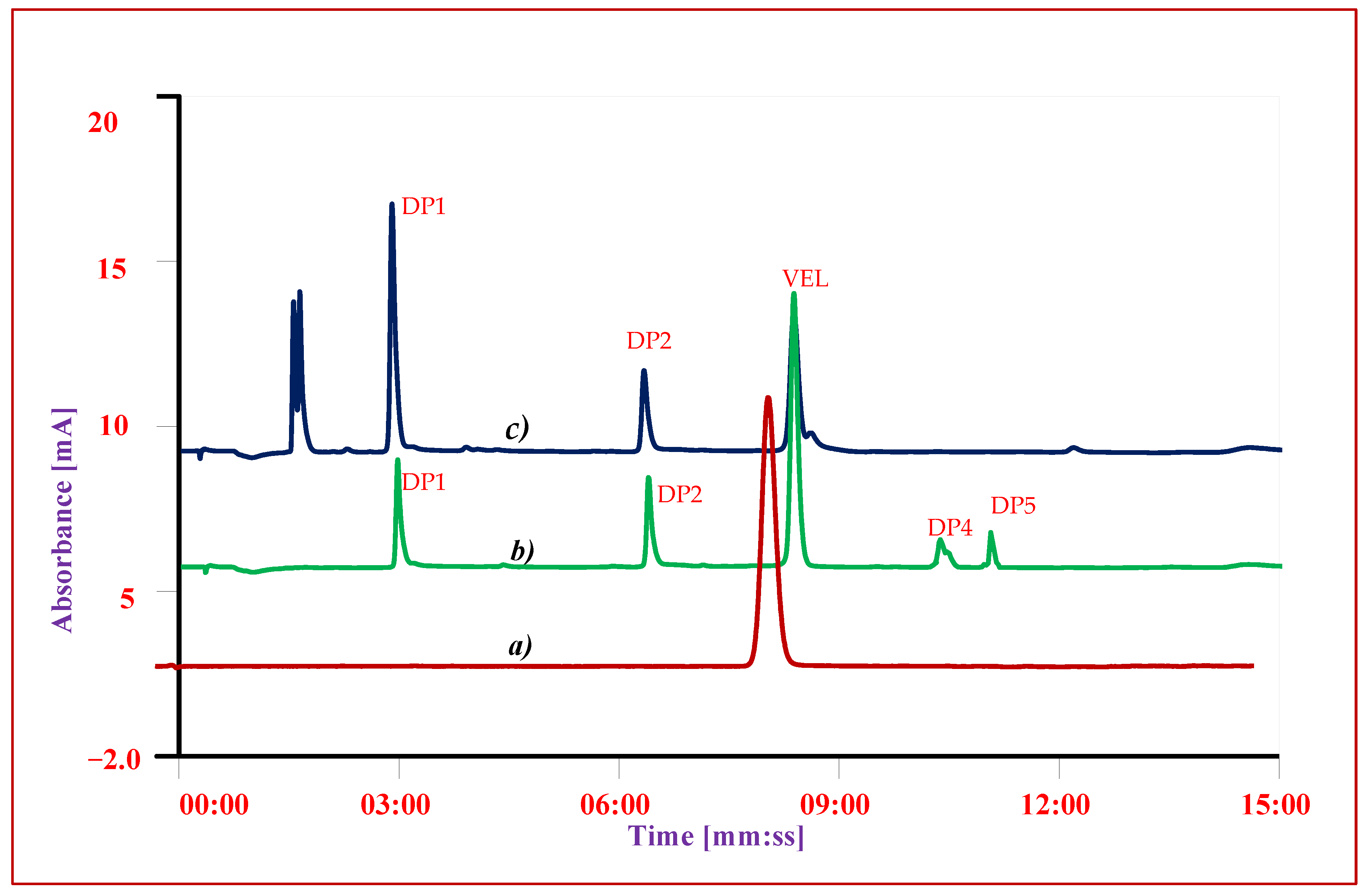
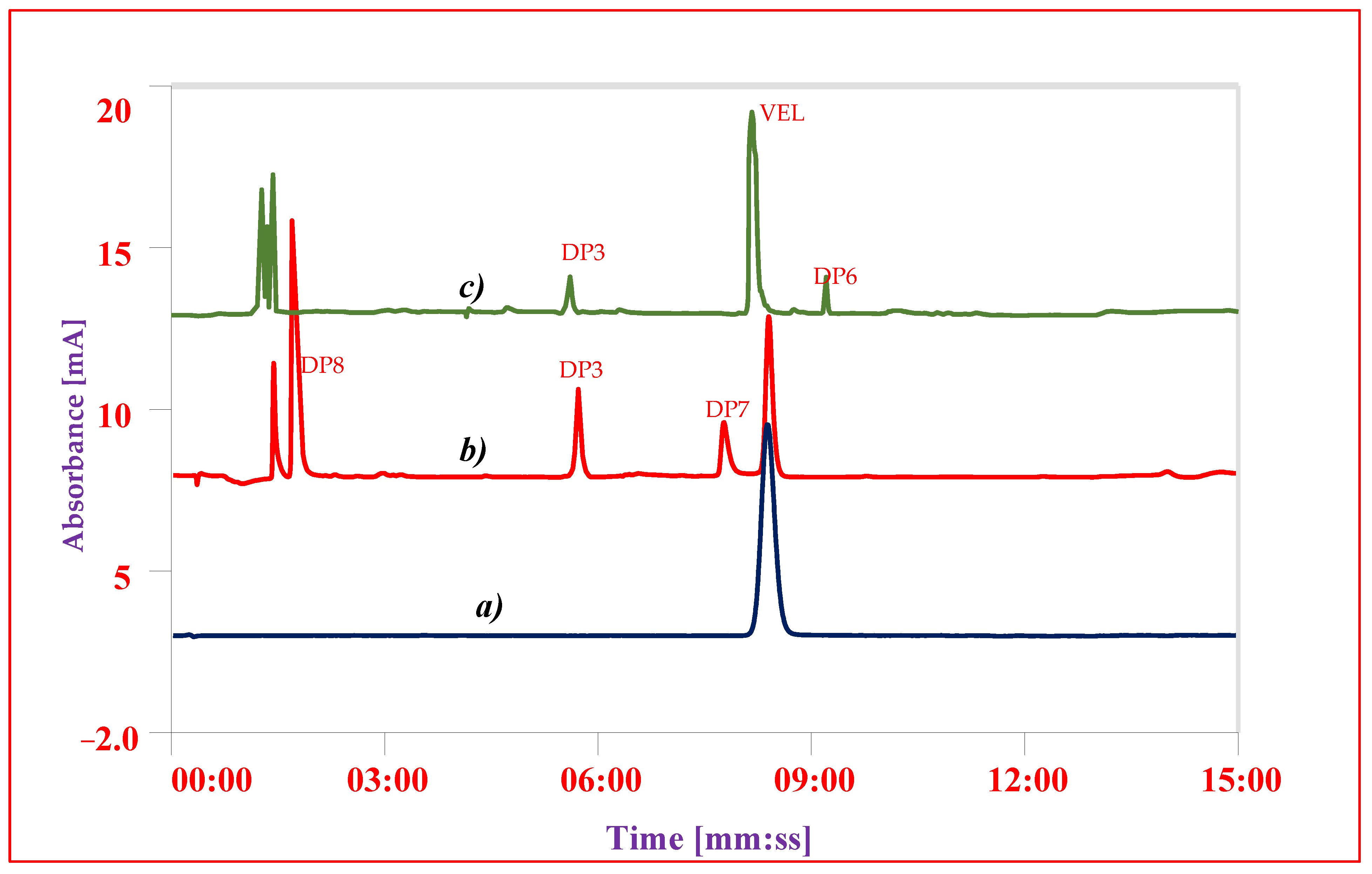
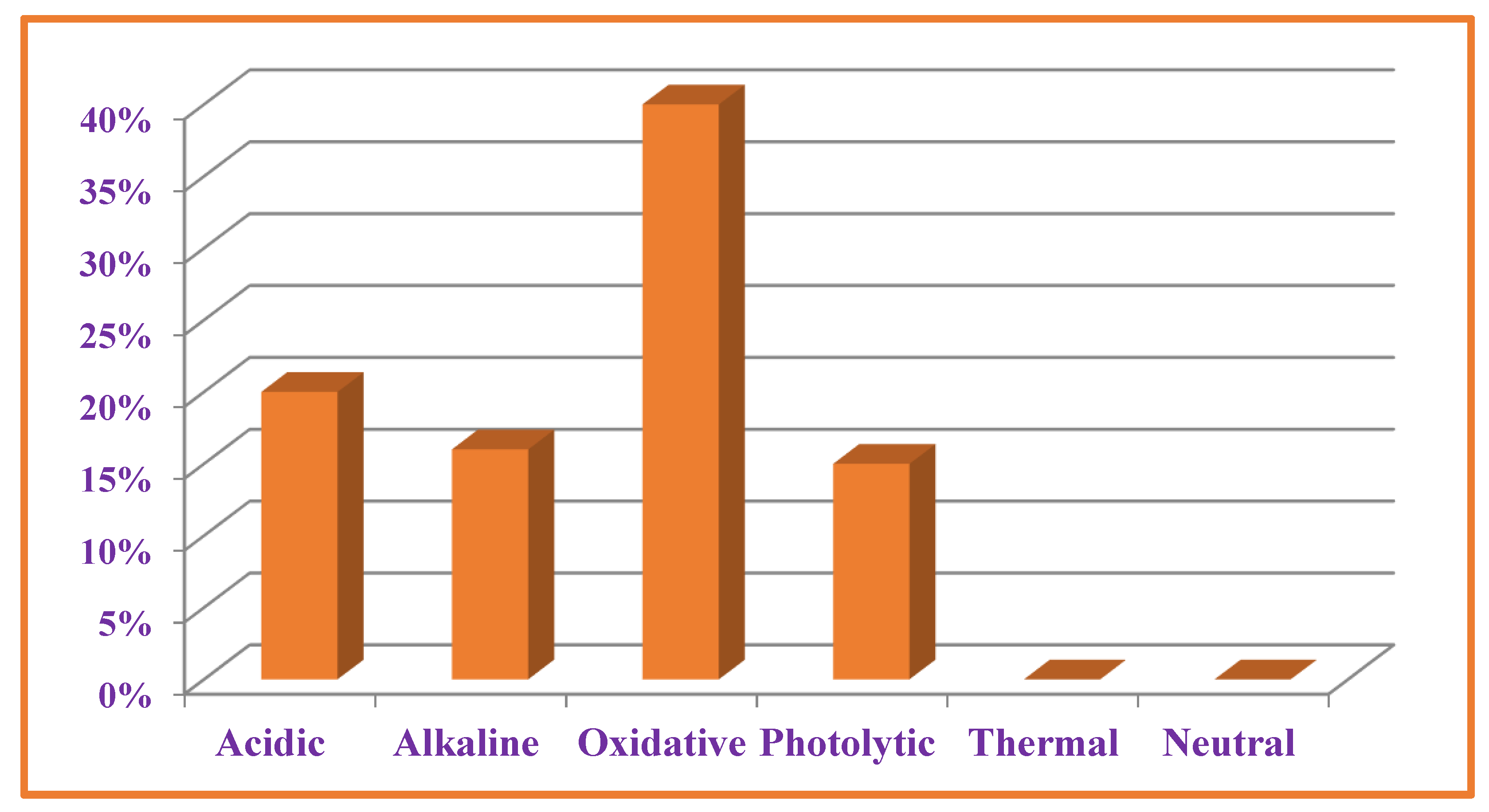
3.7. Forced Degradation Studies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Medium | Material | At Refrigerator (02–08 °C) Protected from Light for 7 Days | At Room Temperature (15–25 °C) Protected from Light for 7 Days | At 80 °C Refluxed for 4 h Protected from Light | At 80 °C Refluxed for 8 h Protected from Light | At 40 °C) Exposed to 1.2 Milli lux h m−2 of Fluorescence Light for 7 Days | At 80 °C/75 ± 5% RH Protected from Light and Moisture for 24 h | At 105 °C Temperature Protected from Light and Moisture for 24 h |
|---|---|---|---|---|---|---|---|---|
| In neutral condition | VEL | 100.13 ± 0.27 0.28% | 98.14 ± 0.74 0.69% | 97.51 ± 0.84 0.74% | 98.27 ± 1.18 1.65% | - | - | - |
| VEL-CSD | 99.44 ± 0.79 0.65% | 99.73 ± 1.14 0.79% | 98.69 ± 1.17 1.20% | 98.66 ± 1.12 1.24% | - | - | - | |
| Acid Hydrolysis (5M HCl) | VEL | - | - | 87.26 ± 3.24 2.47% | 79.84 ± 4.46 5.12% | - | - | - |
| VEL-CSD | - | 90.41 ± 2.37 3.10% | 94.56 ± 3.49 2.18% | - | ||||
| Alkaline hydrolysis (1M sodium hydroxide solutions) | VEL | - | - | 90.51 ± 2.29 1.77% | 87.98 ± 3.87 3.21% | - | - | - |
| VEL-CSD | - | - | 92.36 ± 2.24 1.96% | 89.12 ± 1.14 1.25% | - | - | - | |
| Oxidative conditions (10% H2O2) | VEL | - | - | 76.66 ± 3.21 2.45% | 79.64 ± 5.18 4.75% | - | - | - |
| VEL-CSD | 91.33 ± 6.41 4.82% | 83.45 ± 4.79 2.87 | - | - | - | |||
| Photolytic condition | VEL | - | - | 84.16 ± 2.11 1.98% | - | - | ||
| VEL-CSD | - | - | 91.64 ± 1.75 154% | - | - | |||
| Thermal stress | VEL | - | - | 99.54 ± 0.78 0.78% | 98.71 ± 0.24 0.24% | 98.27 ± 0.67 0.68% | 99.10 ± 0.25 0.25% | 99.16 ± 1.16 1.15% |
