
Computational Design and Development of Benzodioxane-Benzamides as Potent Inhibitors of FtsZ by Exploring the Hydrophobic Subpocket

 , ,
, ,  ,
,  ,
,
Abstract
1. Introduction
2. Results
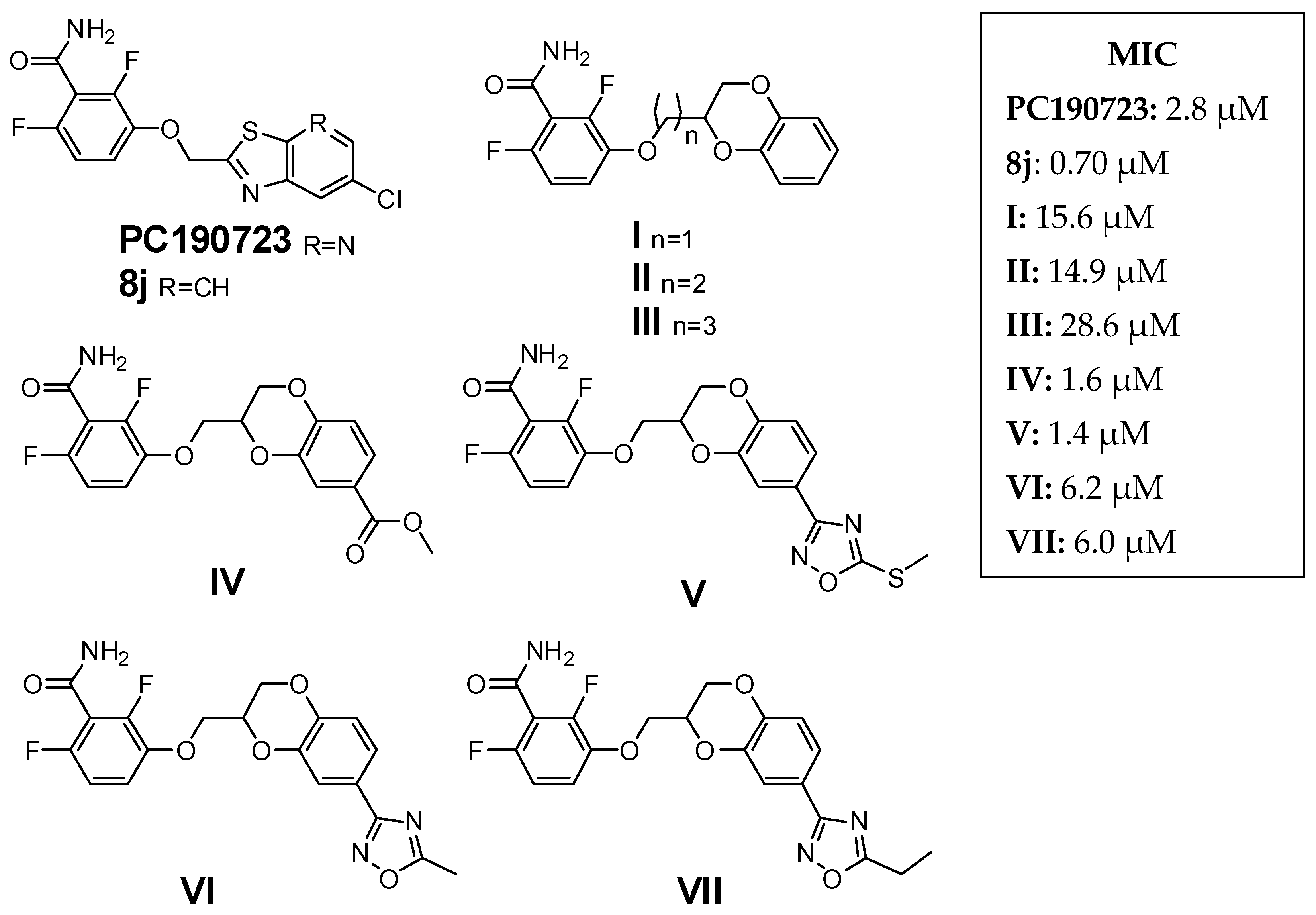
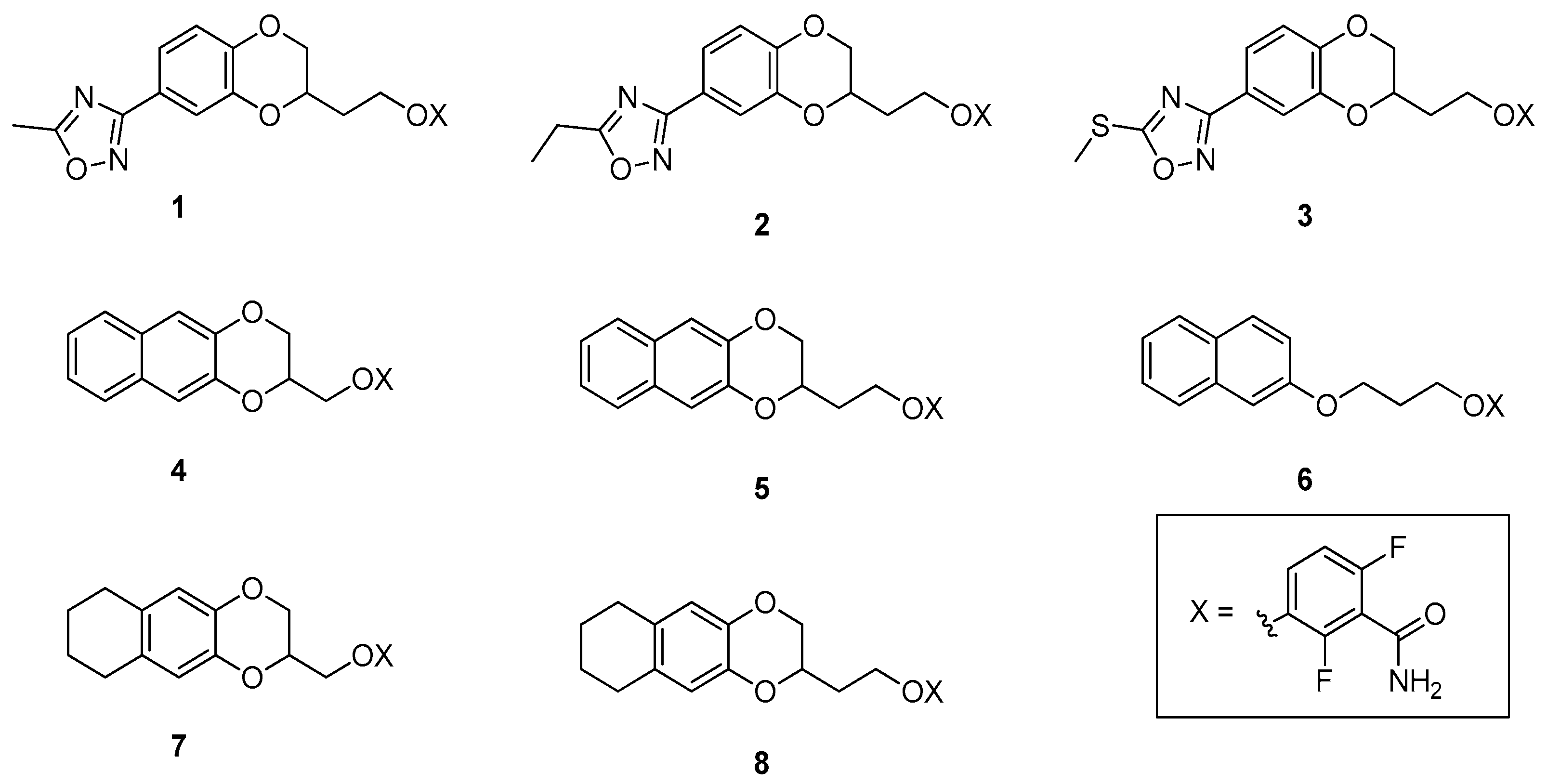
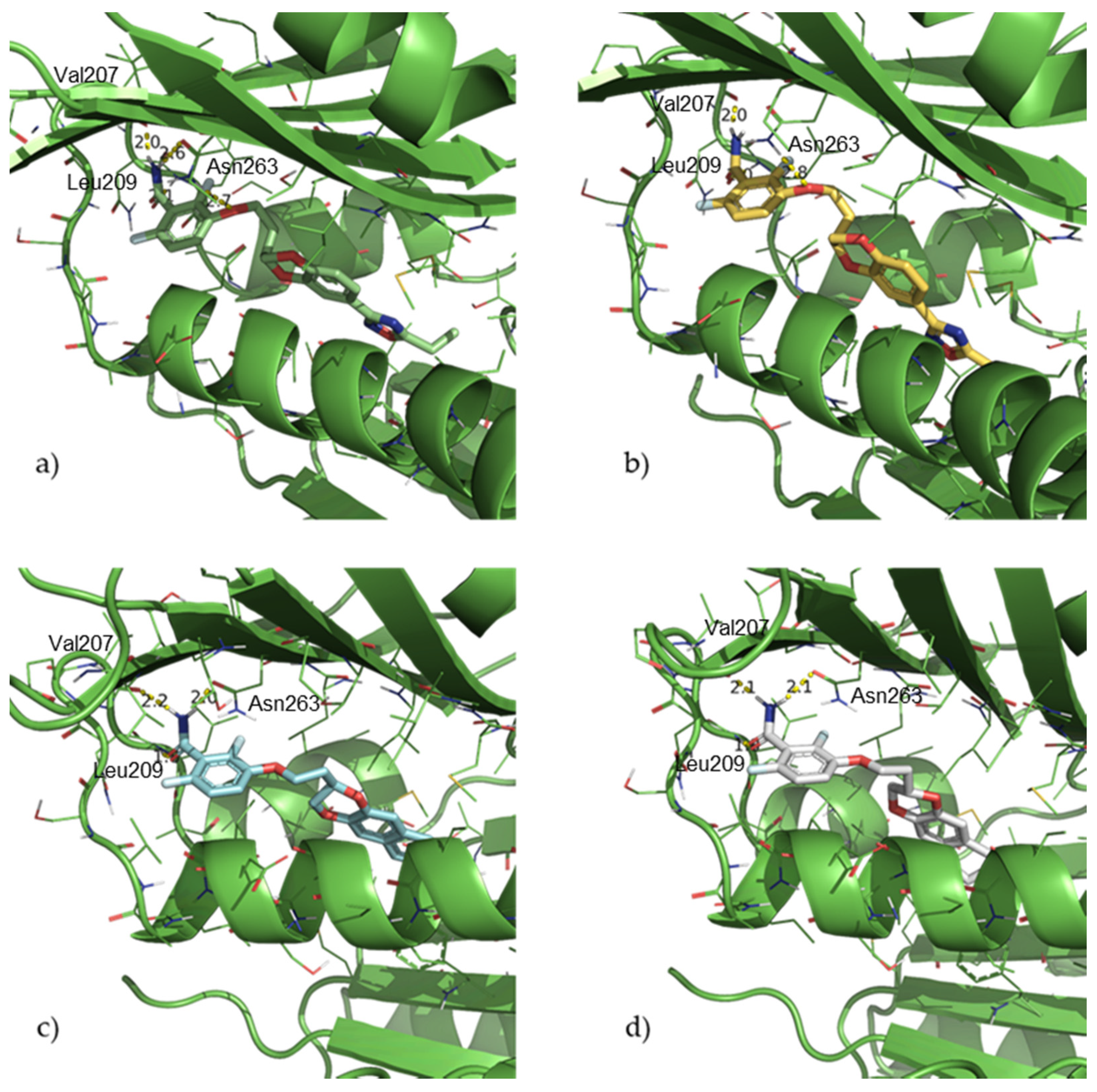
2.1. Design and Computational Studies
2.2. Physicochemical and Drug-Like Profile Calculations
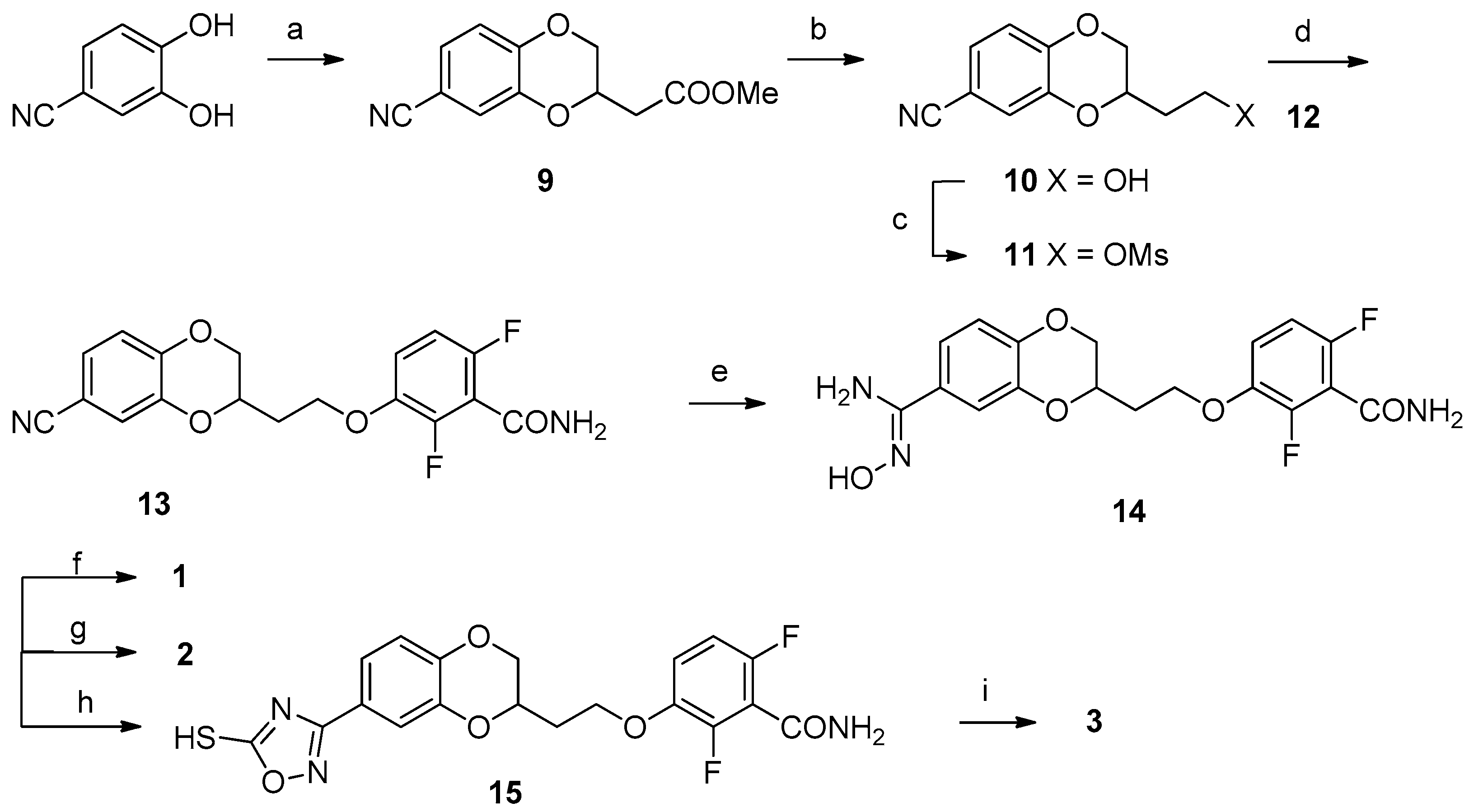
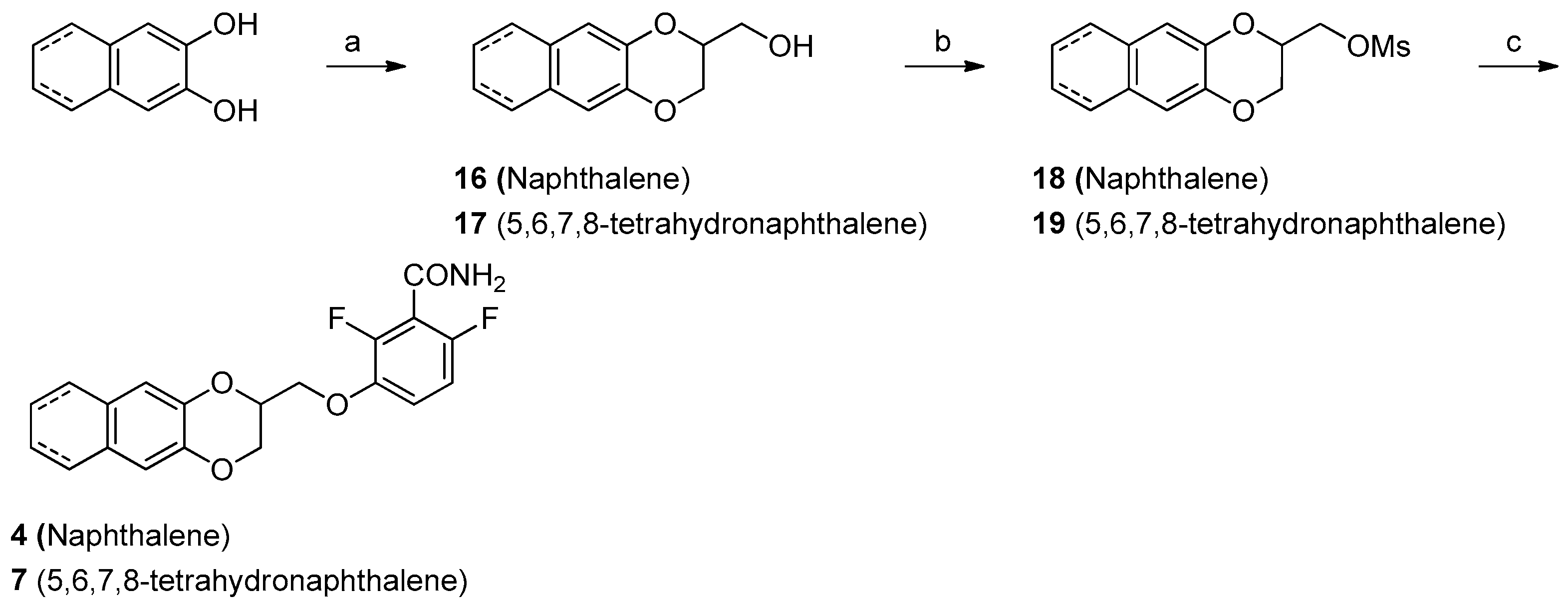
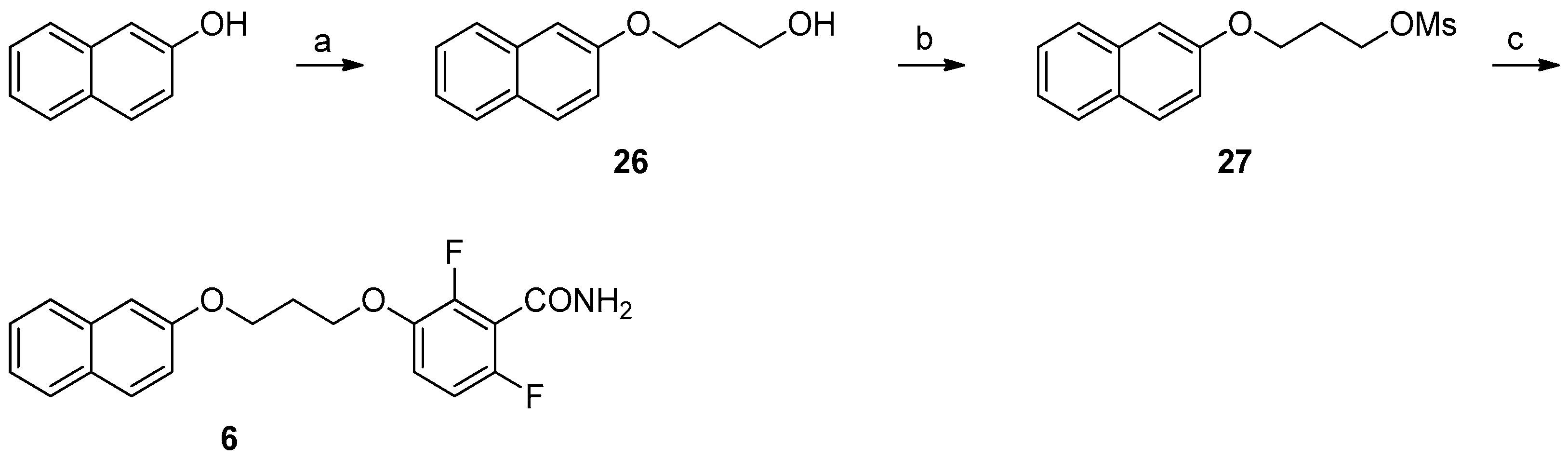
2.3. Chemistry
2.4. Antimicrobial Activity
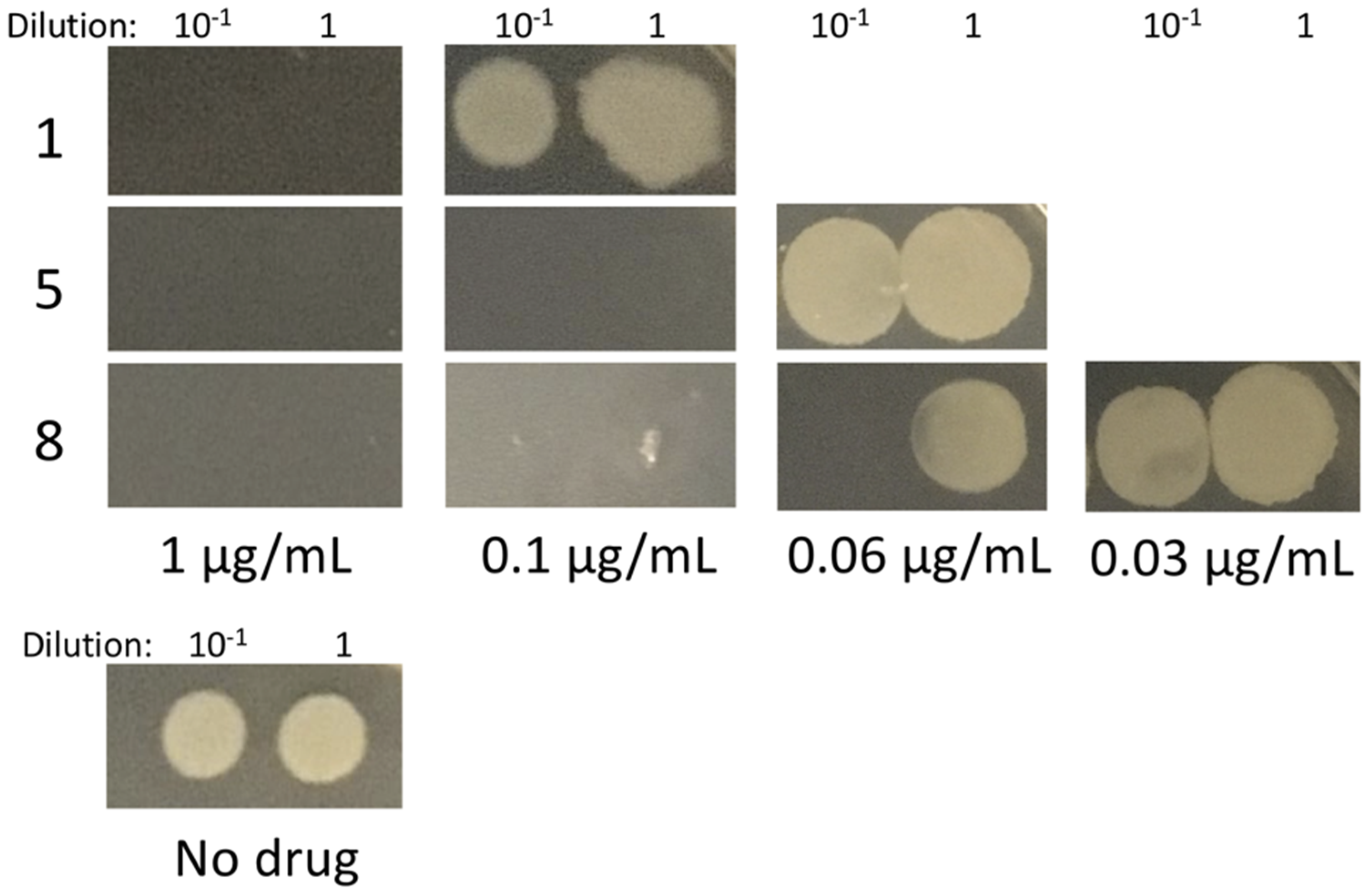
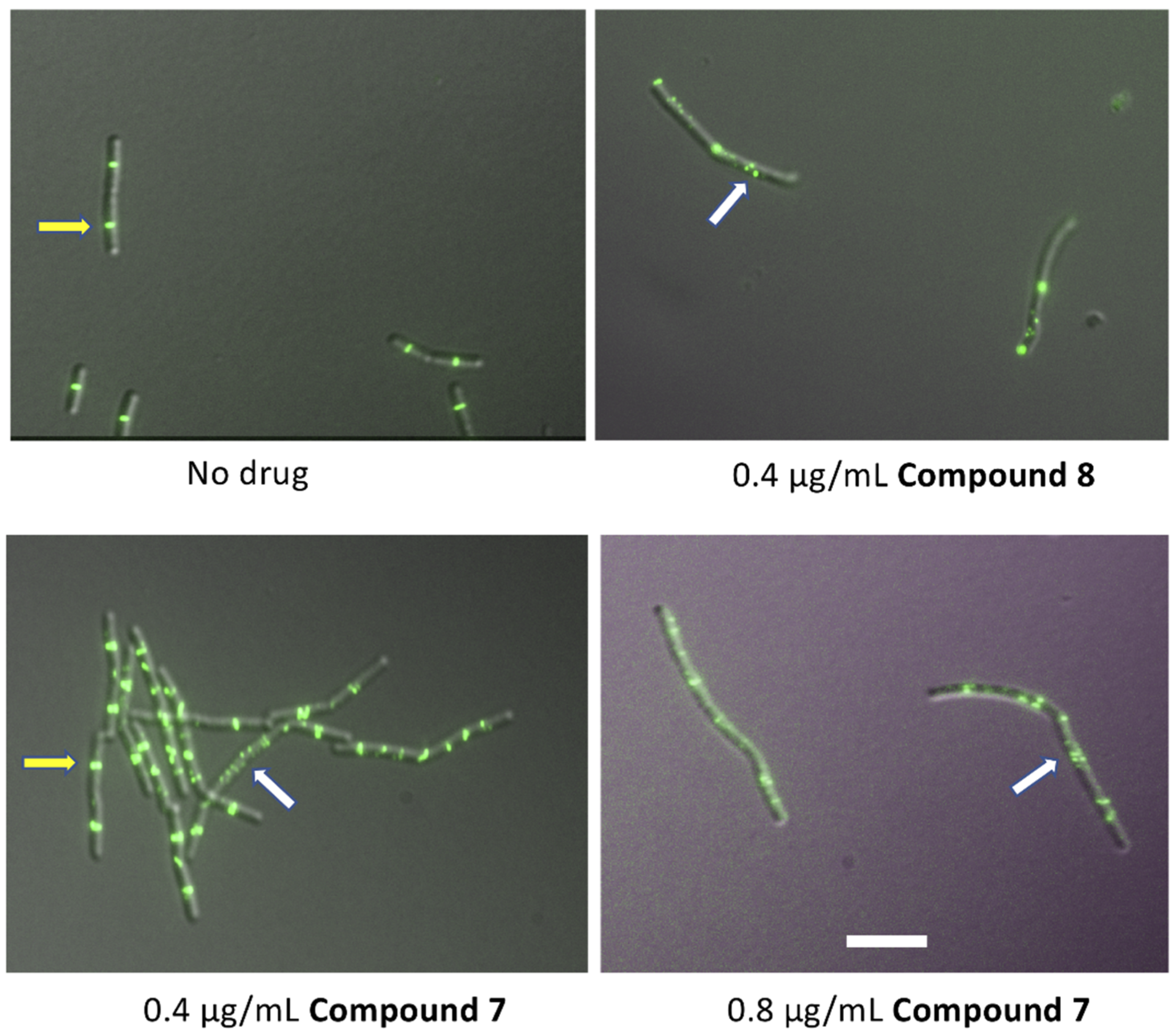
2.5. Effects on B. subtilis
3. Discussion
4. Materials and Methods
4.1. Chemistry
Synthesis
4.2. Cells
4.3. Antibacterial Activity
4.3.1. MSSA and MRSA Protocols
4.3.2. MDRSA
4.3.3. Antibacterial Activity against B. subtilis
4.4. Thiazolyl Blue Tetrazolium Bromide (MTT) Cytotoxicity Assay
4.5. Computational Studies
4.5.1. Ligand Preparation
4.5.2. Molecular Properties and Predictions
4.5.3. Protein Preparation
4.5.4. Induced Fit Docking
4.5.5. Docking Studies
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Giesbrecht, P.; Kersten, T.; Maidhof, H.; Wecke, J. Staphylococcal Cell Wall: Morphogenesis and Fatal Variations in the Presence of Penicillin. Microbiol. Mol. Biol. Rev. 1998, 62, 1371–1414. [Google Scholar] [CrossRef] [PubMed]
- Weidenmaier, C.; Goerke, C.; Wolz, C. Staphylococcus aureus determinants for nasal colonization. Trends Microbiol. 2012, 20, 243–250. [Google Scholar] [CrossRef]
