

Optimization of Pyrazole Compounds as Antibiotic Adjuvants Active against Colistin- and Carbapenem-Resistant Acinetobacter baumannii
 , , , , , ,
, , , , , ,
Abstract
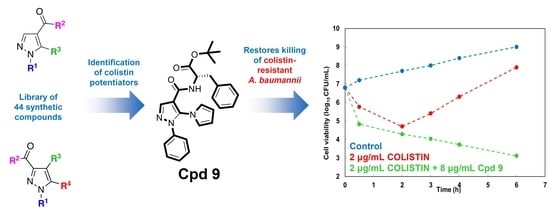
1. Introduction
2. Results
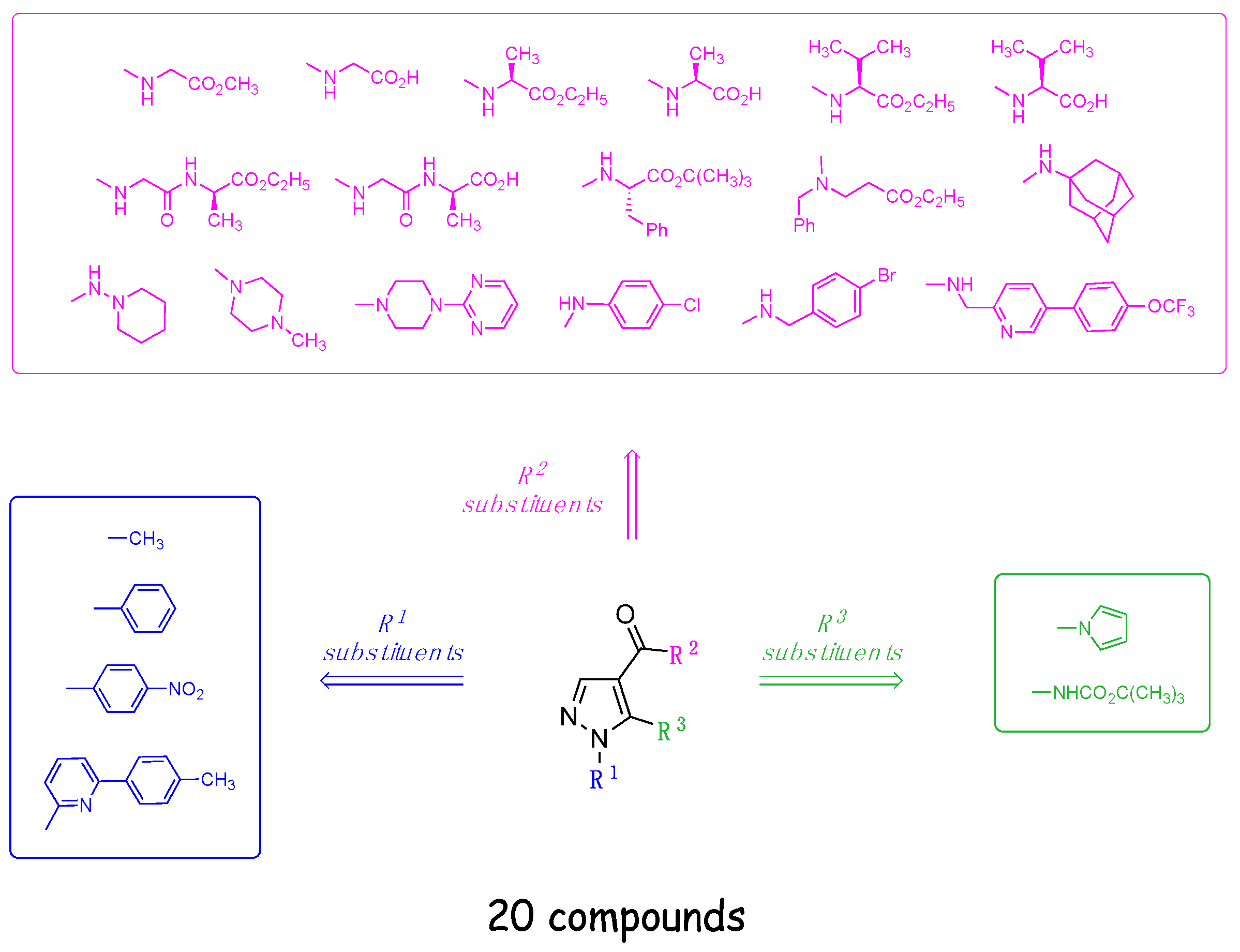
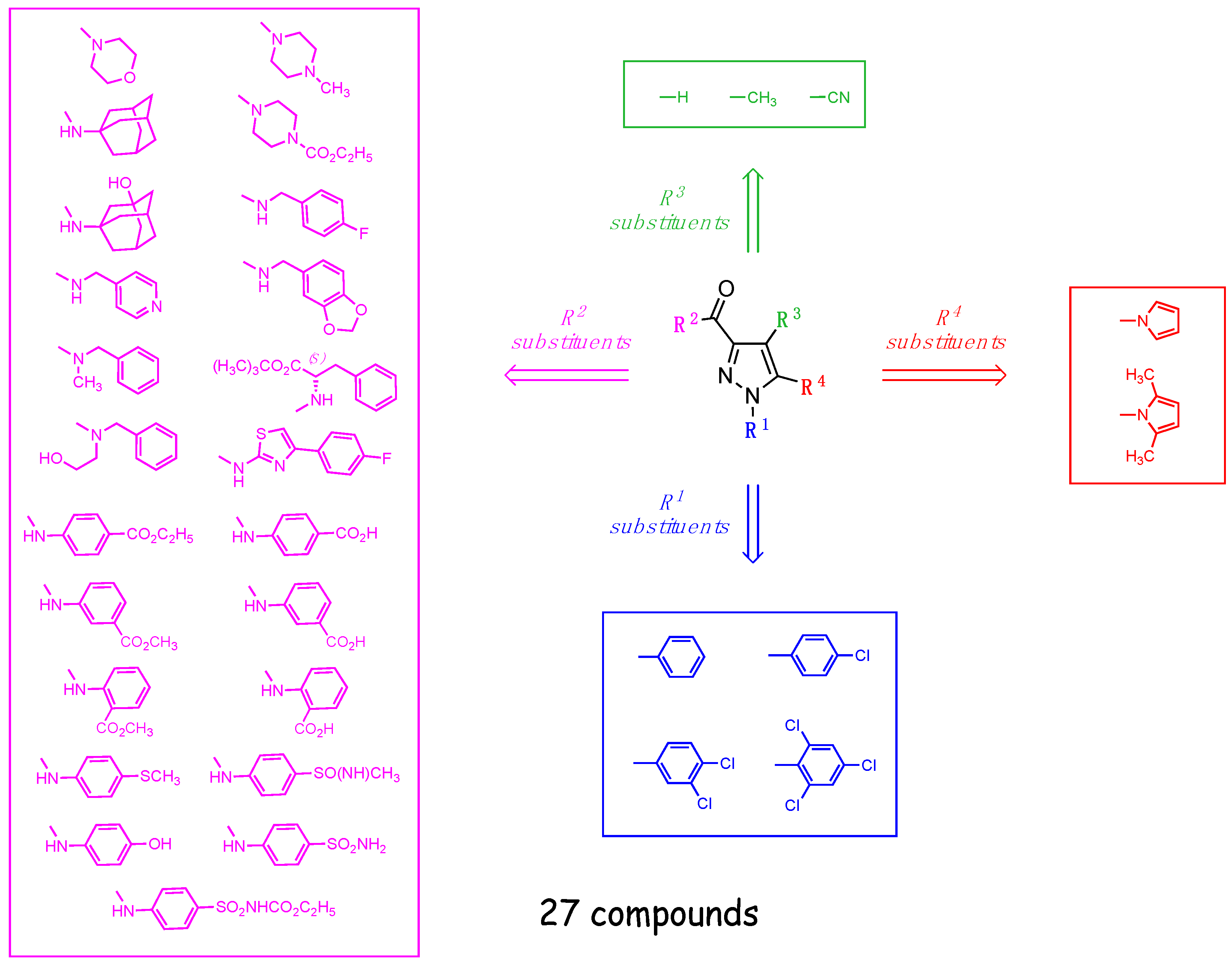
2.1. Chemistry
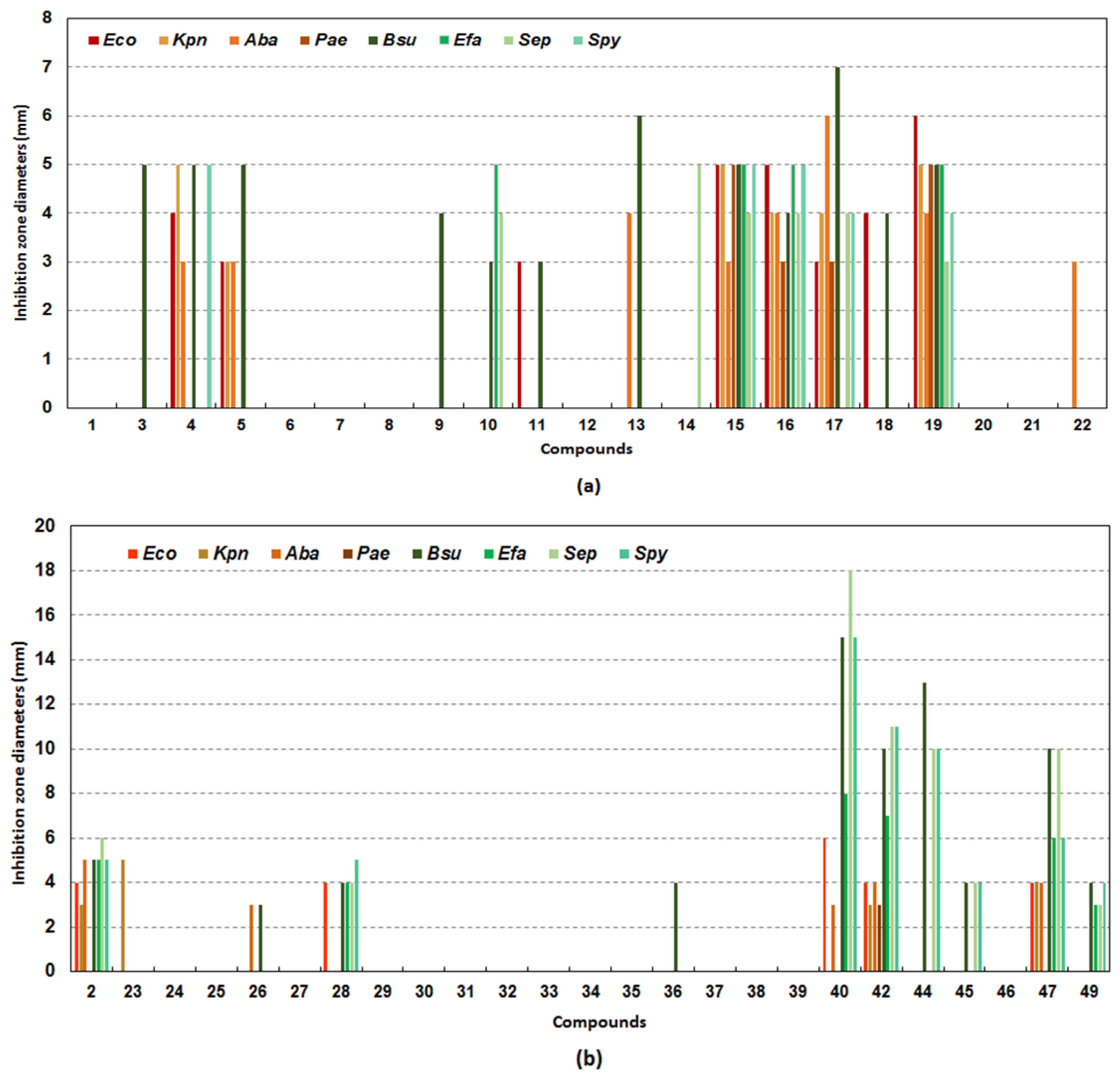
2.2. Biological Evaluation of Pyrazole-4- and Pyrazole-3-Carboxamide Derivatives
3. Discussion and Conclusions
4. Materials and Methods
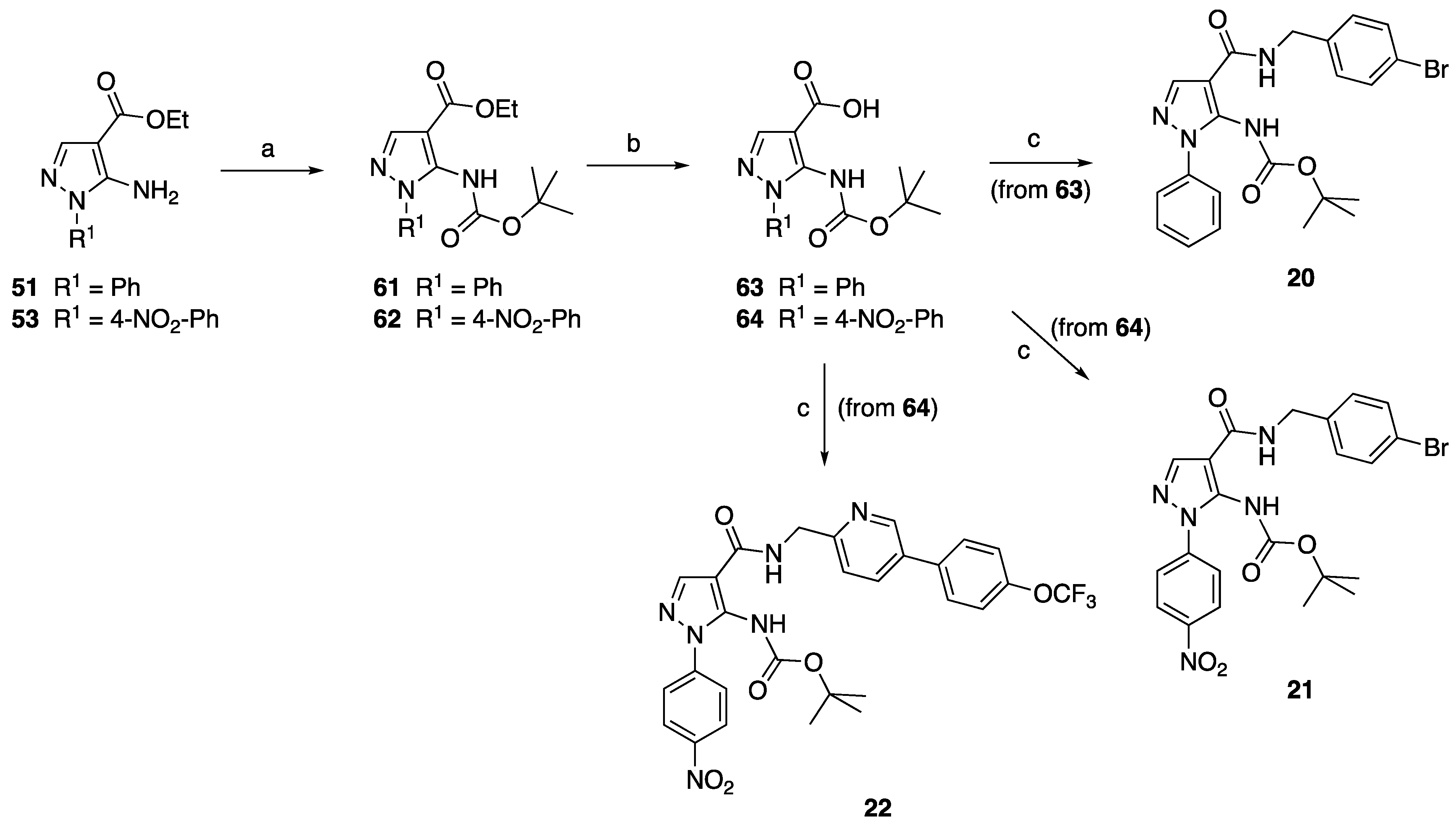
4.1. Chemistry
4.1.1. Synthesis of Aminopyrazole Precursors 50–53
4.1.2. General Procedure for the Synthesis of Ethyl 5-Pyrrol-1-yl-1H-pyrazole-4-carboxylates 54–56
Ethyl 5-Amino-1-(6-bromopyridin-2-yl)-1H-pyrazole-4-carboxylate (56)
4.1.3. Ethyl 5-(1H-Pyrrol-1-yl)-1-[6-(p-tolyl)pyridin-2-yl]-1H-pyrazole-4-carboxylate (57)
4.1.4. General Procedure for the Synthesis of 5-(1H-Pyrrol-1-yl)-1H-pyrazole-4-carboxylic Acids 58–60
4.1.5. General Procedure for the Synthesis of Amide Derivatives 3–14
(4-Methylpiperazin-1-yl)[1-methyl-5-(1H-pyrrol-1-yl)-1H-pyrazol-4-yl]methanone (3)
(4-Methylpiperazin-1-yl)[1-phenyl-5-(1H-pyrrol-1-yl)-1H-pyrazol-4-yl]methanone (4)