| VEL-CSD | - | 98.21 ± 01.31 1.67% | 100.47 ± 1.61 1.52% | 99.44 ± 0.75 0.38% | 98.91 ± 0.31 0.46% | 99.22 ± 0.78 0.80% |
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ugah, U.I.; Alo, M.N.; Gloria, U.C. Epidemiology of hepatitis b virus, hepatitis C virus and human immunodeficiency virus co-infection among assymptomatic persons resident in Alex Ekwueme federal university Ndufu-alike. Sci. Afr. 2021, 14, 00985. [Google Scholar] [CrossRef]
- Thomas, D.L.; Seeff, L.B. Natural history of hepatitis C. Clin. Liver Dis. 2005, 9, 383–398. [Google Scholar] [CrossRef] [PubMed]
- Sandmann, L.; Wedemeyer, H. New Treatments for Chronic Hepatitis B Virus/Hepatitis D Virus Infection. Clin. Liver Dis. 2021, 25, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Maus, A.; Strait, L.; Zhu, D. Nanoparticles as delivery vehicles for antiviral therapeutic drugs. Eng. Regen. 2021, 2, 31–46. [Google Scholar] [CrossRef]
- Naggie, S.; Patel, K.; McHutchison, J. Hepatitis C virus directly acting antivirals: Current developments with NS3/4A HCV serine protease inhibitors. J. Antimicrob. Chemother. 2010, 65, 2063–2069. [Google Scholar] [CrossRef]
- Wei, L.; Lim, S.G.; Xie, Q.; Văn, K.N.; Piratvisuth, T.; Huang, Y.; Wu, S.; Xu, M.; Tang, H.; Cheng, J.; et al. Sofosbuvir–velpatasvir for treatment of chronic hepatitis C virus infection in Asia: A single-arm, open-label, phase 3 trial. Lancet Gastroenterol. Hepatol. 2019, 4, 127–134. [Google Scholar] [CrossRef]
- FDA Approves Epclusa for Treatment of Chronic Hepatitis C Virus Infection. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-epclusa-treatment-chronic-hepatitis-c-virus-infection#:~:text=The%20U.S.%20Food%20and%20Drug,combination%20with%20the%20drug%20ribavirin (accessed on 13 November 2021).