- van Belkum, A.; Verkaik, N.J.; Vogel CP, d.e.; Boelens, H.A.; Verveer, J.; Nouwen, J.L.; Verbrugh, H.A.; Wertheim, H.F.L. Reclassification of Staphylococcus aureus nasal carriage types. J. Infect. Dis. 2009, 199, 1820–1826. [Google Scholar] [CrossRef]
- Gnanamani, A.; Hariharan, P.; Paul-Satyaseela, M. Staphylococcus aureus: Overview of Bacteriology, Clinical Diseases, Epidemiology, Antibiotic Resistance and Therapeutic Approach. In Frontiers in Staphylococcus Aureus; Enany, S., Crotty Alexander, L.E., Eds.; InTech: London, UK, 2017; ISBN 978-953-51-2981-3. [Google Scholar]
- Monaco, M.; Pimentel de Araujo, F.; Cruciani, M.; Coccia, E.M.; Pantosti, A. Worldwide Epidemiology and Antibiotic Resistance of Staphylococcus aureus. Curr. Top. Microbiol. Immunol. 2017, 409, 21–56. [Google Scholar] [CrossRef]
- Lindsay, J.A. Staphylococcus aureus genomics and the impact of horizontal gene transfer. Int. J. Med. Microbiol. 2014, 304, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Chambers, H.F. The changing epidemiology of Staphylococcus aureus? Emerg. Infect. Dis. 2001, 7, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Jevons, M.P. “Celbenin”—resistant Staphylococci. Br. Med. J. 1961, 1, 124–125. [Google Scholar] [CrossRef]
- Lakhundi, S.; Zhang, K. Methicillin-Resistant Staphylococcus aureus: Molecular Characterization, Evolution, and Epidemiology. Clin. Microbiol. Rev. 2018, 31. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Eswara, P.J.; Brzozowski, R.S.; Viola, M.G.; Graham, G.; Spanoudis, C.; Trebino, C.; Jha, J.; Aubee, J.I.; Thompson, K.M.; Camberg, J.L.; et al. An essential Staphylococcus aureus cell division protein directly regulates FtsZ dynamics. Elife 2018, 7. [Google Scholar] [CrossRef]