1-Phenyl-N-(piperidin-1-yl)-5-(1H-pyrrol-1-yl)-1H-pyrazole-4-carboxamide (5)
N-(4-Chlorophenyl)-1-phenyl-5-(1H-pyrrol-1-yl)-1H-pyrazole-4-carboxamide (6)
[1-Phenyl-5-(1H-pyrrol-1-yl)-1H-pyrazol-4-yl][4-(pyrimidin-2-yl)piperazin-1-yl]methanone (7)
N-(Adamantan-1-yl)-1-phenyl-5-(1H-pyrrol-1-yl)-1H-pyrazole-4-carboxamide (8)
tert-Butyl (S)-3-Phenyl-2-[[[1-phenyl-5-(1H-pyrrol-1-yl)-1H-pyrazol-4-yl]carbonyl]amino]propanoate (9)
Ethyl 3-[[N-Benzil-N-[1-phenyl-5-(1H-pyrrol-1-yl)-1H-pyrazol-4-yl]carbonyl]amino]propanoate (10)
Methyl [[[1-Phenyl-5-(1H-pyrrol-1-yl)-1H-pyrazol-4-yl]carbonyl]amino]acetate (11)
Ethyl (S)-2-[[[1-Phenyl-5-(1H-pyrrol-1-yl)-1H-pyrazol-4-yl]carbonyl]amino]propanoate (12)
Ethyl (S)-3-Methyl-2-[[[1-phenyl-5-(1H-pyrrol-1-yl)-1H-pyrazol-4-yl]carbonyl]amino]butanoate (13)
tert-Butyl (S)-3-Phenyl-2-[[[5-(1H-pyrrol-1-yl)-1-[6-(p-tolyl)pyridin-2-yl]-1H-pyrazol-4-yl]carbonyl]amino]propanoate (14)
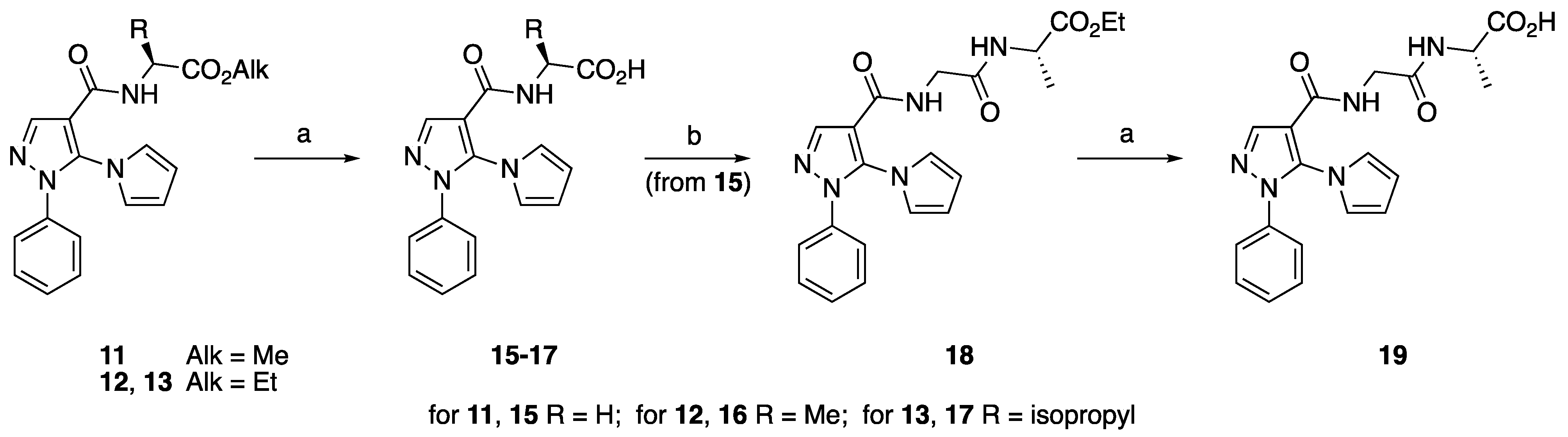
4.1.6. General Procedure for the Synthesis of Acids 15–17
[[[1-Phenyl-5-(1H-pyrrol-1-yl)-1H-pyrazol-4-yl]carbonyl]amino]acetic Acid (15)
(S)-2-[[[1-Phenyl-5-(1H-pyrrol-1-yl)-1H-pyrazol-4-yl]carbonyl]amino]propanoic Acid (16)