- Yang, Y.; Lv, Y.; Shen, C.; Shi, T.; He, H.; Qi, J.; Dong, X.; Zhao, W.; Lu, Y.; Wu, W. In vivo dissolution of poorly water-soluble drugs: Proof of concept based on fluorescence bioimaging. Acta. Pharm. Sin. B 2021, 11, 1056–1068. [Google Scholar] [CrossRef]
- Devane, J.; Butler, J. The impact of in vitro-in vivo relationships on product development. Pharm. Technol. 1997, 21, 146–159. [Google Scholar]
- Bari, S.B.; Kadam, B.R.; Jaiswal, Y.S.; Shirkhedkar, A.A. Impurity profile: Significance in active pharmaceutical ingredient. Eurasian J. Anal. Chem. 2007, 2, 32–53. [Google Scholar] [CrossRef]
- Maggio, R.M.; Calvo, N.L.; Vignaduzzo, S.E.; Kaufman, T.S. Pharmaceutical impurities and degradation products uses and applications of NMR techniques. J. Pharm. Biomed. Anal. 2014, 101, 102–122. [Google Scholar] [CrossRef]
- Singh, S.; Junwal, M.; Modhe, G.; Tiwari, H.; Kurmi, M.; Parashar, N.; Sidduri, P. Forced degradation studies to assess the stability of drugs and products. TrAC Trends Anal. Chem. 2013, 49, 71–88. [Google Scholar] [CrossRef]
- Blessy, M.; Patel, R.D.; Prajapati, P.N.; Agrawal, Y. Development of forced degradation and stability indicating studies of drugs—A review. J. Pharm. Anal. 2014, 4, 159–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamal, R.M.; Gawad, S.A.; Belal, F.F.; Moustapha, M.E. Selective and sensitive spectrofluorimetric quantification of velpatasvir in presence of sofosbuvir. Application to their co-formulated tablet. RSC Adv. 2018, 8, 32909–32915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moustapha, M.E.; El-Gamal, R.M.; Belal, F.F. Two novel UPLC methods utilizing two different analytical columns and different detection approaches for the simultaneous analysis of velpatasvir and sofosbuvir: Application to their co-formulated tablet. BMC Chem. 2019, 13, 118. [Google Scholar] [CrossRef]