- Margolin, W. FtsZ and the division of prokaryotic cells and organelles. Nat. Rev. Mol. Cell Biol. 2005, 6, 862–871. [Google Scholar] [CrossRef]
- Erickson, H.P. FtsZ, a tubulin homologue in prokaryote cell division. Trends Cell Biol. 1997, 7, 362–367. [Google Scholar] [CrossRef]
- Naclerio, G.A.; Sintim, H.O. Multiple ways to kill bacteria via inhibiting novel cell wall or membrane targets. Future Med. Chem. 2020, 12, 1253–1279. [Google Scholar] [CrossRef]
- Tripathy, S.; Sahu, S.K. FtsZ inhibitors as a new genera of antibacterial agents. Bioorg. Chem. 2019, 91, 103169. [Google Scholar] [CrossRef] [PubMed]
- Carro, L. Recent Progress in the Development of Small-Molecule FtsZ Inhibitors as Chemical Tools for the Development of Novel Antibiotics. Antibiotics 2019, 8, 217. [Google Scholar] [CrossRef] [PubMed]
- Casiraghi, A.; Suigo, L.; Valoti, E.; Straniero, V. Targeting Bacterial Cell Division: A Binding Site-Centered Approach to the Most Promising Inhibitors of the Essential Protein FtsZ. Antibiotics 2020, 9, 69. [Google Scholar] [CrossRef]
- Chiodini, G.; Pallavicini, M.; Zanotto, C.; Bissa, M.; Radaelli, A.; Straniero, V.; Bolchi, C.; Fumagalli, L.; Ruggeri, P.; De Giuli Morghen, C.; et al. Benzodioxane-benzamides as new bacterial cell division inhibitors. Eur. J. Med. Chem. 2015, 89, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Straniero, V.; Pallavicini, M.; Chiodini, G.; Zanotto, C.; Volontè, L.; Radaelli, A.; Bolchi, C.; Fumagalli, L.; Sanguinetti, M.; Menchinelli, G.; et al. 3-(Benzodioxan-2-ylmethoxy)-2,6-difluorobenzamides bearing hydrophobic substituents at the 7-position of the benzodioxane nucleus potently inhibit methicillin-resistant Sa and Mtb cell division. Eur. J. Med. Chem. 