(S)-3-Methyl-2-[[[1-phenyl-5-(1H-pyrrol-1-yl)-1H-pyrazol-4-yl]carbonyl]amino]butanoic Acid (17)
4.1.7. N-[N-[[1-Phenyl-5-(1H-pyrrol-1-yl)-1H-pyrazol-4-yl]carbonyl]glycyl]-L-alanine Ethyl Ester (18)
4.1.8. N-[N-[[1-Phenyl-5-(1H-pyrrol-1-yl)-1H-pyrazol-4-yl]carbonyl]glycyl]-L-alanine (19)
4.1.9. General Procedure for the Synthesis of 61 and 62
Ethyl 5-(tert-Butoxycarbonylamino)-1-phenyl-1H-pyrazole-4-carboxylate (61)
Ethyl 5-(tert-Butoxycarbonylamino)-1-(4-nitrophenyl)-1H-pyrazole-4-carboxylate (62)
4.1.10. General Procedure for the Synthesis of Acids 63 and 64
5-(tert-Butoxycarbonylamino)-1-phenyl-1H-pyrazole-4-carboxylic Acid (63)
5-(tert-Butoxycarbonylamino)-1-(4-nitrophenyl)-1H-pyrazole-4-carboxylic Acid (64)
4.1.11. General Procedure for the Synthesis of Amide Derivatives 20–22
N-(4-Bromobenzyl)-5-(tert-butoxycarbonylamino)-1-phenyl-1H-pyrazole-4-carboxamide (20)
N-(4-Bromobenzyl)-5-(tert-butoxycarbonylamino)-1-(4-nitrophenyl)-1H-pyrazole-4-carboxamide (21)
5-(tert-Butoxycarbonylamino)-1-(4-nitrophenyl)-N-[[5-(4-trifluoromethoxyphenyl)pyridin-2-yl]methyl]-1H-pyrazole-4-carboxamide (22)
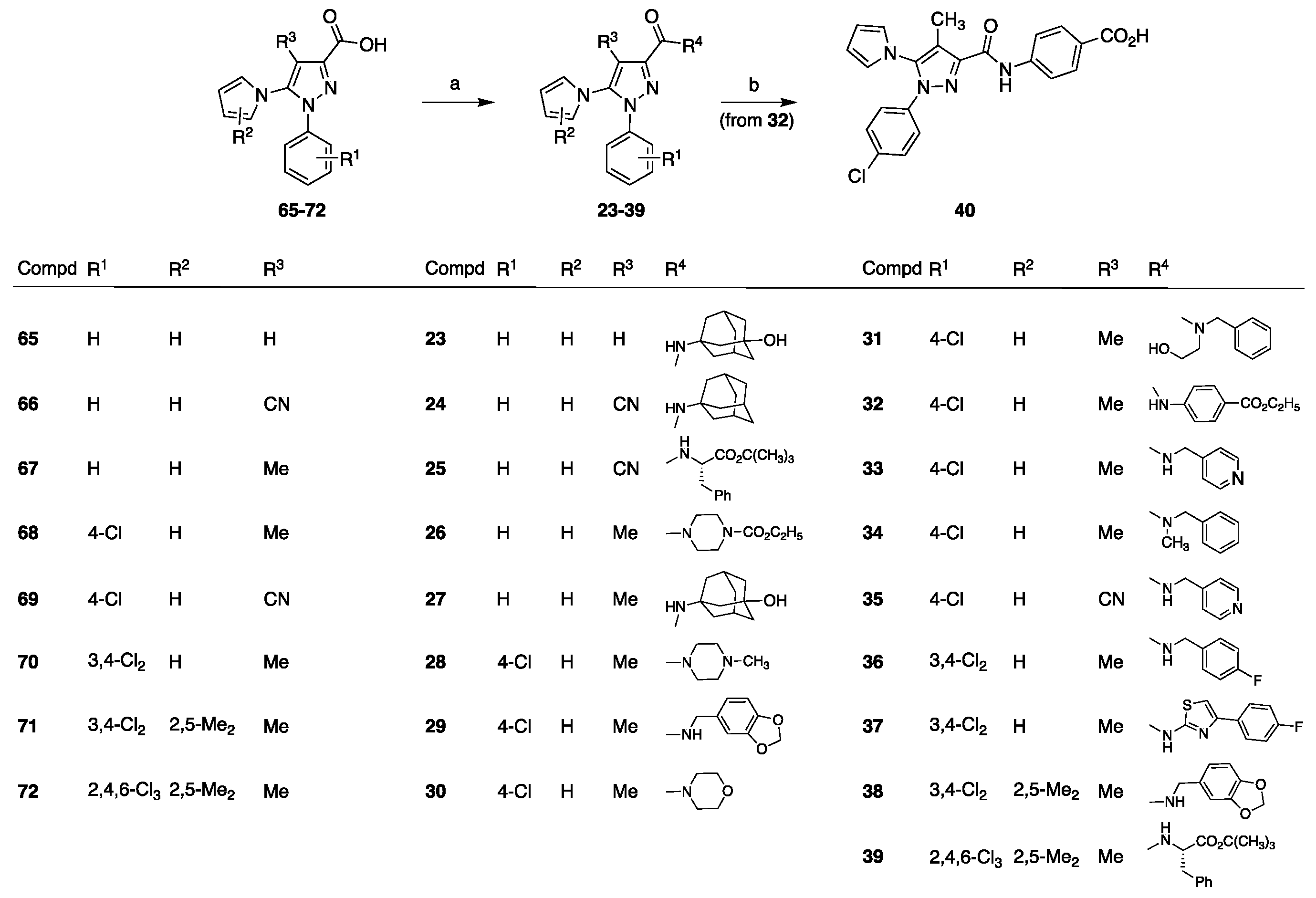
4.1.12. General Procedure for the Synthesis of Amides 23–39