- Kamal, A.H.; Mabrouk, M.M.; Bebawy, L.I.; Mekky, M.A. Spectrophotometric and robust UPLC methods for simultaneous determination of velpatasvir and sofosbuvir in their tablet. Microchem. J. 2019, 149, 103996. [Google Scholar] [CrossRef]
- Rezk, M.R.; Monir, H.H.; Marzouk, H.M. Spectrophotometric assessment of the brand-new antiviral combination: Sofosbuvir and velpatasvir in their pure forms and pharmaceutical formulation. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2019, 213, 159–166. [Google Scholar] [CrossRef]
- Zaman, B.; Waseem, H. Development of Stability Indicating HPLC-UV Method for Determination of Process Impurities and Degradation Products in Sofosbuvir and Velpatasvir Tablets. Pharm. Chem. J. 2021, 54, 1295–1305. [Google Scholar] [CrossRef]
- Bandla, J.; Ganapaty, S. Stability indicating RP-HPLC method development and validation for the simultaneous determination of Sofosbuvir and Velpatasvir in tablet dosage forms. Indian J. Pharm. Biol. Res. 2017, 5, 10–16. [Google Scholar] [CrossRef]
- Omar, M.A.; Abdel-Lateef, M.A.; Ali, R.; Derayea, S.M. Study on fluorescence properties of HCV antiviral (velpatasvir) and its fluorimetric determination in presence of sofosbuvir; application to stability study and human plasma. J. Lumin. 2018, 33, 1249–1256. [Google Scholar] [CrossRef]
- Priyanka, K.; Vinutha, K.; Sridevi, P.; Ramya, B.; Bhagavan Raju, M. A Stability Indicating RP-HPLC method for simultaneous estimation of Velpatasvir and Sofosbuvir in its bulk and tablet dosage form. Am. J. Pharmtech Res. 2018, 8, 129–139. [Google Scholar] [CrossRef]
- Rao, P.V.; Rao, A.L.; Prasad, S. Validated Stability Indicating RP-HPLC method for estimation of antiviral class of drugs Sofosbuvir and Velpatasvir in combination and its comparison with reported methods. Res. J. Pharm. Technol. 2018, 11, 5425–5430. [Google Scholar] [CrossRef]
