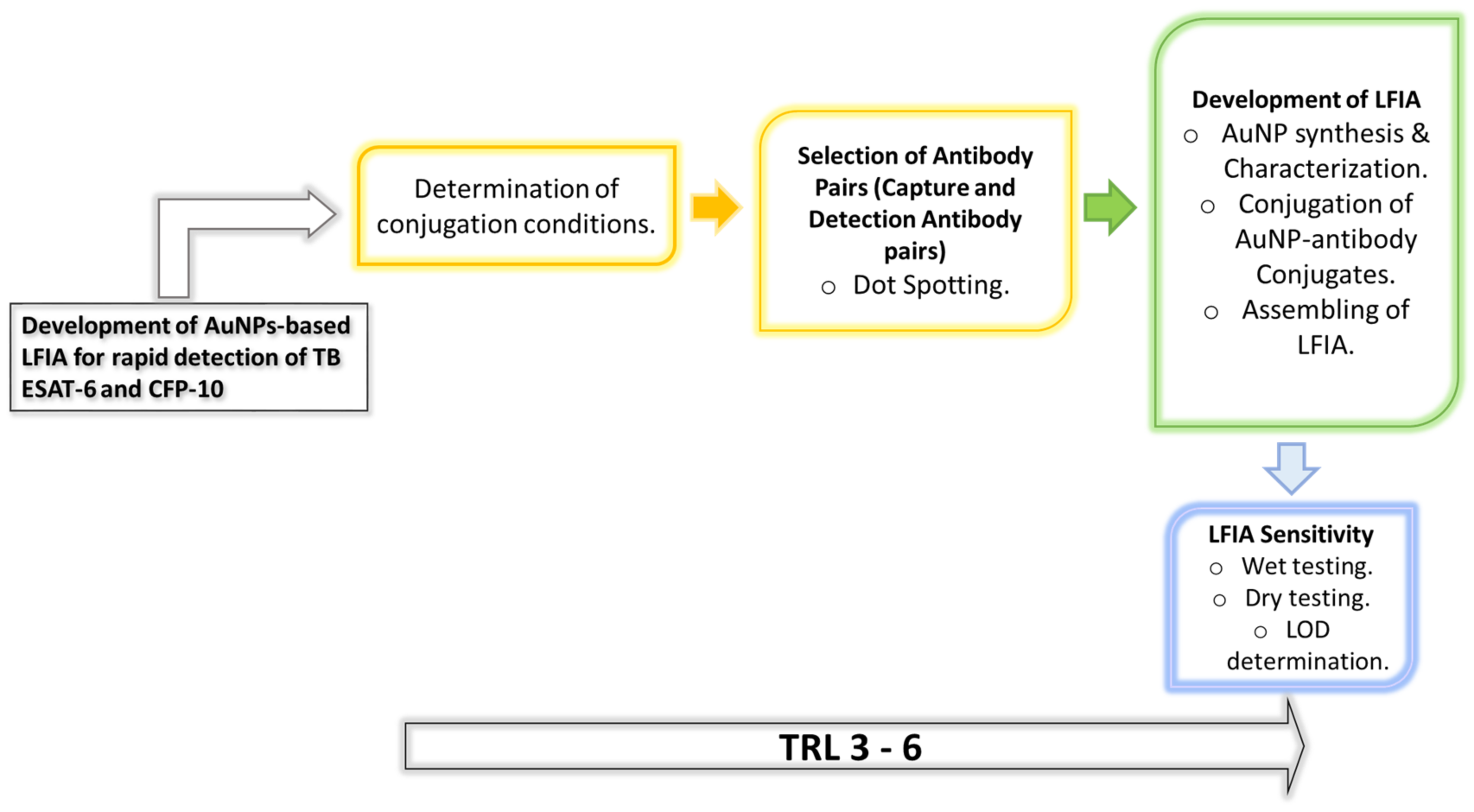
1. Introduction
Tuberculosis (TB) is an infectious disease caused by
Mycobacterium tuberculosis (
M.
tb), which when detected timeously and with appropriate therapeutic intervention can be curable. The global representation of this crisis is massive, as 3.6 million of TB cases can go undiagnosed or unreported in low–middle income countries. In sub-Saharan Africa, 5 out of 10 active TB cases go undiagnosed, and in South Africa (SA) alone, an estimated 150,000 of cases are not diagnosed, which poses a risk for increasing the rates of transmission, morbidity and mortality [
1]. The lack of rapid, accurate diagnostic tools at a point of care (PoC), including the inaccessibility of easily collectible samples, is central to the missed diagnosis and subsequent spread of the disease. In the current study, the immunodominant TB antigens ESAT-6 and CFP-10 were explored as targets for the development of TB-antigen rapid diagnostic test (RDT) prototypes which were based on the lateral flow immunoassays (LFIAs) technology.