N-(Adamantan-3-ol-1-yl)-1-phenyl-5-(1H-pyrrol-1-yl)-1H-pyrazole-3-carboxamide (23)
N-(Adamantan-1-yl)-4-cyano-1-phenyl-5-(1H-pyrrol-1-yl)-1H-pyrazole-3-carboxamide (24)
tert-Butyl (S)-3-Phenyl-2-[[[4-cyano-1-phenyl-5-(1H-pyrrol-1-yl)-1H-pyrazol-3-yl]carbonyl]amino]propanoate (25)
(4-Ethoxycarbonylpiperazin-1-yl)[4-methyl-1-phenyl-5-(1H-pyrrol-1-yl)-1H-pyrazol-3-yl]methanone (26)
N-(Adamantan-3-ol-1-yl)-4-methyl-1-phenyl-5-(1H-pyrrol-1-yl)-1H-pyrazole-3-carboxamide (27)
[1-(4-Chlorophenyl)-4-methyl-5-(1H-pyrrol-1-yl)-1H-pyrazol-3-yl](4-methylpiperazin-1-yl)methanone (28)
N-(Benzo[d][1,3]dioxol-5-ylmethyl)-1-(4-chlorophenyl)-4-methyl-5-(1H-pyrrol-1-yl)-1H-pyrazole-3-carboxamide (29)
[1-(4-Chlorophenyl)-4-methyl-5-(1H-pyrrol-1-yl)-1H-pyrazol-3-yl](morpholin-4-yl)methanone (30)
N-Benzyl-1-(4-chlorophenyl)-N-(2-hydroxyethyl)-4-methyl-5-(1H-pyrrol-1-yl)-1H-pyrazole-3-carboxamide (31)
Ethyl 4-[[[1-(4-Chlorophenyl)-4-methyl-5-(1H-pyrrol-1-yl)-1H-pyrazol-3-yl]carbonyl]amino]benzoate (32)
1-(4-Chlorophenyl)-4-methyl-N-(pyridin-4-ylmethyl)-5-(1H-pyrrol-1-yl)-1H-pyrazole-3-carboxamide (33)
N-Benzyl-1-(4-chlorophenyl)-N,4-dimethyl-5-(1H-pyrrol-1-yl)-1H-pyrazole-3-carboxamide (34)