2016, 120, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Straniero, V.; Sebastián Pérez, V.; Hrast, M.; Zanotto, C.; Casiraghi, A.; Suigo, L.; Zdovc, I.; Radaelli, A.; Giuli Morghen C, d.e.; Valoti, E. Benzodioxane-benzamides as antibacterial agents: Computational and SAR studies to evaluate the influence of the 7-substitution in FtsZ interaction. ChemMedChem 2020, 2, 195–209. [Google Scholar] [CrossRef]
- Straniero, V.; Zanotto, C.; Straniero, L.; Casiraghi, A.; Duga, S.; Radaelli, A.; De Giuli Morghen, C.; Valoti, E. 2,6-Difluorobenzamide Inhibitors of Bacterial Cell Division Protein FtsZ: Design, Synthesis, and Structure-Activity Relationships. ChemMedChem 2017, 12, 1303–1318. [Google Scholar] [CrossRef]
- Straniero, V.; Suigo, L.; Casiraghi, A.; Sebastián-Pérez, V.; Hrast, M.; Zanotto, C.; Zdovc, I.; De Giuli Morghen, C.; Radaelli, A.; Valoti, E. Benzamide Derivatives Targeting the Cell Division Protein FtsZ: Modifications of the Linker and the Benzodioxane Scaffold and Their Effects on Antimicrobial Activity. Antibiotics 2020, 9, 160. [Google Scholar] [CrossRef]
- Haydon, D.J.; Bennett, J.M.; Brown, D.; Collins, I.; Galbraith, G.; Lancett, P.; Macdonald, R.; Stokes, N.R.; Chauhan, P.K.; Sutariya, J.K.; et al. Creating an antibacterial with in vivo efficacy: Synthesis and characterization of potent inhibitors of the bacterial cell division protein FtsZ with improved pharmaceutical properties. J. Med. Chem. 2010, 53, 3927–3936. [Google Scholar] [CrossRef]