To date, microbiological testing such as sputum smear microscopy and sputum culture test remain the most widely used methods of TB diagnosis [
2]. The major limitation of the sputum smear microscopy test which exploits staining of acid-fast bacteria (AFB) is that it requires 10,000 bacilli/mL from a sputum sample for a positive TB detection [
2], making it less sensitive for paucibacillary cases in HIV positive patients, children who often present with extra-pulmonary TB (EPTB) and very ill patients who cannot expectorate [
2,
3,
4]. The sputum culture test has a long turnaround time of 2 to 4 weeks. These tests are expensive and need to be carried out by experienced personnel in specialized facilities and with costly instrumentation. Likewise, molecular tests such as the PCR-based GeneXpert MTB/RIF (Cepheid Inc., Sunnyvale, CA, USA) and the TB loop-mediated isothermal amplification (LAMP) (Eiken; Tokyo, Japan) are costly, and not readily accessible to low-income countries nor suitable for PoC testing [
5,
6,
7]. The blood-based immune response tuberculin skin test (TST) and the IFN-γ release assays (IGRAs) (QuantiFERON-TB Gold, Cellestis Ltd., Australia) have uncircumventable limitations such as their inability to accurately distinguish between active and latent infections [
4]. In addition, TST does not differentiate
M.
tb from nontuberculous
Mycobacterium and the Bacille Calmette–Guérin (BCG) vaccine, making it unsuitable for TB testing in a South African cohort [
2].
Lateral flow immunoassays are user friendly, produce results within 10 to 15 min and are suitable for PoC use either at medical facilities or at home by the end user [
8,
9]. The Alere Determine™ LFIA was introduced in South Africa (SA) in 2018, and used to detect the
Mycobacterial lipoarabinomannan (LAM) antigen in urine samples within 25 min. This test, however, was found to be more suitable for TB testing in HIV-positive patients. The commercially available LFIA-based TB RDTs have limited endorsement from the World Health Organization (WHO) [
10], and in some studies, the reported sensitivity and specificity of the diagnostic kits were inconsistent [
11]. The poor specificity may be attributed to cross-reactivity with the BCG vaccine and other
Mycobacterium species [
12].
In contrast, the current TB-antigen RDT explores the immunodominant TB secreted antigens, ESAT-6 and CFP-10, as targets in detecting TB following the research strategy summarized in
Scheme 1. In the globally used BCG vaccine, these antigens are not expressed subsequent to the deletion of the region of difference (RD)1 locus in which they are located [
13,
14]. As a result, cross-reactivity in patients previously vaccinated with BCG and/or those infected with other
Mycobacterium species will be minimized [
15,
16,
17]. Additionally, these biomarkers have been previously detected in the serum from HIV-positive and HIV-negative pulmonary TB (PTB) and extra-pulmonary TB (EPTB) patients with similar sensitivities. The antigens were detectable at low concentrations <10 nM in the serum, which was useful for diagnosis and evaluating anti-TB response [
14]. To expedite the Stop TB Strategy, TB RDTs are now adopted to detect TB biomarkers in easily accessible samples such as urine [
8] and blood samples, and sensing of volatile organic compounds from the breath [
9,
10]. The presence of ESAT-6 and CFP-10 in urine [
15,
16,
17,
18] and blood [
14,
15] samples makes them ideal TB biomarkers, especially for RDTs. However, the methods used to detect antigens such as mass spectrometry (MS) and ELISA are not suitable for POC testing, as the very long sample processing and analysis require highly trained personnel. Therefore, the proposed AuNP-based LFIAs will be optimized for rapid detection of ESAT-6 and CFP-10 from non-sputum samples which will be beneficial for patients who cannot expectorate.
2. Materials and Methods
2.1. Materials
The ESAT-6 and CFP-10 antibodies were obtained from manufacturers outlined in
Table 1. The antibodies were given specific codes in the current study, as indicated in the table. The gold (III) chloride (HAuCl
4.3H
2O), trisodium citrate (Na
3C
6H
5O
7), and anti-Mouse IgM (μ-chain specific) antibody produced in goat were bought from Sigma-Aldrich (St Louis, Missouri, USA). The anti-Rabbit Affinity purified Goat IgG was acquired from R&D systems (Minneapolis, Minnesota, USA). The nitrocellulose membrane CN95 was purchased from Sartorius (PTY) LTD (Midrand, South Africa), whilst the Millipore conjugate pad G041 and Millipore C083 absorbent pad were bought from Sigma-Aldrich and the backing cards from Diagnostic Consulting Network (Carlsbad, California, USA).
2.2. Synthesis of 14 nm AuNPs
The 14 nm AuNPs were synthesized using the Turkevich method (Turkevich et al., 1951), which entails the reduction of HAuCl
4 with trisodium citrate as described previously. Trisodium citrate and HAuCl
4.3H
2O solutions were prepared at a concentration of 1% and 0.0289 M, respectively. The solutions were each filtered using 0.2 µm filters and thereafter used in the synthesis of the 14 nm AuNPs [
15,
16].