- Vanaja, B.; Vageesh, N.M.; Kistayya, C.; Urukundu, V. RP-HPLC method development and validation for simultaneous estimation of sofosbuvir and velpatasvir in pure and pharmaceutical dosage form. Innov. Int. J. Med. Pharm. Sci. 2018, 3, 45–48. [Google Scholar]
- European Medicine Agency, Assessment Report Sofosbuvir/Velpatasvir. Available online: https://www.ema.europa.eu/en/documents/assessment-report/epclusa-epar-public-assessment-report_en.pdf (accessed on 14 October 2021).
- U.S. Food and Drug Administration. Chemistry Review Sofosbuvir/Velpatasvir. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/208341Orig1s000TOC.cfm (accessed on 14 October 2021).
- Swain, D.; Yadav, A.S.; Sasapu, C.; Akula, V.; Samanthula, G. UPLC Separation of forced degradation and process related impurities of Velpatasvir and structure elucidation by online LC-Quadrupole-Time of flight-Tandem mass Spectrometry. Microchem. J. 2020, 155, 104657. [Google Scholar] [CrossRef]
- Stability Testing of New Drug Substances and Products. Available online: https://database.ich.org/sites/default/files/Q1A%28R2%29%20Guideline.pdf (accessed on 7 February 2021).
- Impurities in New Drug Products. Q3B (R2), Current Step. Available online: https://database.ich.org/sites/default/files/Q3B%28R2%29%20Guideline.pdf (accessed on 7 February 2021).
- Guideline IHT. Photostability testing of new drug substance and products. Fed. Regist. 1996, 62, 27115–27122. [Google Scholar]
- Zaman, B.; Hassan, W. Development of stability indicating HPLC–UV method for determination of daclatasvir and characterization of forced degradation products. Chromatographia 2018, 81, 785–797. [Google Scholar] [CrossRef]
