
Pan and Core Genome Analysis of 183 Mycobacterium tuberculosis Strains Revealed a High Inter-Species Diversity among the Human Adapted Strains




Abstract
1. Introduction
2. Materials and Methods
2.1. Genome Sequencing and Annotation
2.2. Orthologous Gene Prediction and Genome Sequence Comparison
2.3. Phylogenetic Tree Construction
3. Results
3.1. General Features
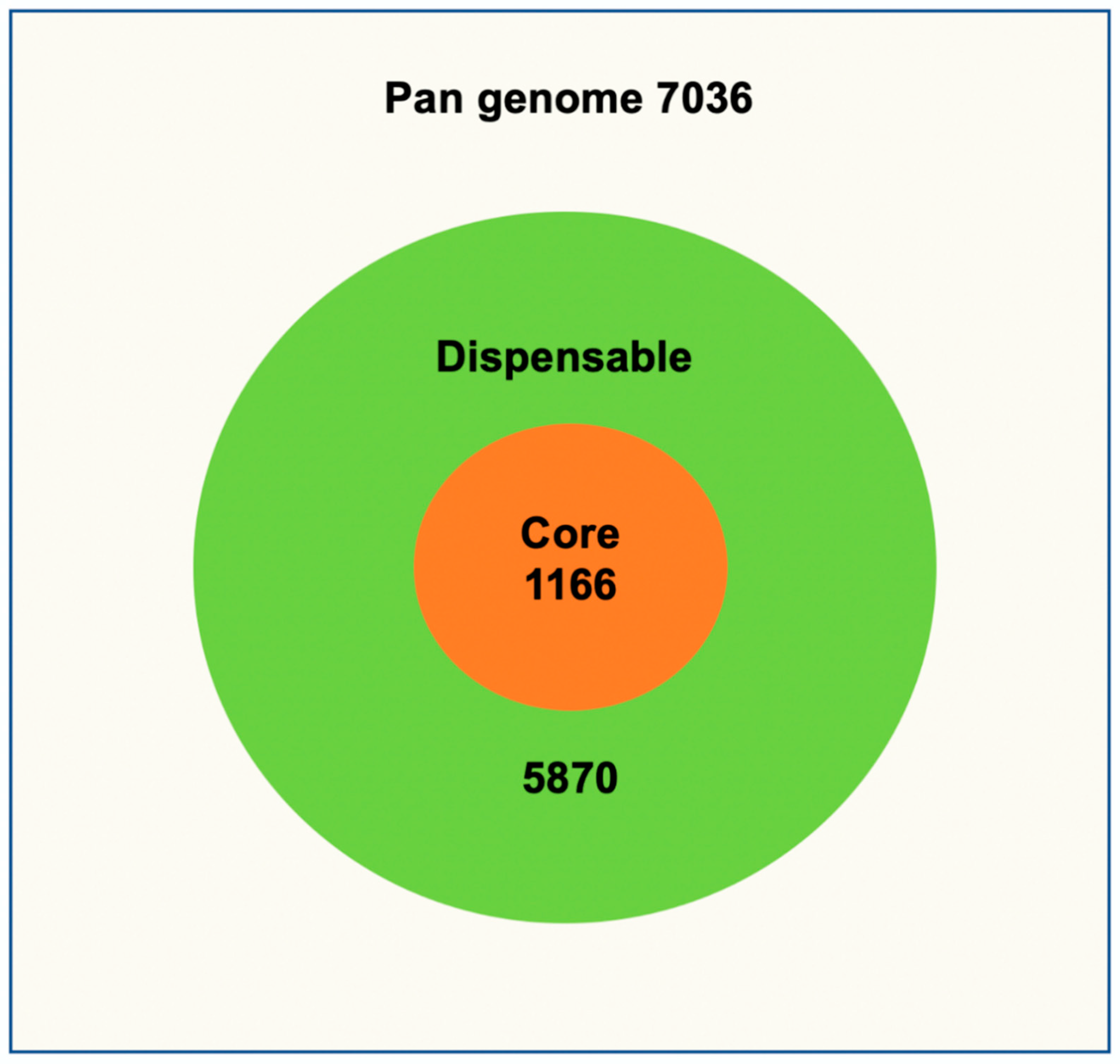
3.2. Core and Pan Genome
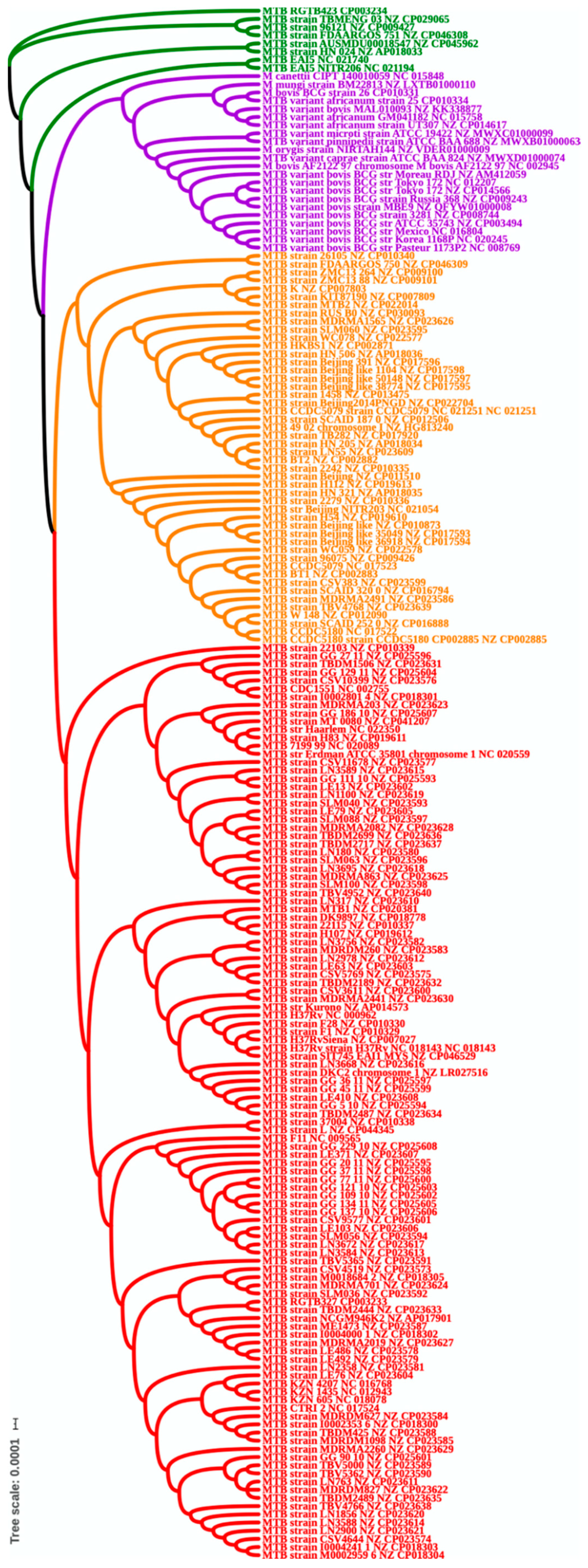
3.3. Phylogenetic Analysis
3.4. Clade Specific Analysis Based on Phylogenetic Tree
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Banuls, A.L.; Sanou, A.; Van Anh, N.T.; Godreuil, S. Mycobacterium tuberculosis: Ecology and evolution of a human bacterium. J. Med. Microbiol. 2015, 64, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Brites, D.; Gagneux, S. Co-evolution of Mycobacterium tuberculosis and Homo sapiens. Immunol Rev. 2015, 264, 6–24. [Google Scholar] [CrossRef]
- WHO. Global Tuberculosis Report; World Health Organization: Geneva, Switherland, 2019; pp. 1–284. [Google Scholar]
- Orgeur, M.; Brosch, R. Evolution of virulence in the Mycobacterium tuberculosis complex. Curr. Opin. Microbiol. 2018, 41, 68–75. [Google Scholar] [CrossRef]
- Brites, D.; Loiseau, C.; Menardo, F.; Borrell, S.; Boniotti, M.B.; Warren, R.; Dippenaar, A.; Parsons, S.D.C.; Beisel, C.; Behr, M.A.; et al. A New Phylogenetic Framework for the Animal-Adapted Mycobacterium tuberculosis Complex. Front. Microbiol. 2018, 9, 2820. [Google Scholar] [CrossRef]
- Ngabonziza, J.C.S.; Loiseau, C.; Marceau, M.; Jouet, A.; Menardo, F.; Tzfadia, O.; Antoine, R.; Niyigena, E.B.; Mulders, W.; Fissette, K.; et al. A sister lineage of the Mycobacterium tuberculosis complex discovered in the African Great Lakes region. Nat. Commun. 2020, 11, 2917. [Google Scholar] [CrossRef] [PubMed]
- WHO; FAO; OIE. Road Map of Zoonotic Tuberculosis; World Health Organization (WHO); Food and Agriculture Organization of the United Nations (FAO); World Organisation for Animal Health (OIE): 2017. Available online: https://www.oie.int/fileadmin/Home/eng/Our_scientific_expertise/docs/pdf/Tuberculosis/Roadmap_zoonotic_TB.pdf (accessed on 2 February 2020).