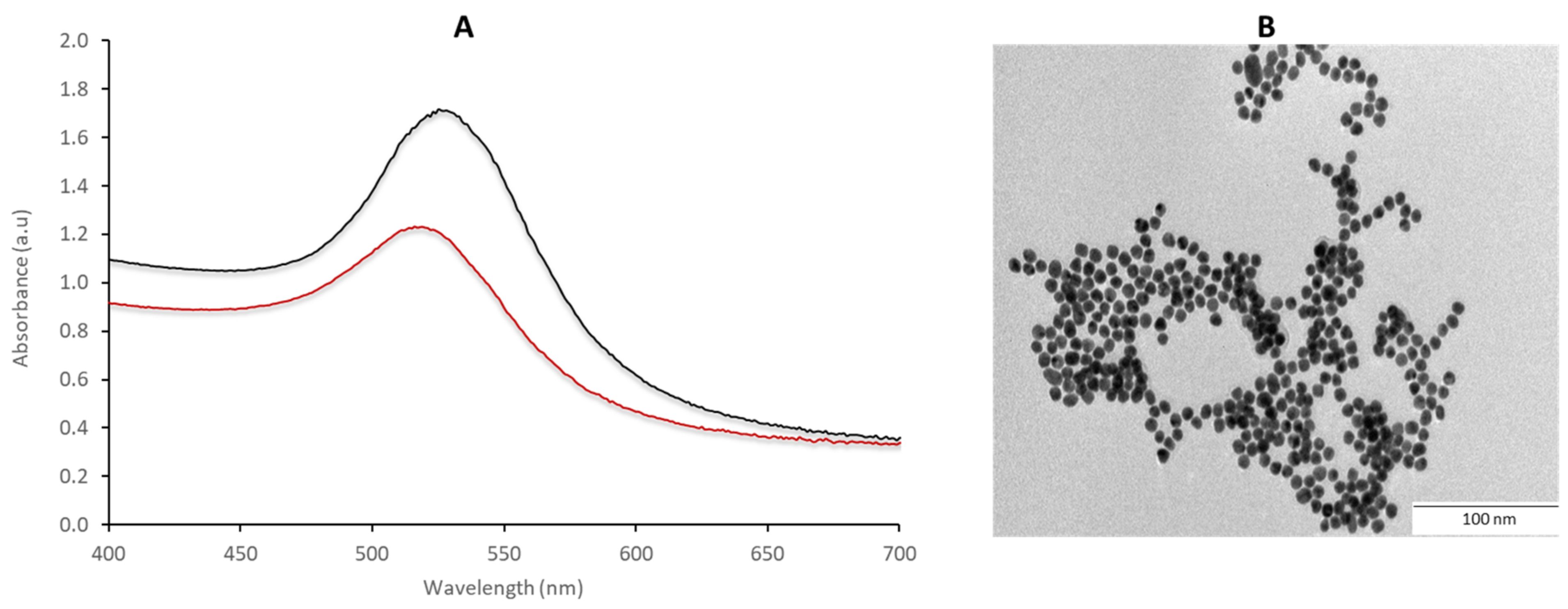
2.3. Characterization of the AuNPs Using UV-vis Spectrophotometer and TEM
The optical properties of the 14 nm AuNPs were measured using the UV-vis spectrophotometer (Multiskan
TM GO plate reader (Thermo Fischer Scientific, Waltham, Massachusetts, USA)) within a wavelength range of 200 to 800 nm. Transmission electron microscopy (TEM) analysis was carried out on the JEOL JEM-2100F following previous procedures [
19,
20].
2.4. Conjugation of Antibodies to AuNPs
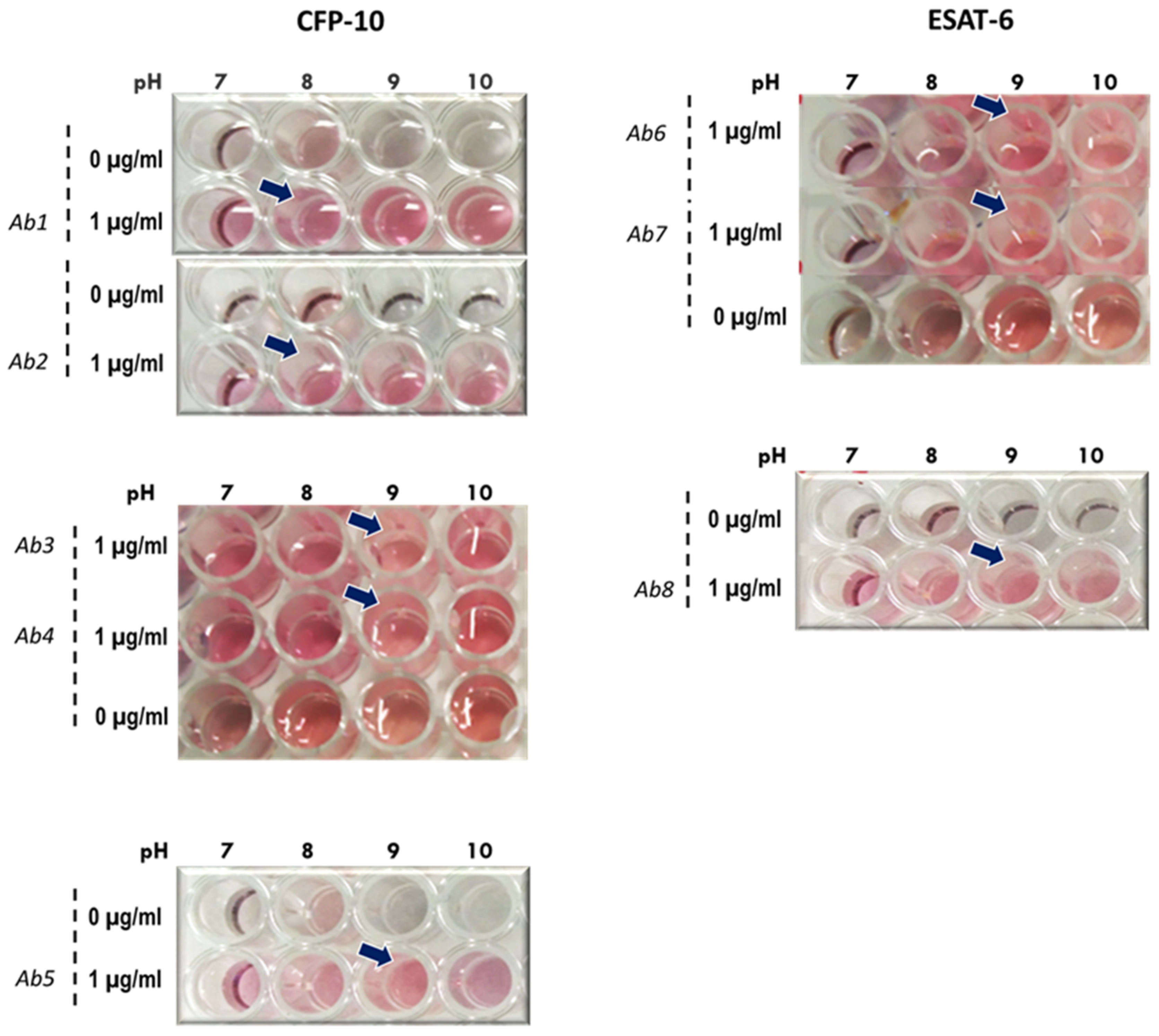
2.4.1. Stability of AuNP–Antibody Conjugates
Stability of 14 nm AuNP–antibody conjugates was assessed at different pH levels. The pH levels of AuNPs were adjusted to pH 7, 8, 9 and 10 using 0.1 M K2CO3. Thereafter, 100 µL of each AuNP solutions was dispensed into a 96-well plate. The antibodies were added to the AuNPs to a final concentration of 10 µg/mL. In negative controls, phosphate-buffered saline (PBS) was added in place of the antibodies. The reactions were incubated for 5 min at room temperature, then 50 µL of 0.1 M NaCl was added and color changes, if any, were recorded.