- Haydon, D.J.; Stokes, N.R.; Ure, R.; Galbraith, G.; Bennett, J.M.; Brown, D.R.; Baker, P.J.; Barynin, V.V.; Rice, D.W.; Sedelnikova, S.E.; et al. An inhibitor of FtsZ with potent and selective anti-staphylococcal activity. Science 2008, 321, 1673–1675. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Inc. QikProp Descriptors and Properties; Schrödinger Inc.: New York, NY, USA, 2012. [Google Scholar]
- Li, P.; Yang, S.; Zhu, R.; Sun, B.; Li, Z.; Huang, P.; Buser, J.Y.; Miguel Minguez, J.; Ryan, S.J. Continuous Flow Conditions for High Temperature Formation of a Benzodioxan Pharmaceutical Intermediate: Rapid Scaleup for Early Phase Material Delivery. Org. Process Res. Dev. 2020, 24, 1938–1947. [Google Scholar] [CrossRef]
- Bolchi, C.; Catalano, P.; Fumagalli, L.; Gobbi, M.; Pallavicini, M.; Pedretti, A.; Villa, L.; Vistoli, G.; Valoti, E. Structure-affinity studies for a novel series of homochiral naphtho and tetrahydronaphtho analogues of alpha 1 antagonist WB-4101. Bioorg. Med. Chem. 2004, 12, 4937–4951. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.W.; Wu, L.J.; Czaplewski, L.G.; Errington, J. Multiple effects of benzamide antibiotics on FtsZ function. Mol. Microbiol. 2011, 80, 68–84. [Google Scholar] [CrossRef]
- Gueiros-Filho, F.J.; Losick, R. A widely conserved bacterial cell division protein that promotes assembly of the tubulin-like protein FtsZ. Genes Dev. 2002, 16, 2544–2556. [Google Scholar] [CrossRef]
- Bhambhani, A.; Iadicicco, I.; Lee, J.; Ahmed, S.; Belfatto, M.; Held, D.; Marconi, A.; Parks, A.; Stewart, C.R.; Margolin, W.; et al. Bacteriophage SP01 Gene Product 56 Inhibits Bacillus subtilis Cell Division by Interacting with FtsL and Disrupting Pbp2B and FtsW Recruitment. J. Bacteriol. 2020, 203. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Schrödinger Inc. LigPrep; Schrödinger Release 2015-4; Schrödinger Inc.: New York, NY, USA, 2015. [Google Scholar]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Inc. QikProp; Schrödinger Inc.: New York, NY, USA, 2014. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings 1PII of original article: S0169-409X(96)00423-1. Adv. Drug Deliv. Rev. 1997, 23, 3–25, reprinted in Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Fujita, J.; Maeda, Y.; Mizohata, E.; Inoue, T.; Kaul, M.; Parhi, A.K.; LaVoie, E.J.; Pilch, D.S.; Matsumura, H. Structural Flexibility of an Inhibitor Overcomes Drug Resistance Mutations in Staphylococcus aureus FtsZ. ACS Chem. Biol. 2017, 12, 1947–1955. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Farid, R.; Day, T.; Friesner, R.A.; Pearlstein, R.A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem. 2006, 14, 3160–3173. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Inc. Glide; Schrödinger Release 2015-4; Schrödinger Inc.: New York, NY, USA, 2015. [Google Scholar]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Compound | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
| #stars | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| #rotor | 5 | 6 | 6 | 4 | 5 | 7 | 4 | 5 |
| mol MW | 417.36 | 431.39 | 449.42 | 371.34 | 385.36 | 365.33 | 375.37 | 389.39 |
| Dipole | 8.655 | 6.737 | 6.781 | 6.875 | 7.728 | 8.626 | 7.237 | 6.919 |
| Donor-HB | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Acceptor-HB | 7.75 | 7.75 | 7.75 | 4.75 | 4.75 | 6.45 | 4.75 | 4.75 |
| QPlogPo/w | 2.985 | 3.402 | 3.578 | 3.91 | 4.351 | 3.258 | 3.959 | 4.324 |
| QPlogS | −5.771 | −6.235 | −6.326 | −5.623 | −6.048 | −4.89 | −5.967 | −6.214 |