- Esteban, J.; Munoz-Egea, M.C. Mycobacterium bovis and Other Uncommon Members of the Mycobacterium tuberculosis Complex. Microbiol. Spectr. 2016, 4, 753–765. [Google Scholar] [CrossRef] [PubMed]
- Palmer, M.V. Mycobacterium bovis: Characteristics of wildlife reservoir hosts. Transbound. Emerg. Dis. 2013, 60, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Malone, K.M.; Gordon, S.V. Mycobacterium tuberculosis Complex Members Adapted to Wild and Domestic Animals. Adv. Exp. Med. Biol. 2017, 1019, 135–154. [Google Scholar] [CrossRef] [PubMed]
- WHO. Safety of BCG Vaccine in HIV-Infected Children; Global Vaccine Safety 2006; Weekly Epidemiological Record; World Health Organization: Geneve, Switzerland, 2007; Volume 82, pp. 17–24. [Google Scholar]
- Chisholm, R.H.; Tanaka, M.M. The emergence of latent infection in the early evolution of Mycobacterium tuberculosis. Proc. Biol. Sci. 2016, 283, 20160499. [Google Scholar] [CrossRef]
- Gagneux, S. Ecology and evolution of Mycobacterium tuberculosis. Nat. Rev. Microbiol. 2018, 16, 202–213. [Google Scholar] [CrossRef]
- Zakham, F.; Aouane, O.; Ussery, D.; Benjouad, A.; Ennaji, M.M. Computational genomics-proteomics and Phylogeny analysis of twenty one mycobacterial genomes (Tuberculosis & non Tuberculosis strains). Microb. Inform. Exp. 2012, 2, 1–9. [Google Scholar] [CrossRef]
- Faksri, K.; Xia, E.; Tan, J.H.; Teo, Y.-Y.; Ong, R.T.-H. In silico region of difference (RD) analysis of Mycobacterium tuberculosis complex from sequence reads using RD-Analyzer. BMC Genom. 2016, 17, 847. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.V.; Brosch, R.; Billault, A.; Garnier, T.; Eiglmeier, K.; Cole, S.T. Identification of variable regions in the genomes of tubercle bacilli using bacterial artificial chromosome arrays. Mol. Microbiol. 1999, 32, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Coscolla, M.; Gagneux, S. Consequences of genomic diversity in Mycobacterium tuberculosis. Semin. Immunol. 2014, 26, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Bottai, D.; Frigui, W.; Sayes, F.; Di Luca, M.; Spadoni, D.; Pawlik, A.; Zoppo, M.; Orgeur, M.; Khanna, V.; Hardy, D.; et al. TbD1 deletion as a driver of the evolutionary success of modern epidemic Mycobacterium tuberculosis lineages. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Kroesen, V.M.; Madacki, J.; Frigui, W.; Sayes, F.; Brosch, R. Mycobacterial virulence: Impact on immunogenicity and vaccine research. F1000Research 2019, 8, 2025. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, Y.; Zheng, H.; Pan, Y.; Liu, H.; Du, P.; Wan, L.; Liu, J.; Zhu, B.; Zhao, G.; et al. Genome sequencing and analysis of BCG vaccine strains. PLoS ONE 2013, 8, e71243. [Google Scholar] [CrossRef]
- Blom, J.; Albaum, S.P.; Doppmeier, D.; Pühler, A.; Vorhölter, F.J.; Zakrzewski, M.; Goesmann, A. EDGAR: A software framework for the comparative analysis of prokaryotic genomes. BMC Bioinform. 2009, 10, 154. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Felsenstein, J. PHYLIP—Phylogeny Inference Package, Version 3.6 Seattle: Department of Genome Sciences; 3.6 Seattle: Department of Genome Sciences, University of Washington: Seattle, WA, USA, 2005. [Google Scholar]
- Forrellad, M.A.; Klepp, L.I.; Gioffre, A.; Sabio y Garcia, J.; Morbidoni, H.R.; Santangelo, M.d.l.P.; Cataldi, A.A.; Bigi, F. Virulence factors of the Mycobacterium tuberculosis complex. Virulence 2013, 4, 3–66. [Google Scholar] [CrossRef]
- Riojas, M.A.; McGough, K.J.; Rider-Riojas, C.J.; Rastogi, N.; Hazbon, M.H. Phylogenomic analysis of the species of the Mycobacterium tuberculosis complex demonstrates that Mycobacterium africanum, Mycobacterium bovis, Mycobacterium caprae, Mycobacterium microti and Mycobacterium pinnipedii are later heterotypic synonyms of Mycobacterium tuberculosis. Int. J. Syst. Evol. Microbiol. 2018, 68, 324–332. [Google Scholar] [CrossRef]
- Yang, T.; Zhong, J.; Zhang, J.; Li, C.; Yu, X.; Xiao, J.; Jia, X.; Ding, N.; Ma, G.; Wang, G.; et al. Pan-Genomic Study of Mycobacterium tuberculosis Reflecting the Primary/Secondary Genes, Generality/Individuality, and the Interconversion Through Copy Number Variations. Front. Microbiol. 2018, 9, 1886. [Google Scholar] [CrossRef]
- Wan, X.; Koster, K.; Qian, L.; Desmond, E.; Brostrom, R.; Hou, S.; Douglas, J.T. Genomic analyses of the ancestral Manila family of Mycobacterium tuberculosis. PLoS ONE 2017, 12, e0175330. [Google Scholar] [CrossRef]