2.4.2. Conjugation of Antibodies to the AuNPs
To conjugate the antibodies to the AuNPs, 1 mL of AuNP solutions (OD 1 for the 14 nm AuNPs) at the selected optimal pH and 1 µg/mL antibody were added to an Eppendorf tube and incubated for 20 min with agitation. Then, 100 µL of conjugate blocking buffer (10% bovine serum albumin (BSA) prepared in borate buffer, pH 9) was added to the conjugate mixture and further incubated for 30 min. The conjugate mixture was then centrifuged at 15 000 rpm for 20 min at 4 °C. The supernatant was discarded and the AuNPs were re-suspended in diluent buffer and stored at 4 °C until further analysis.
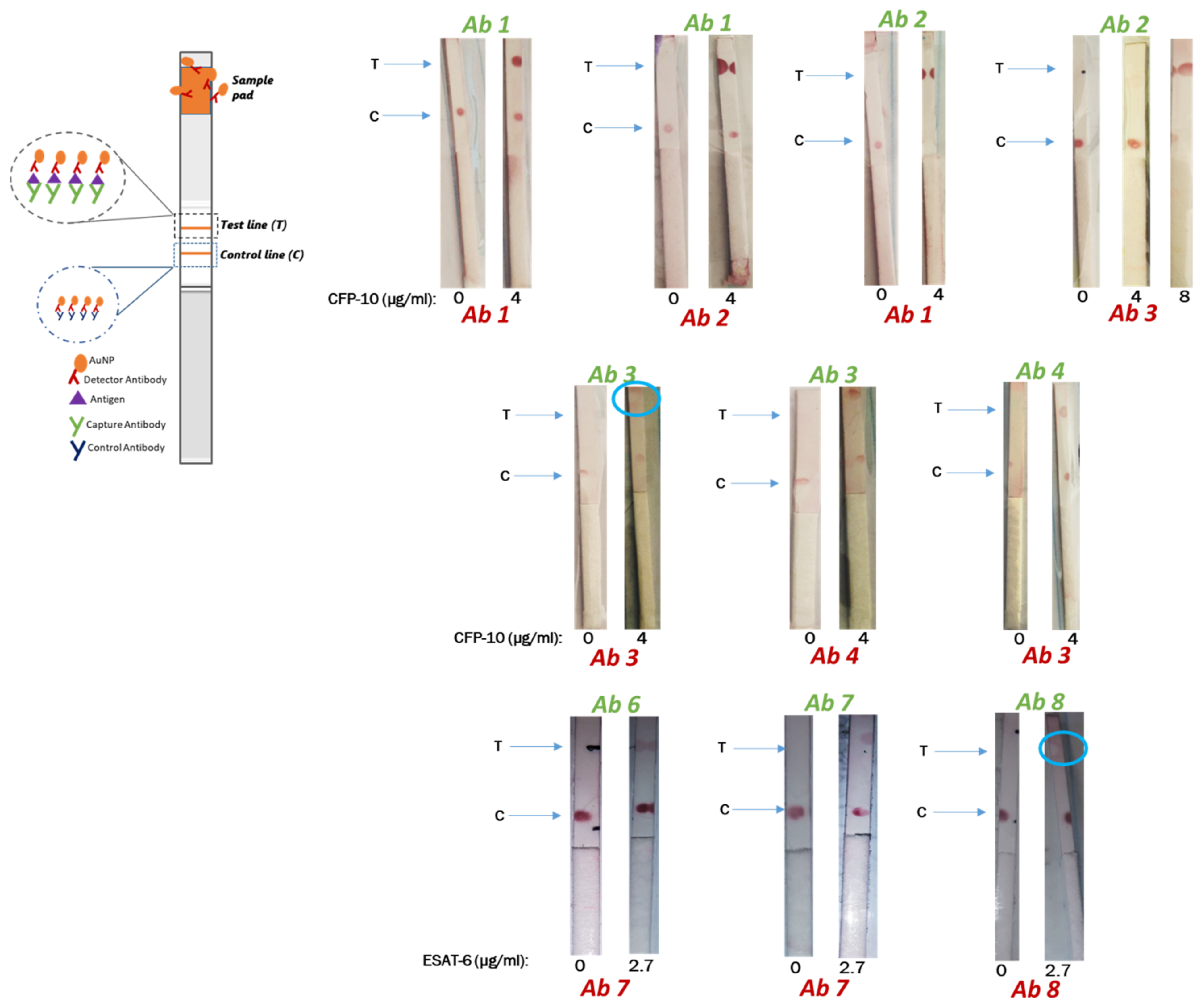
2.5. Selection of Antibody Pairs by Dot Spotting
The detection and capture antibody pairs for ESAT-6 and CFP-10 were tested for compatibility using dot spotting following a sandwich assay. Immobilization of antibodies was carried out by dot spotting 1 µL of 1 mg/mL of antibodies at about 6 mm of spacing between the control and the test line antibody. Thereafter, the membranes were dried for 15 min in an oven at 37 °C. Blocking of the membranes was carried out for 5 min in 0.1% BSA blocking buffer and dried for 45 min at 37 °C in an oven. Strips were cut into 3 mm and analyzed accordingly. Binding of the detection antibodies to the antigen was analyzed by immersing the strips in an Eppendorf tube containing the AuNP–antibody conjugate with buffer (negative test) or AuNP–antibody conjugate with buffer containing antigen (test sample) (refer to
Scheme 1). The experiment was allowed to run for 15 min, and signals were then visualized by a red dot.