| CIQPlogS | −5.703 | −5.98 | −6.338 | −5.857 | −6.138 | −4.87 | −5.678 | −5.96 |
| QPlogHERG | −6.225 | −6.356 | −6.247 | −6.296 | −6.459 | −6.137 | −5.581 | −5.574 |
| QPPCaco | 242.42 | 296.467 | 294.572 | 1003.662 | 1004.009 | 996.155 | 996.137 | 996.13 |
| QPPMDCK | 274.89 | 341.549 | 606.764 | 1292.396 | 1210.755 | 1267.327 | 1284.186 | 1199.828 |
| #metab | 3 | 3 | 2 | 2 | 2 | 2 | 4 | 4 |
| QPlogKhsa | 0.189 | 0.29 | 0.294 | 0.419 | 0.566 | 0.038 | 0.522 | 0.645 |
| HumanOralAbsorption | 3 | 1 | 1 | 3 | 3 | 3 | 3 | 1 |
| %HumanOralAbsorptio | 87.1 | 91.11 | 92.091 | 100 | 100 | 100 | 100 | 100 |
| PSA | 114.154 | 113.006 | 111.857 | 76.188 | 74.716 | 82.813 | 76.249 | 74.709 |
| #NandO | 8 | 8 | 8 | 5 | 5 | 6 | 5 | 5 |
| RuleOfFive | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| RuleOfThree | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 1 |
| #ringatoms | 21 | 21 | 21 | 20 | 20 | 16 | 20 | 20 |
| #in34 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| #in56 | 21 | 21 | 21 | 20 | 20 | 16 | 20 | 20 |
| #noncon | 2 | 2 | 2 | 2 | 2 | 2 | 6 | 6 |
| #nonHatm | 30 | 31 | 31 | 27 | 28 | 26 | 27 | 28 |
| Cpd | MSSA ATCC 29213 and MRSA ATCC 43300 | MRC-5 | TI | MDRSA 12.1 | MDRSA 11.7 | |
|---|---|---|---|---|---|---|
| MIC (μM) | MBC (μM) | TD90 (μM) | MIC (μM) | MIC (μM) | ||
| I | 15.6 | 249 | 49.8 | 3.2 | 24.9 | 49.8 |
| II | 14.9 | 14.9 | 226 | 15.2 | 11.9 | 11.9 |
| V | 1.4 | 1.4 | 1700 | 1231 | <2.3 | <2.3 |
| VI | 6.2 | 6.2 | >1983 | >320 | 5.0 | 9.9 |
| VII | 6.0 | 6.0 | >1916 | >320 | 4.8 | 9.6 |
| 1 | 1.2 | 1.2 | >1910 | >1591 | 1.2 | 1.2 |
| 2 | 0.6 | 0.6 | 1854 | 3090 | 0.6 | 0.6 |
| 3 | 0.5 | 0.5 | 1668 | 3034 | 1.1 | 1.1 |
| 4 | 7.1 | 7.1 | >2276 | >320 | 11.4 | 11.4 |