1-(4-Chlorophenyl)-4-cyano-N-(pyridin-4-ylmethyl)-5-(1H-pyrrol-1-yl)-1H-pyrazole-3-carboxamide (35)
1-(3,4-Dichlorophenyl)-N-(4-fluorobenzyl)-4-methyl-5-(1H-pyrrol-1-yl)-1H-pyrazole-3-carboxamide (36)
1-(3,4-Dichlorophenyl)-N-[4-(4-fluorophenyl)thiazol-2-yl]-4-methyl-5-(1H-pyrrol-1-yl)-1H-pyrazole-3-carboxamide (37)
N-(Benzo[d][1,3]dioxol-5-ylmethyl)-1-(3,4-dichlorophenyl)-4-methyl-5-(2,5-dimethyl-1H-pyrrol-1-yl)-1H-pyrazole-3-carboxamide (38)
tert-Butyl (S)-3-Phenyl-2-[[[4-methyl-1-(2,4,6-trichlorophenyl)-5-(2,5-dimethyl-1H-pyrrol-1-yl)-1H-pyrazol-3-yl]carbonyl]amino]propanoate (39)
4.1.13. 4-[[[1-(4-Chlorophenyl)-4-methyl-5-(1H-pyrrol-1-yl)-1H-pyrazol-3-yl]carbonyl]amino]benzoic Acid (40)
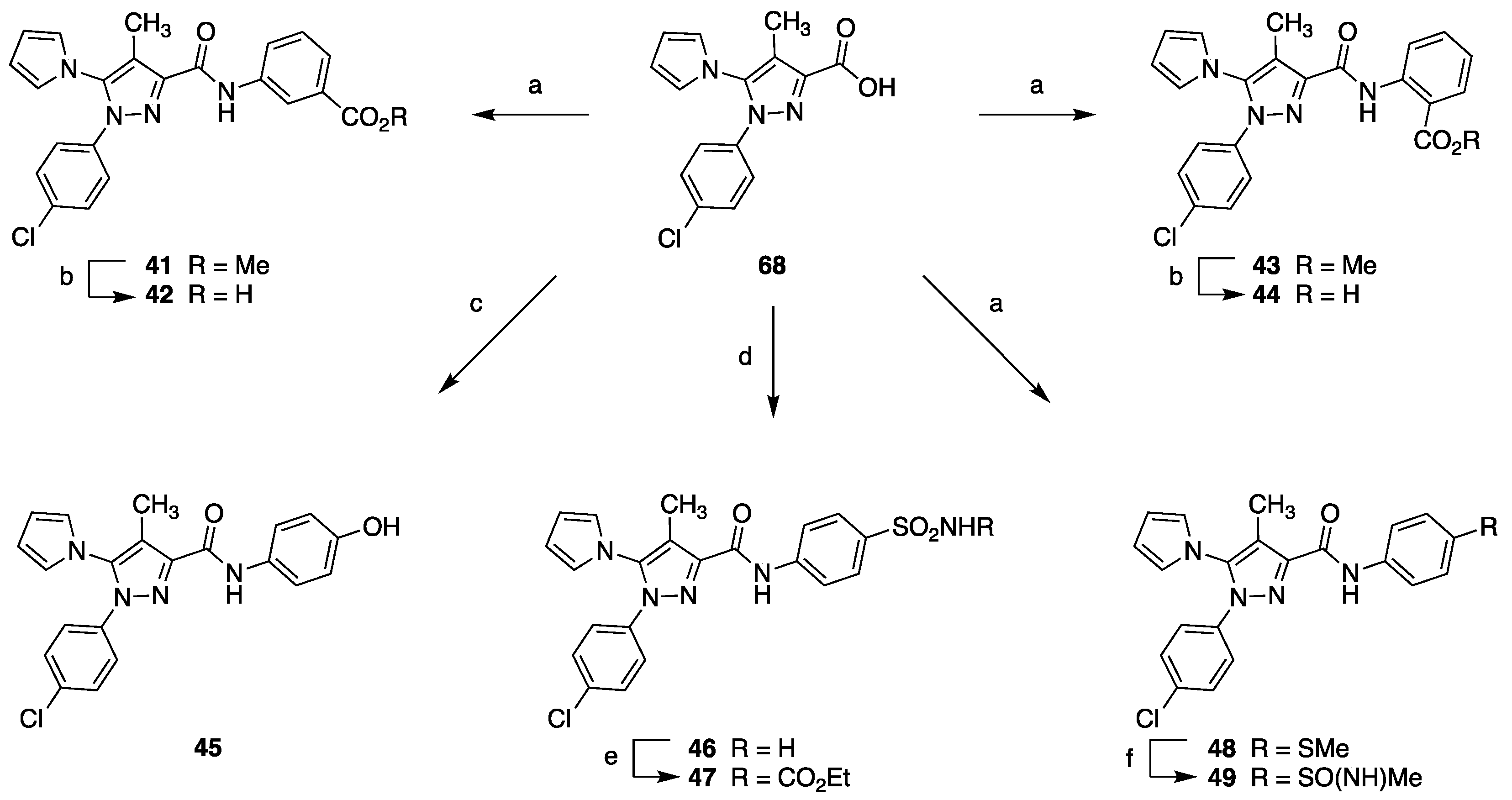
4.1.14. General Procedure for the Synthesis of Amides 41, 43, and 48
Methyl 3-[[[1-(4-Chlorophenyl)-4-methyl-5-(1H-pyrrol-1-yl)-1H-pyrazol-3-yl]carbonyl]amino]benzoate (41)
Methyl 2-[[[1-(4-Chlorophenyl)-4-methyl-5-(1H-pyrrol-1-yl)-1H-pyrazol-3-yl]carbonyl]amino]benzoate (43)
1-(4-Chlorophenyl)-N-(4-methylthiophenyl)-4-methyl-5-(1H-pyrrol-1-yl)-1H-pyrazole-3-carboxamide (48)
4.1.15. General Procedure for the Synthesis of Acids 42 and 44
3-[[[1-(4-Chlorophenyl)-4-methyl-5-(1H-pyrrol-1-yl)-1H-pyrazol-3-yl]carbonyl]amino]benzoic Acid (42)