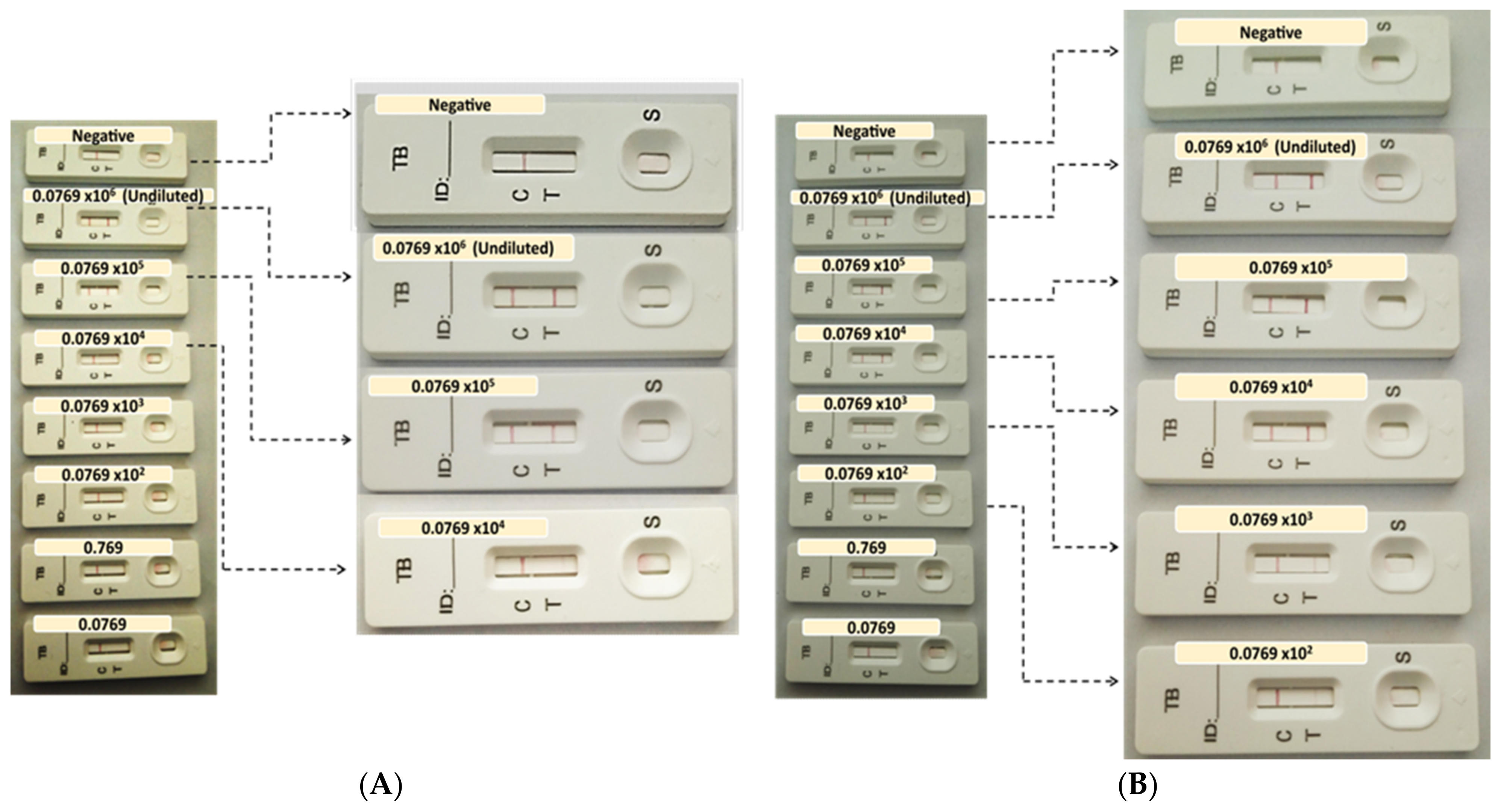
2.6. Development of the AuNP-Based LFIA and Determination of the Limit of Detection (LOD)
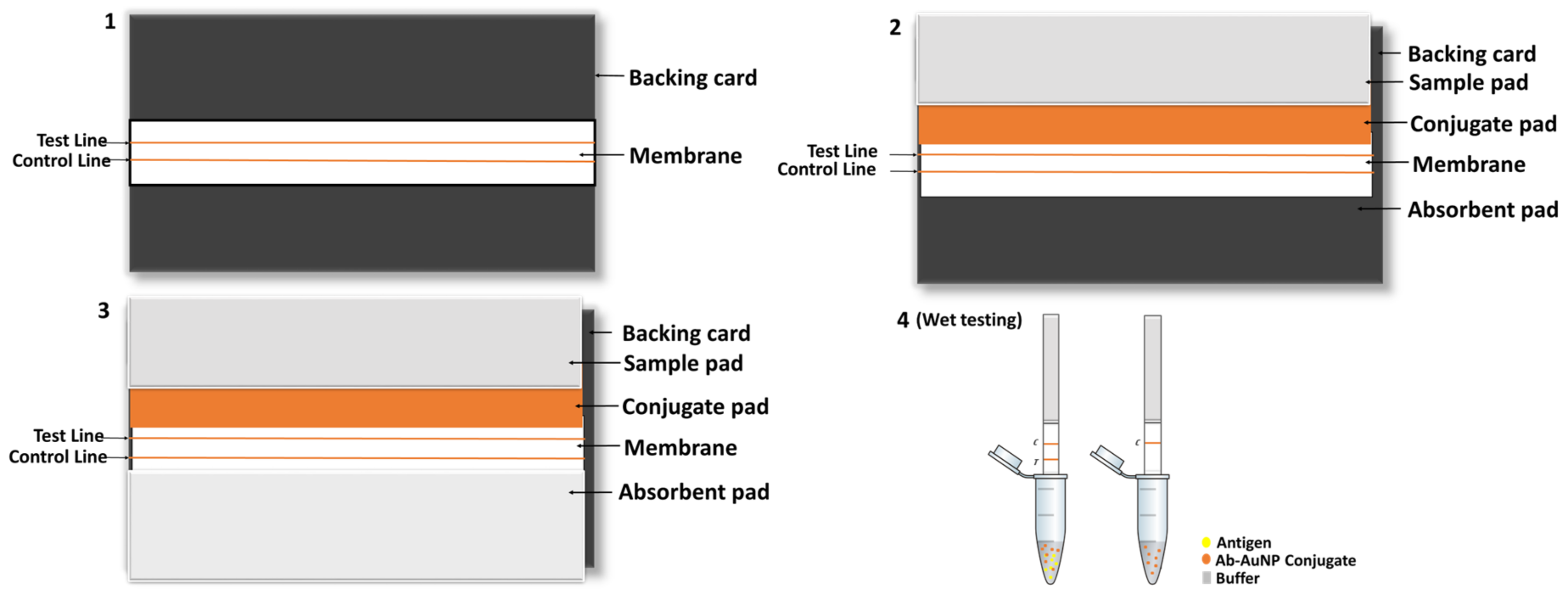
The selected control and test line antibodies were dispensed using XYZ (X = 25 mm; Y = 30 mm and Z = 8 mm) Platform Dispenser HM3030 (Shanghai Kinbio Tech.Co, LTD, Pudong New Strict, Shanghai, China) at a rate of 1 µL/cm on to the nitrocellulose membrane which was laminated onto a backing card. The card was then dried for 30 min at 37 °C in an oven, and the membrane was blocked with 0.1% BSA in phosphate buffer for 5 min with agitation. Drying was carried out for 45 min at 37 °C.
Meanwhile, the conjugate pad was prepared by immersing it in conjugate pad blocking buffer (3% BSA in borate buffer) for 15 min with gentle agitation. Once the conjugate pad had been dried, the AuNP conjugate of choice was sprayed using the KinBio Dispenser instrument (Kinbio Tech.Co, LTD, Pudong New Strict, Shanghai, China) and again dried for 45 min at 37 °C. Finally, the conjugate pad and absorbent pad were laminated on the backing card with the membrane attached and cut into 3 mm strips using the High Speed Cutter ZQ4500 (Kinbio Tech.Co, LTD, Pudong New Strict, Shanghai, China) prior to assembling into cassettes (schematic representation of the assembling process in
Scheme 2).
To determine the LOD of the prototypes, the recombinant antigens were serially diluted with 1 x PBS at concentrations ranging between 0.0769 to 0.0769 × 106 ng/mL for CFP-10, and 0.0000625 to 62.5 × 102 ng/mL for ESAT-6. The recombinant antigens were provided at different concentrations by suppliers; hence, the starting concentrations dictated the working solution’s dilution series. A volume (65 µL) of the prepared samples was loaded onto the assembled test strips and allowed to run for 15 min prior to the analysis of the results.
2.7. Wet and Dry Conjugate Testing of the LFIA
Wet testing: To perform wet conjugate testing, 30 µL of running buffer and 10 µL of AuNP–antibody conjugate were added to an Eppendorf tube. For a test sample, commercial recombinant M. tb CFP-10 (227-20144, Biodisc Raybiotech Life, Inc., Peachtree Corners, Georgia, USA) or M. tb ESAT-6 (Creative diagnostics PTY (Ltd), Shirley, NY, USA) was also added to separate tubes, after which the sample pad part of the strip was cut and immersed into those tubes. The test was allowed to run for 15 min prior to analysis.