- Validation of Analytical Procedures: Text and Methodology Q2 (R1). Available online: https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf (accessed on 7 February 2021).
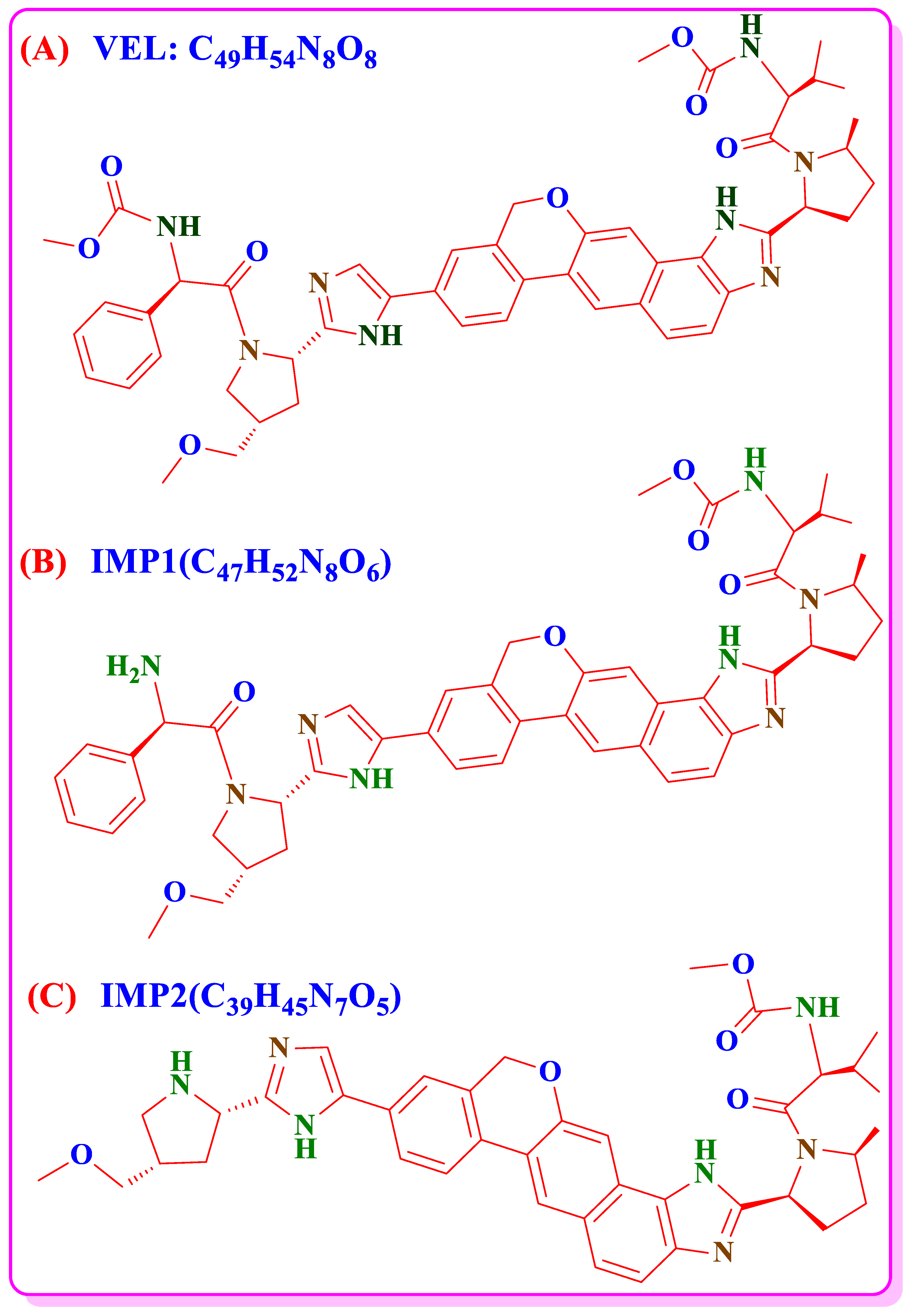

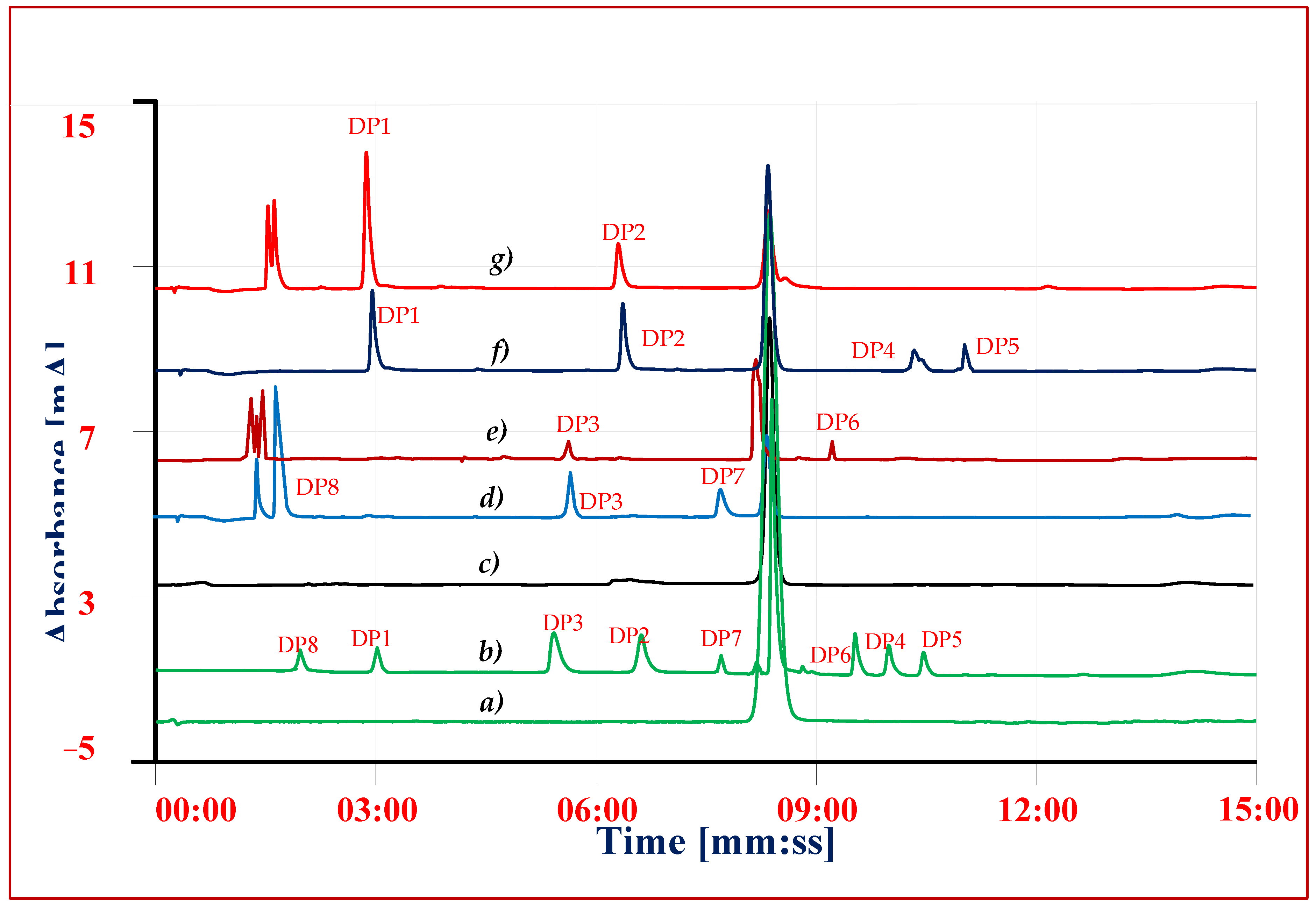
| Parameters | VEL | IMP1 | IMP2 | DP-1 | DP2 | DP3 | DP-4 | DP-5 | DP-6 | DP-7 | DP-8 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Peak Area (A) mAs | 1653.94 ± 25.8 | 123.51 ± 13.713 | 112.54 ± 11.9 | 197.36 ± 18.3 | 221.76 ± 23.2 | 245.7 ± 16.7 | 197.3 ± 19.7 | 178.9 ± 12.6 | 213.5 ± 13.5 | 195.6 ± 16.4 | 237.9 ± 11.3 |
| Relative standard deviation (RSD) | 0.44% | 9.41% | 9.28% | 7.81% | 6.98% | 5.74% | 8.4% | 6.87% | 7.44% | 8.15% | 5.41% |
| Retention Time (tR) | 8.5 ± 0.1 | 8.91 ± 0.1 | 8.3 ± 0.11 | 3.1 ± 0.1 | 7.6 ± 0.1 | 5.7 ± 0.1 | 10.4 ± 0.1 | 11.1 ± 0.1 | 9.7 ± 0.1 | 8.3 ± 0.1 | 2.7 ± 0.1 |
| Relative Retention Time (tRR) | - | 1.11375 | 1.0375 | 0.3875 | 0.95 | 0.7125 | 1.3 | 1.3875 | 1.2125 | 1.0375 | 0.3375 |
| Theoretical Plates (N) | 14,456 | 3403 | 4412 | 5256 | 7642 | 6323 | 3423 | 3672 | 4217 | 4822 | 3201 |
| Symmetry Factor (AS) | 1.021 | 0.841 | 0.907 | 1.07 | 0.922 | 0.925 | 0.923 | 0.976 | 1.02 | 0.921 | 0.916 |
| Retention factor K’ | 8.96 | 9.93 | 8.98 | 4.45 | 7.55 | 5.89 | 9.12 | 10.74 | 8.91 | 7.84 | 2.11 |
| Analyte | Conc. Level | 20% | 40% | 60% | 80% | 100% | 120% |
|---|---|---|---|---|---|---|---|
| VEL | Recovery% | 99.11 ± 0.35 | 99.45 ± 0.21 | 101 ± 0.23 | 101.16 ± 0.46 | 99.87 ± 0.41 | 100.61 ± 0.77 |
| RSD% | 0.24 | 0.35 | 0.24 | 0.45 | 0.39 | 0.61 | |
| IMP1 | Recovery% | 85.12 ± 3.25 | 89.1 ± 2.25 | 87.41 ± 2.64 | 81.16 ± 4.54 | 89.75 ± 3.63 | 89.14 ± 5.61 |
| RSD% | 3.31 | 2.78 | 2.67 | 5.12 | 4.13 | 6.12 | |