- Periwal, V.; Patowary, A.; Vellarikkal, S.K.; Gupta, A.; Singh, M.; Mittal, A.; Jeyapaul, S.; Chauhan, R.K.; Singh, A.V.; Singh, P.K.; et al. Comparative whole-genome analysis of clinical isolates reveals characteristic architecture of Mycobacterium tuberculosis pangenome. PLoS ONE 2015, 10, e0122979. [Google Scholar] [CrossRef] [PubMed]
- Kavvas, E.S.; Catoiu, E.; Mih, N.; Yurkovich, J.T.; Seif, Y.; Dillon, N.; Heckmann, D.; Anand, A.; Yang, L.; Nizet, V.; et al. Machine learning and structural analysis of Mycobacterium tuberculosis pan-genome identifies genetic signatures of antibiotic resistance. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Ioerger, T.R.; Huttenhower, C.; Long, J.E.; Sassetti, C.M.; Sacchettini, J.C.; Rubin, E.J. Global assessment of genomic regions required for growth in Mycobacterium tuberculosis. PLoS Pathog. 2012, 8, e1002946. [Google Scholar] [CrossRef] [PubMed]
- Sassetti, C.M.; Rubin, E.J. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. USA 2003, 100, 12989–12994. [Google Scholar] [CrossRef] [PubMed]
- Millington, K.A.; Fortune, S.M.; Low, J.; Garces, A.; Hingley-Wilson, S.M.; Wickremasinghe, M.; Kon, O.M.; Lalvani, A. Rv3615c is a highly immunodominant RD1 (Region of Difference 1)-dependent secreted antigen specific for Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. USA 2011, 108, 5730–5735. [Google Scholar] [CrossRef] [PubMed]
- Domenech, P.; Reed, M.B. Rapid and spontaneous loss of phthiocerol dimycocerosate (PDIM) from Mycobacterium tuberculosis grown in vitro: Implications for virulence studies. Microbiology (Read. Engl.) 2009, 155, 3532–3543. [Google Scholar] [CrossRef] [PubMed]
- Chiner-Oms, Ã.; Sanchez-Buso, L.; Corander, J.; Gagneux, S.; Harris, S.R.; Young, D.; Gonzalez-Candelas, F.; Comas, I. Genomic determinants of speciation and spread of the Mycobacterium tuberculosis complex. Sci. Adv. 2019, 5, eaaw3307. [Google Scholar] [CrossRef]
- Brosch, R.; Gordon, S.V.; Garnier, T.; Eiglmeier, K.; Frigui, W.; Valenti, P.; Dos Santos, S.; Duthoy, S.P.; Lacroix, C.L.; Garcia-Pelayo, C.; et al. Genome plasticity of BCG and impact on vaccine efficacy. Proc. Natl. Acad. Sci. USA 2007, 104, 5596–5601. [Google Scholar] [CrossRef]