Dry testing: The backing card was assembled with the conjugate pad which contained the AuNP–antibody conjugate. To conduct a negative test, 40 µL of running buffer (without any antigens/proteins) was added to the sample pad on the test strip and allowed to flow for 15 min. The same procedure was followed for a test sample by using a buffer containing the commercial recombinant proteins/antigens. Various concentrations of antigen were tested until a red line/dot was visible. Concentrations of 4 and 2.7 µg/mL for CFP-10 and ESAT-6, respectively, were used as a reference point, as determined in the Dot spotting results. Where necessary and where false negatives were observed higher concentrations were also tested.
4. Discussion
The attenuation of Mycobacterium bovis to the widely used BCG vaccine resulted in the deletion of the RD1 loci. We postulated that exploiting biomarkers within this region as targets for the TB-antigen RDT may minimize and/or eliminate false positives from previously BCG vaccinated individuals and/or those infected with other Mycobacteria species. The CFP-10 and ESAT-6 are two of the immunodominant antigens that are expressed by the RD1 loci and were explored as targets for the development of AuNP-based LFIA. The stability of antibodies with the 14 nm AuNPs at various pHs was tested, and overall the antibodies were stable at most pHs. Interestingly, Wu and colleagues found their CFP-10 and ESAT-6 antibodies to be suitable for conjugation with AuNPs sized between 18 and 20 nm [
11], suggesting that the smaller-sized AuNPs are more stable with these biomarkers.
Initially, Dot spotting was used to determine compatible pairs of detection and capture antibodies for CFP-10 and ESAT-6 from different suppliers. Suitable antibody pairs will then be sourced from the same supplier to maintain consistency. In cases where the test line was visualized but with the faint control line, the antigen could be causing steric hindrance, blocking the binding site for the immobilized secondary antibody of the control line. The kinetics of solid-phase immobilized antibodies have demonstrated that equilibrium between antigen, capture and detection antibodies do not occur simultaneously, suggesting that steric hindrance plays a role in the detection-antigen-capture sandwich formation [
22]. However in some instances, antibodies are able to re-organize to allow for further binding, and a high binding affinity may prevent this re-organization, causing saturation of binding sites at a faster rate [
22,
23,
24]. Thus, in addition to target epitope compatibility, the association (k
ass) and dissociation (k
off) rate constants of the detection and capture antibodies may have a crucial role in defining suitable binding pairs.
The false negatives for the other pairs suggest that the capture and detection antibodies are competing for the same epitope on the antigen. It is possible that the detection antibody, which is the first to be encountered by the antigen in solution during testing, saturates the same epitope that is recognized and required for binding by the capture antibody. This effect was observed by Cavalera and colleagues, which they referred to as the ‘antigen hook effect’, and they showed that lowering the concentration of the detection antibody can enhance the sensitivity of LFIA. The ‘hook effect’ has been previously described and was initially observed for the LFIA-based pregnancy test, which measures the concentration of human chorionic gonadotropin (hCG). High concentrations of hCG led to disappearance of the test line, which yielded false negatives [
25,
26]. Cavalera et al. also reported that the distance of the test line from the sample pad affects the sensitivity of the LFIA. The further the test line was from the sample pad meant that there was an increase in contact time between the detection of the AuNP conjugate and the antigen which increased the saturation effect, thus augmenting the ‘antigen hook effect’ [
27,
28].
False positives were also observed when BSA-based buffers were used, such as when testing the Ab8/Ab8 pair in the 0.1 to 1% BSA concentration. Since BSA is a protein like the antigens, it may present with peptides that act as epitopes which may be causing non-specific binding. This effect became less pronounced at a BSA concentration of 0.1% versus at 1%. Using casein as an alternate buffer eliminated the false positives (results not shown), and this may be because casein is a less structured protein (mostly random coiled or disordered) than BSA (67% helix, 10% turns and 23% extended) [
29,
30], making it less stable and more prone to denaturation effects by the surfactant than BSA. It is possible that if a higher concentration of surfactant was used with the BSA buffers no false positives would appear. ‘Ghost lines’ are an artefact characterized by a red background encircling a white dot where an antibody was immobilized. The appearance of ‘ghost lines’ has been proposed to be as a result of high concentrations of capture antibody on the test line repelling the conjugate. Since only the Tris-based buffers caused the ‘ghost line’ effect with the Ab8/Ab8 pair, a plausible reason may be that the buffer confers an overall charge to the Ab8–antigen complex that caused it to be repelled by the immobilized capture antibody.
Although CFP-10’s Ab1/Ab1 pair showed potential, the Ab1/Ab2 pairing had an LOD that was 100 times more sensitive to the same antigen. Generally, for sandwich-type LFIA, a pair of mAbs targeting different epitopes of the antigen are often used over a single epitope-targeting mAb as both the capture and detection antibody [
23,
27,
28]. The sensitivity of the LFIA is often lower with single epitope-targeting pairs. This does not mean that successful LFIAs have not been developed using the same mAb as both the capture and detection antibody, but some considerations and optimizations have to be performed [
27,
28]. Wu and colleagues determined the LOD of their CFP-10 test strip to be 2.4 ng/mL and 6.0 ng/mL for ESAT-6 [
11], suggesting that the current study’s CFP-10 LFIA prototype is only slightly weaker in detecting the antigen. In contrast, the ESAT-6 prototype was 96-fold more sensitive than the one produced by Wu and colleagues, suggesting the prospect of a potentially better performance in clinical sample evaluation.