2-[[[1-(4-Chlorophenyl)-4-methyl-5-(1H-pyrrol-1-yl)-1H-pyrazol-3-yl]carbonyl]amino]benzoic Acid (44)
4.1.16. 1-(4-Chlorophenyl)-N-(4-hydroxyphenyl)-4-methyl-5-(1H-pyrrol-1-yl)-1H-pyrazole-3-carboxamide (45)
4.1.17. 1-(4-Chlorophenyl)-4-methyl-5-(1H-pyrrol-1-yl)-N-(4-sulfamoylphenyl)-1H-pyrazole-3-carboxamide (46)
4.1.18. Ethyl [[4-[1-(4-Chlorophenyl)-4-methyl-5-(1H-pyrrol-1-yl)-1H-pyrazole-3-carboxamido]phenyl]sulfonyl]carbamate (47)
4.1.19. 1-(4-Chlorophenyl)-4-methyl-N-[4-(S-methylsulfonimidoyl)phenyl]-5-(1H-pyrrol-1-yl)-1H-pyrazole-3-carboxamide (49)
4.2. Antibacterial Susceptibility Testing
4.2.1. Agar Diffusion-Based Methods
4.2.2. Broth Microdilution Methods and MIC Determinations
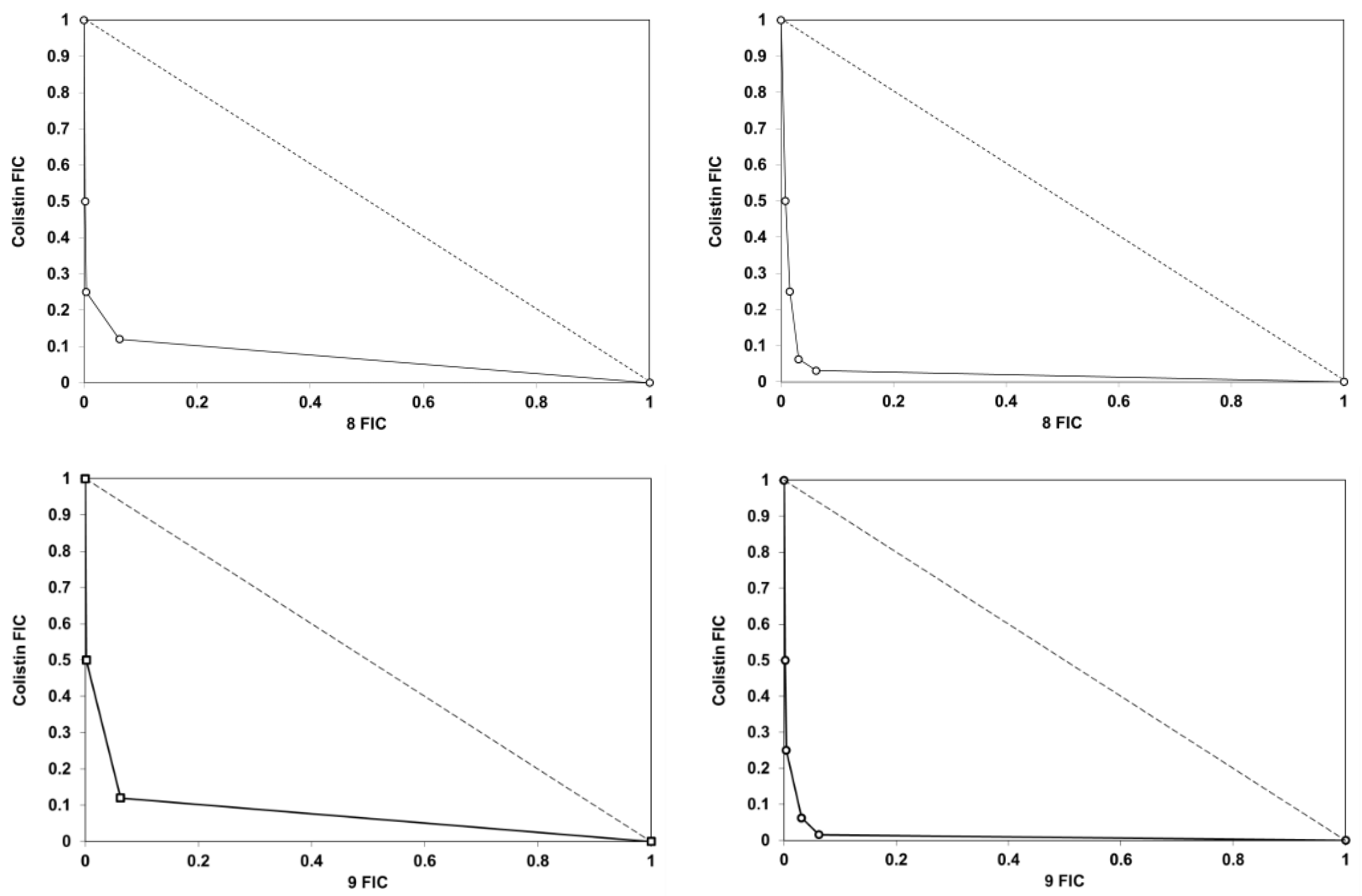
4.2.3. Chequerboard Analysis and FIC Determinations
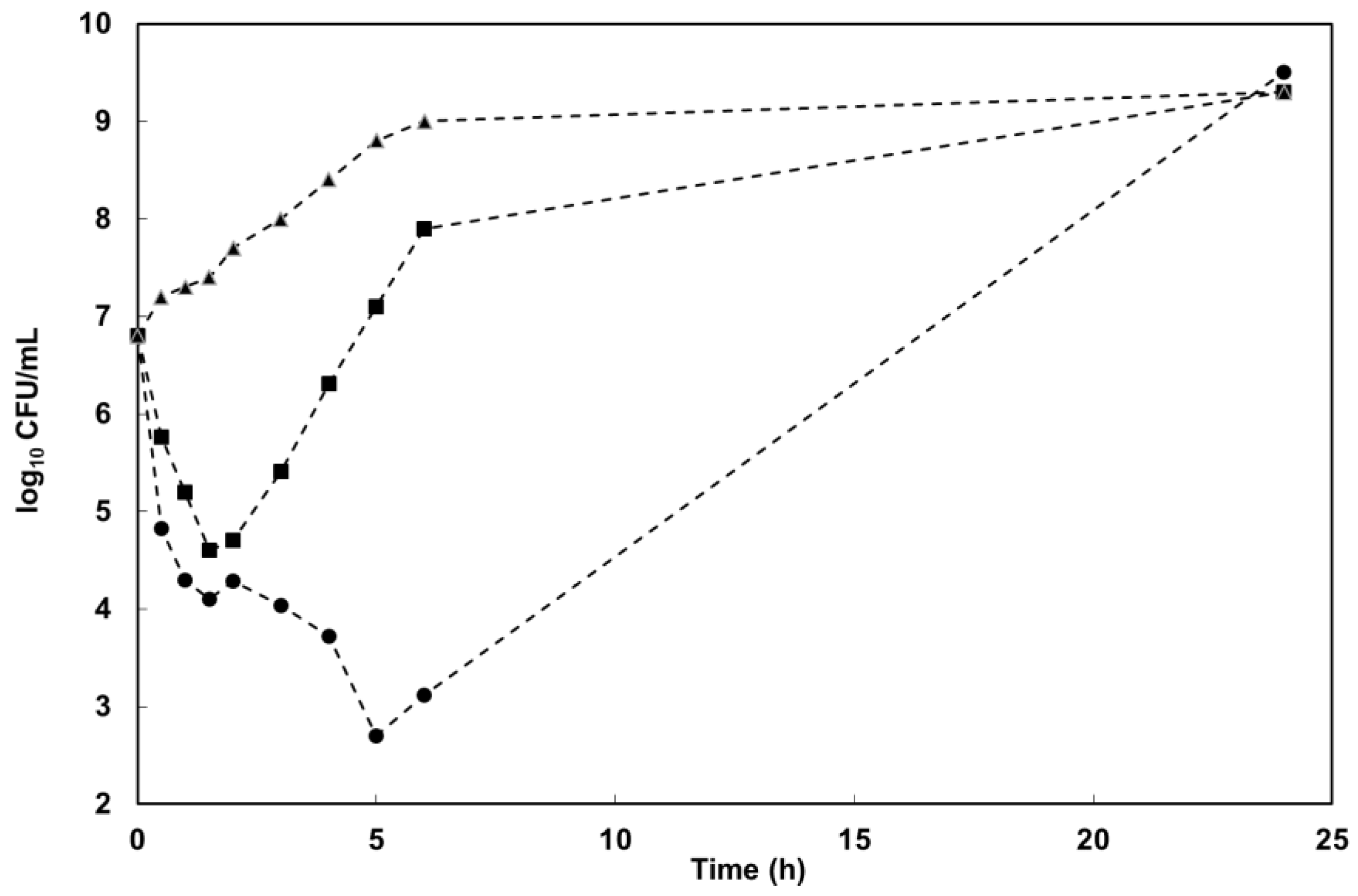
4.2.4. Kill-Curve Analysis
4.3. Cytotoxicity Assays
4.3.1. Membrane Integrity Assays
4.3.2. Cell Viability and Proliferation Assays
4.3.3. Hemolytic Activity
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Antibiotic Resistance. Available online: https://www.who.int/news-room/fact-sheets/detail/antibiotic-resistance (accessed on 24 October 2022).