- Miller, A.; Reandelar, M.J.; Fasciglione, K.; Roumenova, V.; Li, Y.; Otazu, G.H. Correlation between universal BCG vaccination policy and reduced morbidity and mortality for COVID-19: An epidemiological study. MedRxiv 2020. [Google Scholar] [CrossRef]
- Larsen, B.T.; Smith, M.L.; Grys, T.E.; Vikram, H.R.; Colby, T.V. Histopathology of Disseminated Mycobacterium bovis Infection Complicating Intravesical BCG Immunotherapy for Urothelial Carcinoma. Int. J. Surg. Pathol. 2015, 23, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Meije, Y.; Martinez-Montauti, J.; Cayla, J.A.; Loureiro, J.; Ortega, L.; Clemente, M.; Sanz, X.; Ricart, M.; Santoma, M.J.; Coll, P.; et al. Healthcare-Associated Mycobacterium bovis-Bacille Calmette-Guérin (BCG) Infection in Cancer Patients Without Prior BCG Instillation. Clin. Infect. Dis. 2017, 65, 1136–1143. [Google Scholar] [CrossRef][Green Version]
- Bottai, D.; Frigui, W.; Clark, S.; Rayner, E.; Zelmer, A.; Andreu, N.; de Jonge, M.I.; Bancroft, G.J.; Williams, A.; Brodin, P.; et al. Increased protective efficacy of recombinant BCG strains expressing virulence-neutral proteins of the ESX-1 secretion system. Vaccine 2015, 33, 2710–2718. [Google Scholar] [CrossRef] [PubMed]
- Ru, H.; Liu, X.; Lin, C.; Yang, J.; Chen, F.; Sun, R.; Zhang, L.; Liu, J. The Impact of Genome Region of Difference 4 (RD4) on Mycobacterial Virulence and BCG Efficacy. Front. Cell. Infect. Microbiol. 2017, 7, 239. [Google Scholar] [CrossRef]
- Bentley, S.D.; Comas, I.; Bryant, J.M.; Walker, D.; Smith, N.H.; Harris, S.R.; Thurston, S.; Gagneux, S.; Wood, J.; Antonio, M.; et al. The genome of Mycobacterium africanum West African 2 reveals a lineage-specific locus and genome erosion common to the M. tuberculosis complex. PLoS Negl. Trop. Dis. 2012, 6, e1552. [Google Scholar] [CrossRef]
- Rouli, L.; Merhej, V.; Fournier, P.E.; Raoult, D. The bacterial pangenome as a new tool for analysing pathogenic bacteria. New Microbes New Infect. 2015, 7, 72–85. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| RV Number or Gene in the Core Genome | Role of Virulence Factor |
|---|---|
| Rv3615c (espC), Rv3867 (espH), Rv1539 (lspA), Rv1791 | Secretion system |
| Rv2936 (DrrA), Rv2937 (DrrB), Rv2938 (drrC): daunorubicin ABC transporter | Synthesis of Phthiocerol dimycocerosates (PDIM) |