Other technologies have been developed to improve the sensitivity for detection of ESAT-6, such as the magnetic-bead-coupled AuNP-based immuno-PCR assay (MB GNP-I-PCR assay). Comparing the MB GNP-I-PCR assay to the ELISA, the authors reported an LOD of 10 fg/mL, which was 105-fold higher in sensitivity compared to the 1 ng/mL displayed by ELISA [
31]. Simultaneously exploiting different sized AuNPs, 20 and 60 nm, to capture the ESAT-6 antigen may improve the LOD. However, this technique remains unsuitable for PoCs, as it requires additional steps that need skilled personnel and laboratory equipment and is more costly than the lateral flow assays. Previous studies have indicated that LOD values are not the ultimate predictor and/or determinant of sensitivity in clinical sample testing. Despite the reported LODs of 2.4 ng/mL and 6.0 ng/mL for CFP-10 and ESAT-6, respectively, Wu and colleagues found that the positive detection rate was 29.4% for CFP-10 and 41.2% for ESAT-6 in TB-positive plasma samples [
11]. Accordingly, test conditions and LOD studies need to be carried out and optimized in the intended clinical sample of choice. Another type of immunoassay previously used for ESAT-6 detection in plasma/serum was electrochemiluminescence (ECL)-based, with a reported LOD of 6 pg/mL and 56% sensitivity [
3]. There can be various contributing factors to this phenomenon, including the quantity of the biomarkers or analytes in the samples. For example, the status of expression and/or dysregulation of a potential biomarker is influenced by different factors, including the geography of the sampled population. Genetic variations in the host and of the Mycobacterium can contribute to the differential regulation and expression repertoire of biomarkers in the host [
25,
28]. Such factors need to be considered in biomarker discovery for TB.
Mehaffy and colleagues used mass spectroscopy (MS) to identify
M.
tb peptides in human serum. Their experimental approach included an exosome enrichment step from the plasma of TB-positive patients originating from four different countries: South Africa, Bangladesh, Peru and Vietnam. By using Multiple Reaction Monitoring MS, the group discovered that their representative peptides for CFP-10 and ESAT-6 were detected in 3% and 15% of the total sample population, respectively [
32]. Out of the 10 South Africans, only one was smear positive who was also co-infected with HIV; none of these patients had the CFP-10 peptide in their serum, and the 3 who had the ESAT-6 peptide were also HIV positive. With respect to the South African cohort, these results were inconclusive because of the small sample size tested and due to that 90% of the samples that were collected were smear-negative samples [
32].
In an independent study, Nanodisk-MS was used to detect CFP-10 and ESAT-6 in serum. The method involved the enrichment of selected CFP-10 and ESAT-6 peptides using antibody-conjugated Nanodisks prior to MS analysis. The LODs of the recombinant CFP-10 and ESAT-6 in the TB-free serum were 50 pM and 200 pM, respectively [
14]. In HIV-negative TB patients’ serum, the sensitivity was 100% and 91% in smear-positive and smear-negative patients, respectively. The specificity was 87.1% in healthy controls and 90.6% in nontuberculous Mycobacteria patients. HIV co-infected TB patients exhibit EPTB, therefore, smear-negative and -positive EPTB and PTB patients were tested. In PTB cases the sensitivities were 91.3% and 82.4% in smear-positive and smear-negative patients, respectively, and 92.3% and 75.0% in EPTB patients [
14]. The similarity in sensitivities between PTB and EPTB patients was an important outcome of the study, as it illustrated that serum has great potential to be used as an alternative sample for testing TB including EPTB. The key lesson learned from these studies is that a trend exists, wherein ESAT-6 shows a higher LOD compared to CFP-10 but with apparently superior sensitivity in clinical samples. Additionally, the specificity of the immunodominant antigens in healthy controls and more importantly in nontuberculous Mycobacteria patients is essential for developing a device that reduces cross-reactivity with nontuberculous Mycobacteria patients. These studies, including ours, demonstrated that nanotechnology-based immunoassays can greatly improve the simplicity, sensitivity and robustness of diagnostic methods.
5. Conclusions
The immunodominant antigens CFP-10 and ESAT-6 that are expressed by the RD1 loci of M. tb are potential biomarkers for developing a TB-antigen RDT that can discriminate previously active TB cases from previously BCG vaccinated and nontuberculous Mycobacteria cases. The pairs that were selected for preliminary testing with commercial recombinant protein in the current study showed great promise, with LODs that were either comparable or outperforming ones from those previously reported the in literature. An exhaustive study into biomarker discovery and research is required prior to choosing an appropriate target analyte in developing a diagnostic device for TB. The process is complicated and delayed by a spectrum of factors, such as the disease state, that is, active or latent disease, which can determine the level of expression and/or differential regulation of the targeted biomarker. Other factors such as the TB vaccination status, HIV status, age, and geography of the population tested need to be considered as well. Thus, future studies will explore other targets from the RD1 loci. Additionally, previous studies indicate that the methods used to improve sensitivity required additional steps, such as an enrichment step alongside exploiting immunoassays in combination with nanotechnology. These approaches need to be greatly refined in order to provide rapid results at the point of need/care, hence, our approach still has superiority. Proof-of-concept investigations for the rapid detection of TB in easily accessible clinical samples such as blood and saliva will be carried out both internally and subsequently validated externally to confirm clinical utility. This will greatly improve the diagnostic and therapeutic outcomes in low-income countries.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}