| IMP2 | Recovery% | 83.15 ± 4.35 | 91.3 ± 3.66 | 87.33 ± 4.12 | 87.17 ± 3.76 | 87.41 ± 3.15 | 92.15 ± 4.71 |
| RSD% | 4.39 | 4.21 | 4.75 | 4.16 | 3.41 | 5.02 | |
| DP1 | Recovery% | 84.09 ± 3.27 | 86.17 ± 6.28 | 83.81 ± 6.15 | 92.53 ± 4.12 | 93.56 ± 4.75 | 95.51 ± 3.45 |
| RSD% | 4.12 | 7..02 | 7.21 | 5.06 | 4.95 | 4.17 | |
| DP2 | Recovery% | 86.34 ± 5.11 | 87.69 ± 3.59 | 91.12 ± 3.86 | 92.08 ± 6.65 | 93.86 ± 2.84 | 94.21 ± 1.76 |
| RSD% | 6.15 | 3.96 | 4.09 | 7.31 | 3.11 | 2.92 | |
| DP3 | Recovery% | 84.12 ± 6.12 | 89.22 ± 4.15 | 92.42 ± 2.86 | 94.45 ± 6.12 | 94.86 ± 1.31 | 96.39 ± 1.87 |
| RSD% | 7.29 | 5.34 | 3.03 | 7.15 | 1.45 | 2.11 | |
| DP4 | Recovery% | 82.15 ± 7.23 | 98.44 ± 6.35 | 90.27 ± 3.97 | 92.18 ± 5.1 | 94.56 ± 2.11 | 93.15 ± 1.64 |
| RSD% | 8.21 | 7.38 | 4.10 | 5.52 | 2.24 | 1.71 | |
| DP5 | Recovery% | 84.1 ± 6.51 | 82.17 ± 3.21 | 93.37 ± 3.26 | 95.62 ± 4.45 | 93.31 ± 1.33 | 92.21 ± 1.29 |
| RSD% | 7.92 | 3.97 | 3.31 | 5.12 | 1.95 | 1.86 | |
| DP6 | Recovery% | 85.94 ± 8.11 | 91.23 ± 8.12 | 94.72 ± 2.51 | 94.68 ± 1.68 | 96.11 ± 3.14 | 94.56 ± 1.59 |
| RSD% | 9.12 | 8.07 | 1.75 | 2.17 | 4.09 | 2.22 | |
| DP7 | Recovery% | 81.23 ± 6.44 | 83.54 ± 5.34 | 83.17 ± 1.18 | 89.6 ± 1.68 | 86.25 ± 1.35 | 81.97 ± 3.67 |
| RSD% | 7.31 | 6.24 | 1.23 | 1.91 | 1.78 | 4.02 | |
| DP8 | Recovery% | 86.31 ± 7.53 | 94.39 ± 8.12 | 91.56 ± 5.41 | 91.37 ± 4.75 | 93.34 ± 2.79 | 95.19 ± 3.34 |
| RSD% | 8.22 | 8.41 | 5.60 | 5.13 | 3.19 | 3.46 |
| Parameters | VEL | IMP1 | IMP2 | DP-1 | DP2 | DP3 | DP-4 | DP-5 | DP-6 | DP-7 | DP-8 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Precision | ±0.22 | ±9.12 | ±8.71 | ±2.71 | ±3.24 | ±6.34 | ±5.96 | ±6.23 | ±5.17 | ±7.11 | ±6.32 |
| Robustness | ±0.76 | ±8.45 | ±6.39 | ±3.86 | ±6.19 | ±4.37 | ±7.34 | ±5.14 | ±4.19 | ±6.51 | ±6.84 |