| 5 | 0.6 | 0.6 | 233 | 360 | 0.6 | 1.3 |
| 6 | 14.0 | 14.0 | >2238 | >160 | 11.2 | 11.2 |
| 7 | 13.3 | 13.3 | 200 | 15.0 | 5.3 | 10.7 |
| 8 | 0.25 | 0.25 | 2054 | 8200 | 0.3 | 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Straniero, V.; Sebastián-Pérez, V.; Suigo, L.; Margolin, W.; Casiraghi, A.; Hrast, M.; Zanotto, C.; Zdovc, I.; Radaelli, A.; Valoti, E. Computational Design and Development of Benzodioxane-Benzamides as Potent Inhibitors of FtsZ by Exploring the Hydrophobic Subpocket. Antibiotics 2021, 10, 442. https://doi.org/10.3390/antibiotics10040442
Straniero V, Sebastián-Pérez V, Suigo L, Margolin W, Casiraghi A, Hrast M, Zanotto C, Zdovc I, Radaelli A, Valoti E. Computational Design and Development of Benzodioxane-Benzamides as Potent Inhibitors of FtsZ by Exploring the Hydrophobic Subpocket. Antibiotics. 2021; 10(4):442. https://doi.org/10.3390/antibiotics10040442
Chicago/Turabian StyleStraniero, Valentina, Victor Sebastián-Pérez, Lorenzo Suigo, William Margolin, Andrea Casiraghi, Martina Hrast, Carlo Zanotto, Irena Zdovc, Antonia Radaelli, and Ermanno Valoti. 2021. "Computational Design and Development of Benzodioxane-Benzamides as Potent Inhibitors of FtsZ by Exploring the Hydrophobic Subpocket" Antibiotics 10, no. 4: 442. https://doi.org/10.3390/antibiotics10040442
APA StyleStraniero, V., Sebastián-Pérez, V., Suigo, L., Margolin, W., Casiraghi, A., Hrast, M., Zanotto, C., Zdovc, I., Radaelli, A., & Valoti, E. (2021). Computational Design and Development of Benzodioxane-Benzamides as Potent Inhibitors of FtsZ by Exploring the Hydrophobic Subpocket. Antibiotics, 10(4), 442. https://doi.org/10.3390/antibiotics10040442