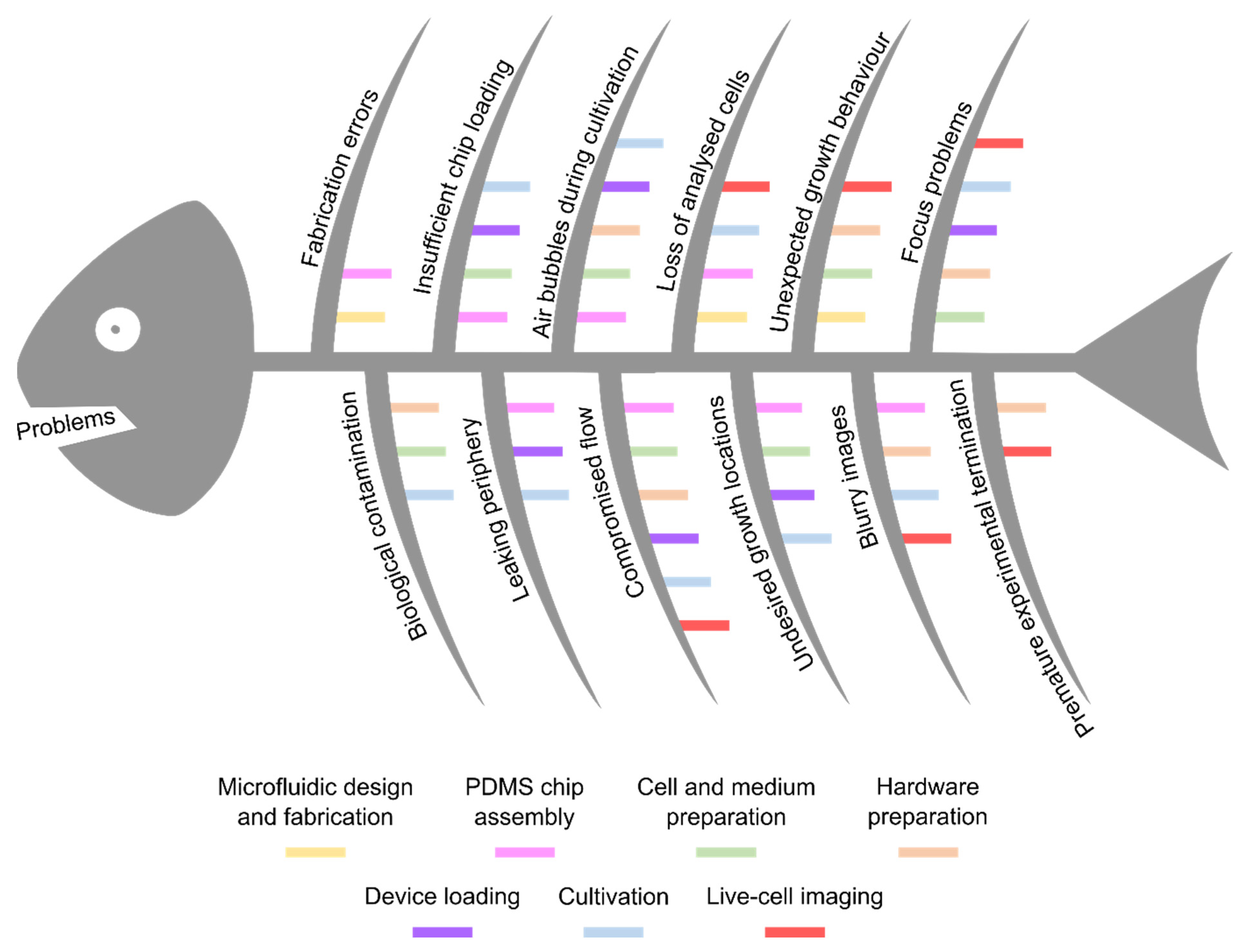
In the following, selected examples of these challenges in microfluidic single-cell cultivation (MSCC) approaches (
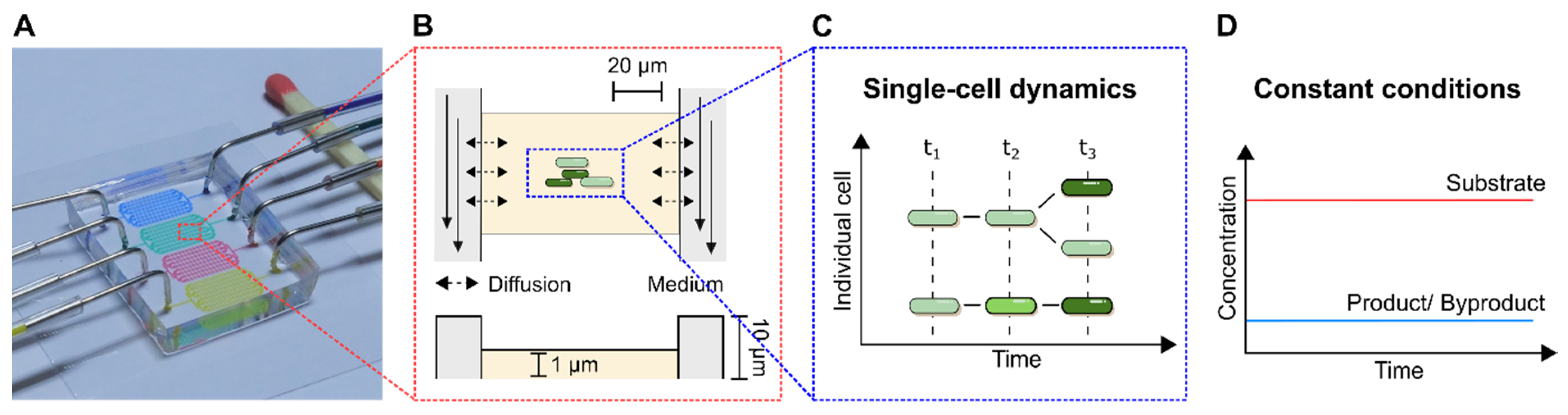
Figure 4A) are presented and suitable troubleshooting strategies are proposed in detail. As a subclass of MC, MSCC allows the cultivation of cells with single-cell resolution and thereby the investigation of single-cell dynamics or population heterogeneities with high spatio-temporal resolution. For this purpose, most commonly 2D cultivation chamber designs are utilised (
Figure 4B), that result in monolayer growth of the applied organisms [
14]. Due to the resulting spatial restriction, cultivated cells stay in one focal plane during live-cell imaging, which allows time resolved analysis of single-cell behaviour in contrast to population average measurements (
Figure 4C). Additionally, cultivation volumes range from nanolitre to picolitre scale, resulting in constant nutrient supply and defined cultivation conditions over the whole cultivation time (
Figure 4D).
4.1. Case Study I—Establishing MSCC for a New Organism
It is not unusual that cells, when exposed to a different cultivation environment, need time for adaption, which in most cases results in a distinct lag-phase. Likewise, only a fraction of cells might restart growth when discrepancies between the former and the new cultivation environment are given [
75]. Therefore, this behaviour especially occurs when cultivation protocols of well-established organisms in lab-scale are transferred to MSCC. With minimising the cultivation volume in microfluidic devices, formerly dispensable physical properties like surface tension, surface-to-volume ratio, laminar flow, and diffusive mass exchange become crucial. At the same time, running MSCC under constant perfusion holds unique challenges ready, that do not arise in other cultivation scales and operating modes like the constant wash-out of secreted (by-)products and a constant oversupply of nutrients (
Figure 4D) [
14]. Recently, we developed a novel MSCC platform for the long-term cultivation of Chinese hamster ovary (CHO) suspension cells [
63] and therefore had to overcome several of these challenges.