| Rv0642c (mmaA4) | Mycolic acid synthesis (hydroxymycolate synthase) |
| Rv3568c (hsaC), Rv3541c | Catabolism of cholesterol |
| Rv0167, Rv0170, Rv0173 (mce operon 1), Rv0589 (part of operon Mce2A), Rv0199 | Cell wall and conserved membrane proteins |
| Rv1410c, Rv1235 (lpqY) | Lipoproteins |
| Rv2031c, acr1 alpha-crystallin (hspX) | Inhibition of macrophage effectors |
| Rv1941, Rv1932 (tpX), Rv2234 (ptpA) | Oxidative/nitrosative stresses |
| Rv3133c (devR), Rv3132c (dosR), Rv2745c (clgR), Rv0353 (hspR), Rv3416 (whiB3), Rv0348 (mosR), Rv3082c (virS), Rv0821c PhoY2, Rv0990c, Rv0491 (regX3) | transcriptional regulators |
| Rv2115c (mpa) | Mycobacterial proteasome ATPase |
| Rv0758 (phoR), sensor kinase of phosphate regulon | Gene Expression Regulator |
| Rv2069 (sigC), Rv3414c (sigD), Rv1221 (sigE), Rv3223c (sigH), Rv0735 (sigL) | Sigma factors: RNA polymerase sigma factors |
| Rv2711 (ideR) iron-dependent repressor and activator Rv1811 (mgtC) Mg2+ transport P-type ATPase | Metal importers |
| Rv0990c conserved heat shock protein (hsp22.5) | Other mycobacterial virulence factors |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zakham, F.; Sironen, T.; Vapalahti, O.; Kant, R. Pan and Core Genome Analysis of 183 Mycobacterium tuberculosis Strains Revealed a High Inter-Species Diversity among the Human Adapted Strains. Antibiotics 2021, 10, 500. https://doi.org/10.3390/antibiotics10050500
Zakham F, Sironen T, Vapalahti O, Kant R. Pan and Core Genome Analysis of 183 Mycobacterium tuberculosis Strains Revealed a High Inter-Species Diversity among the Human Adapted Strains. Antibiotics. 2021; 10(5):500. https://doi.org/10.3390/antibiotics10050500
Chicago/Turabian StyleZakham, Fathiah, Tarja Sironen, Olli Vapalahti, and Ravi Kant. 2021. "Pan and Core Genome Analysis of 183 Mycobacterium tuberculosis Strains Revealed a High Inter-Species Diversity among the Human Adapted Strains" Antibiotics 10, no. 5: 500. https://doi.org/10.3390/antibiotics10050500
APA StyleZakham, F., Sironen, T., Vapalahti, O., & Kant, R. (2021). Pan and Core Genome Analysis of 183 Mycobacterium tuberculosis Strains Revealed a High Inter-Species Diversity among the Human Adapted Strains. Antibiotics, 10(5), 500. https://doi.org/10.3390/antibiotics10050500