| Slope | 74.69 | 876.4 | 655.9 | 74.5 | 653.9 | 899.6 | 742.7 | 654.5 | 955.9 | 1012 | 844 |
| Correlation r | 0.9999 | 0.9982 | 0.9987 | 0.9991 | 0.9993 | 0.9924 | 0.9996 | 0.9985 | 0.9994 | 0.9997 | 0.9993 |
| Intercept | −2.962 | −1.073 | −7.026 | −7.309 | −6.541 | −15.800 | −8.116 | −6.481 | −6.995 | −6.48159 | −6.99547 |
| LOD (µg mL−1) | 0.016 | 0.003 | 0.017 | 0.09 | 0.021 | 0.016 | 0.015 | 0.020 | 0.016 | 0.013 | 0.019 |
| LOQ (µg mL−1) | 0.05 | 0.010 | 0.053 | 0.27 | 0.064 | 0.05 | 0.044 | 0.06 | 0.05 | 0.05 | 0.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaman, B.; Hassan, W.; Khan, A.; Mushtaq, A.; Ali, N.; Bilal, M.; Ahmed, D.A. Forced Degradation Studies and Development and Validation of HPLC-UV Method for the Analysis of Velpatasvir Copovidone Solid Dispersion. Antibiotics 2022, 11, 897. https://doi.org/10.3390/antibiotics11070897
Zaman B, Hassan W, Khan A, Mushtaq A, Ali N, Bilal M, Ahmed DA. Forced Degradation Studies and Development and Validation of HPLC-UV Method for the Analysis of Velpatasvir Copovidone Solid Dispersion. Antibiotics. 2022; 11(7):897. https://doi.org/10.3390/antibiotics11070897
Chicago/Turabian StyleZaman, Bakht, Waseem Hassan, Adnan Khan, Ayesha Mushtaq, Nisar Ali, Muhammad Bilal, and Dina A. Ahmed. 2022. "Forced Degradation Studies and Development and Validation of HPLC-UV Method for the Analysis of Velpatasvir Copovidone Solid Dispersion" Antibiotics 11, no. 7: 897. https://doi.org/10.3390/antibiotics11070897
APA StyleZaman, B., Hassan, W., Khan, A., Mushtaq, A., Ali, N., Bilal, M., & Ahmed, D. A. (2022). Forced Degradation Studies and Development and Validation of HPLC-UV Method for the Analysis of Velpatasvir Copovidone Solid Dispersion. Antibiotics, 11(7), 897. https://doi.org/10.3390/antibiotics11070897