- Ferri, M.; Ranucci, E.; Romagnoli, P.; Giaccone, V. Antimicrobial Resistance: A Global Emerging Threat to Public Health Systems. Crit. Rev. Food Sci. Nutr. 2017, 57, 2857–2876. [Google Scholar] [CrossRef] [PubMed]
- Cassini, A.; Högberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; Colomb-Cotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M.; et al. Attributable Deaths and Disability-Adjusted Life-Years Caused by Infections with Antibiotic-Resistant Bacteria in the EU and the European Economic Area in 2015: A Population-Level Modelling Analysis. Lancet Infect. Dis. 2019, 19, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, Research, and Development of New Antibiotics: The WHO Priority List of Antibiotic-Resistant Bacteria and Tu-berculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Trebosc, V.; Gartenmann, S.; Tötzl, M.; Lucchini, V.; Schellhorn, B.; Pieren, M.; Lociuro, S.; Gitzinger, M.; Tigges, M.; Bumann, D.; et al. Dissecting Colistin Resistance Mechanisms in Extensively Drug-Resistant Acinetobacter baumannii Clinical Isolates. mBio 2019, 10, e01083-19. [Google Scholar] [CrossRef] [PubMed]
- Papp-Wallace, K.M. The Latest Advances in β-Lactam/β-Lactamase Inhibitor Combinations for the Treatment of Gram-Negative Bacterial Infections. Expert Opin. Pharmacother. 2019, 20, 2169–2184. [Google Scholar] [CrossRef] [PubMed]
- Docquier, J.-D.; Mangani, S. An Update on β-Lactamase Inhibitor Discovery and Development. Drug Resist. Updat. 2018, 36, 13–29. [Google Scholar] [CrossRef]
- Theuretzbacher, U.; Outterson, K.; Engel, A.; Karlén, A. The Global Preclinical Antibacterial Pipeline. Nat. Rev. Microbiol. 2020, 18, 275–285. [Google Scholar] [CrossRef]
- Melander, R.J.; Melander, C. The Challenge of Overcoming Antibiotic Resistance: An Adjuvant Approach? ACS Infect. Dis. 2017, 3, 559–563. [Google Scholar] [CrossRef]
- Laws, M.; Shaaban, A.; Rahman, K.M. Antibiotic Resistance Breakers: Current Approaches and Future Directions. FEMS Microbiol. Rev. 2019, 43, 490–516. [Google Scholar] [CrossRef]
- Van Giau, V.; An, S.S.A.; Hulme, J. Recent Advances in the Treatment of Pathogenic Infections Using Antibiotics and Nano-Drug Delivery Vehicles. Drug Des. Devel. Ther. 2019, 13, 327–343. [Google Scholar] [CrossRef]
- Mugnaini, C.; Sannio, F.; Brizzi, A.; Del Prete, R.; Simone, T.; Ferraro, T.; De Luca, F.; Corelli, F.; Docquier, J.-D. Screen of Unfocused Libraries Identified Compounds with Direct or Synergistic Antibacterial Activity. ACS Med. Chem. Lett. 2020, 11, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Artico, M.; Massa, S.; Stefancich, G.; Corelli, F.; Ortenzi, G. Heterocyclic Systems. V. Synthesis of 1H,4H,6H-Pyrazolo[3,4-e]pyrrolo[2,1-c][1,4]oxazepine Derivatives. Heterocycles 1985, 23, 1417. [Google Scholar] [CrossRef]
- Massa, S.; Mai, A.; Artico, M.; Corelli, F. Heterocyclic System. XI. Synthesis of 1H,4H-Pyrazolo[4,3-b]pyrrolizine and 2H,4H-Pyrazolo[4,3-b]pyrrolizine Derivatives. J. Heterocycl. Chem. 1990, 27, 1805–1808. [Google Scholar] [CrossRef]
- Silvestri, R.; Cascio, M.G.; La Regina, G.; Piscitelli, F.; Lavecchia, A.; Brizzi, A.; Pasquini, S.; Botta, M.; Novellino, E.; Di Marzo, V.; et al. Synthesis, Cannabinoid Receptor Affinity, and Molecular Modeling Studies of Substituted 1-Aryl-5-(1H-pyrrol-1-yl)-1H-pyrazole-3-carboxamides. J. Med. Chem. 2008, 51, 1560–1576. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Wang, B.; Wu, T.; Wan, J.; Tu, Z.; Njire, M.; Wan, B.; Franzblauc, S.G.; Zhang, T.; Lu, X.; et al. Design, Synthesis, and Biological Evaluation of Pyrazolo[1,5-a]pyridine-3-carboxamides as Novel Antitubercular Agents. ACS Med. Chem. Lett. 2015, 6, 814–818. [Google Scholar] [CrossRef]
- Cannatelli, A.; D’Andrea, M.M.; Giani, T.; Di Pilato, V.; Arena, F.; Ambretti, S.; Gaibani, P.; Rossolini, G.M. In vivo emergence of colistin resistance in Klebsiella pneumoniae producing KPC-type carbapenemases mediated by insertional inactivation of the PhoQ/PhoP mgrB regulator. Antimicrob. Agents Chemother. 2013, 57, 5521–5526. [Google Scholar] [CrossRef]
- D’Andrea, M.M.; Giani, T.; D’Arezzo, S.; Capone, A.; Petrosillo, N.; Visca, P.; Luzzaro, F.; Rossolini, G.M. Characterization of PABVA01, a Plasmid Encoding the OXA-24 Carbapenemase from Italian Isolates of Acinetobacter baumannii. Antimicrob. Agents Chemother. 2009, 53, 3528–3533. [Google Scholar] [CrossRef]
- Kelesidis, T.; Falagas, M.E. The Safety of Polymyxin Antibiotics. Expert Opin. Drug Saf. 2015, 14, 1687–1701. [Google Scholar] [CrossRef]
- Amaya-Villar, R.; Garnacho-Montero, J. How Should We Treat Acinetobacter Pneumonia? Curr. Opin. Crit. Care 2019, 25, 465–472. [Google Scholar] [CrossRef]
- Terminology Relating to Methods for the Determination of Susceptibility of Bacteria to Antimicrobial Agents. Clin. Microbiol. Infect. 2000, 6, 503–508. [CrossRef]
- Wahab Khan, M.; Uddin, M.K.; Ali, M.; Rahman, M.S.; Rashid, M.A.; Chowdhury, R. A Convenient Synthesis of New Annelated Pyrimidines and Their Biological Importance: Synthesis of Annelated Pyrimidines. J. Heterocycl. Chem. 2014, 51, E216–E221. [Google Scholar] [CrossRef]
- Christopher, A.; Takeru, E. Indane Derivatives and the Use Thereof as Soluble Guanylate Cyclase Activators. WIPO (PCT) Patent WO2017103888 (A1), 18 December 2018. [Google Scholar]
- Chen, H.; Wang, B.; Li, P.; Yan, H.; Li, G.; Huang, H.; Lu, Y. The Optimization and Characterization of Functionalized Sulfonamides Derived from Sulfaphenazole against Mycobacterium Tuberculosis with Reduced CYP 2C9 Inhibition. Bioorg. Med. Chem. Lett. 2021, 40, 127924. [Google Scholar] [CrossRef] [PubMed]
- Kopp, M.; Lancelot, J.-C.; Dallemagne, P.; Rault, S. Synthesis of Novel Pyrazolopyrrolopyrazines, Potential Analogs of Sildenafil. J. Heterocycl. Chem. 2001, 38, 1045–1050. [Google Scholar] [CrossRef]
- Approved Standard; Clinical and Laboratory Standards Institute (Eds.) Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically: M07-A10, 10th ed.; Documents/Clinical and Laboratory Standards Institute; Committee for Clinical Laboratory Standards: Wayne, PA, USA, 2015; ISBN 978-1-56238-987-1. [Google Scholar]
- Hall, M.J.; Middleton, R.F.; Westmacott, D. The Fractional Inhibitory Concentration (FIC) Index as a Measure of Synergy. J. Antimicrob. Chemother. 1983, 11, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Bechlars, S.; Wüstenhagen, D.A.; Drägert, K.; Dieckmann, R.; Strauch, E.; Kubick, S. Cell-free synthesis of functional thermostable direct hemolysins of Vibrio parahaemolyticus. Toxicon 2013, 76, 132–142. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
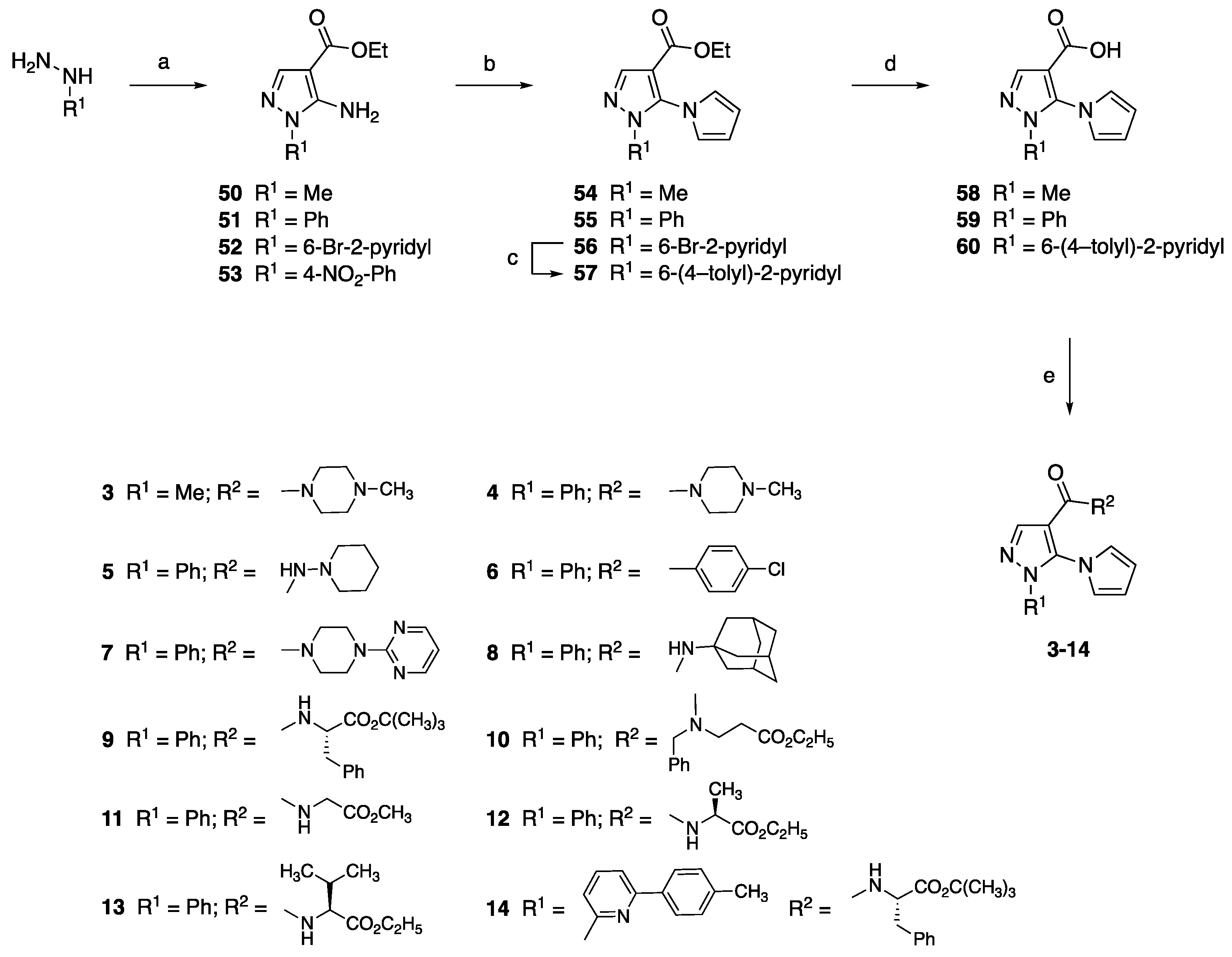{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd a | Synergistic Activity (Growth Inhibition) b | |||
|---|---|---|---|---|
| Eco | Kpn | Pae | Aba | |
| 3 | ||||
| 4 | ||||
| 5 | ||||
| 6 | ||||
| 7 | ||||
| 8 | ■ | ■ | ||
| 9 | ■ | ■ | ■ | |
| 10 | ||||
| 11 | - c | |||
| 12 | ||||
| 13 | ■ | ■ | ||
| 14 | ||||
| 15 | - | |||
| 16 | ||||
| 17 | ||||
| 18 | - | - | ||
| 19 | - | - | ||
| 20 | ■ | ■ | ||
| 21 | - | - | - | |
| 22 | ■ | ■ | ||
| 23 | ||||
| 24 | ||||
| 25 | ||||
| 26 | ||||
| 27 | ■ | ■ | ||
| 28 | ■ | ■ | ■ | |
| 29 | ||||
| 30 | ||||
| 31 | ■ | ■ | ■ | |
| 32 | - | - | - | |
| 33 | - | - | - | |
| 34 | - | - | - | |
| 35 | ||||
| 36 | ||||
| 37 | ||||
| 38 | ||||
| 39 | ■ | |||
| 40 | ||||
| 42 | ||||
| 44 | ||||
| 45 | ■ | ■ | ■ | |
| 46 | ||||
| 47 | ||||
| 49 | ■ | ■ | ■ | |
| Compound | MAC (µg/mL) (MIC/MAC Ratio) | ||
|---|---|---|---|
| Eco | Kpn | Aba | |
| 1 | 64 (8) | 64 (8) | 32 (16) |
| 8 | 32 (16) | - a | 4 (128) |
| 9 | 8 (64) | 8 (64) | 4 (128) |
| 13 | 64 (8) | 32 (16) | - |
| 20 | - | 64 (8) | 64 (8) |
| 22 | - | 64 (8) | 64 (8) |
| 2 | 64 (8) | 32 (16) | 16 (32) |
| 27 | 32 (16) | 32 (16) | - |
| 28 | 64 (8) | 64 (8) | 64 (8) |
| 31 | 32 (16) | 16 (32) | 32 (16) |
| 39 | - | - | 32 (16) |
| 45 | 4 (128) | 4 (128) | 8 (64) |
| 49 | 8 (64) | 4 (128) | 8 (64) |
| Bacterial Strain | Average FIC Index | |||||
|---|---|---|---|---|---|---|
| 1 | 8 | 9 | 2 | 45 | 49 | |
| E. coli CCUGT | 0.5 | 0.38 | 0.33 | 0.31 | 0.31 | 0.32 |
| K. pneumoniae ATCC 13833 | 0.5 | 0.5 | 0.32 | 0.5 | 0.31 | 0.39 |
| K. pneumoniae SI-004Bo a | 1 | 1 | 1 | 1 | 1 | 1 |
| A. baumannii ATCC 17978 | 0.38 | 0.31 | 0.34 | 0.39 | 0.31 | 0.38 |
| A. baumannii N50 b | 0.5 | 0.24 | 0.23 | 0.4 | 0.26 | 0.31 |
| Compound | IC50 (μg/mL) |
|---|---|
| Colistin | >256 |
| 8 | >256 |
| 8 + colistin | 16.4 |
| 9 | >256 |
| 9 + colistin | >256 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sannio, F.; Brizzi, A.; Del Prete, R.; Avigliano, M.; Simone, T.; Pagli, C.; Ferraro, T.; De Luca, F.; Paolino, M.; Corelli, F.; et al. Optimization of Pyrazole Compounds as Antibiotic Adjuvants Active against Colistin- and Carbapenem-Resistant Acinetobacter baumannii. Antibiotics 2022, 11, 1832. https://doi.org/10.3390/antibiotics11121832
Sannio F, Brizzi A, Del Prete R, Avigliano M, Simone T, Pagli C, Ferraro T, De Luca F, Paolino M, Corelli F, et al. Optimization of Pyrazole Compounds as Antibiotic Adjuvants Active against Colistin- and Carbapenem-Resistant Acinetobacter baumannii. Antibiotics. 2022; 11(12):1832. https://doi.org/10.3390/antibiotics11121832
Chicago/Turabian StyleSannio, Filomena, Antonella Brizzi, Rosita Del Prete, Marialuce Avigliano, Tiziana Simone, Carlotta Pagli, Teresa Ferraro, Filomena De Luca, Marco Paolino, Federico Corelli, and et al. 2022. "Optimization of Pyrazole Compounds as Antibiotic Adjuvants Active against Colistin- and Carbapenem-Resistant Acinetobacter baumannii" Antibiotics 11, no. 12: 1832. https://doi.org/10.3390/antibiotics11121832
APA StyleSannio, F., Brizzi, A., Del Prete, R., Avigliano, M., Simone, T., Pagli, C., Ferraro, T., De Luca, F., Paolino, M., Corelli, F., Mugnaini, C., & Docquier, J.-D. (2022). Optimization of Pyrazole Compounds as Antibiotic Adjuvants Active against Colistin- and Carbapenem-Resistant Acinetobacter baumannii. Antibiotics, 11(12), 1832. https://doi.org/10.3390/antibiotics11121832