The development of our MSCC device resulted in successful trapping of CHO cells. After optimizing the loading procedure regarding the cell density and loading strategy, as mammalian cell lines are known to be compromised in physiology by increasing shear stress, in first instance, we obtained cells that did not grow; only residual morphological changes and occasional division events were observable. As performed in shake flask or bioreactor cultivations, our MSCC experiment was set up at a cultivation temperature of 37 °C with steady perfusion of commercially available chemically defined, serum-free medium (TCX6D, Xell AG, Bielefeld, Germany). Since air bubbles constantly entered the system during MSCC, we assumed compromised cultivation conditions regarding nutrient supply to be the reason for the absence of steady cellular growth. To identify whether air was pumped into the chip or arose gradually inside the channels, observing the devices inlets during live-cell imaging proved helpful. Yet, most of the time air bubbles were introduced during connecting the pumping periphery, which made special precautions necessary like connecting needles to wetted inlets or removing stuck air bubbles from the inlet mechanically. Additionally, we increased flow rate from 0.8 µL/min to 2 µL/min to shorten residence time of the perfused medium inside our device and lowered the supply channel’s height. Thereby, we reduced the device’s total volume to compensate evaporation of liquid through the PDMS chip. With a 10-times medium exchange per minute throughout the whole device, we successfully minimised gas formation on-chip.
After adapting the microfluidic design concerning supply channel and chamber height as well as the cultivation protocol, the first MSCC runs again failed after a few days, as CHO cells still did not divide properly inside our device. When vital, the cells show constantly occurring protuberances of their cellular membrane during live-cell imaging, which arise from steadily secretion of vesicles [
63]. Here, we observed a persistent change of cellular morphology to a smooth surface followed by cell death after a few more days. Since cell death did not occur immediately after loading the cells and still sporadic cell division events were observable, we assumed its origin to be not connected to preculture handling but to appear in a later stage of cultivation. To check whether PDMS, which is often described as harmful for cell culture [
76], might cause cell death, we washed the PDMS chips with n-Pentane to remove uncrosslinked monomers, but cells did not grow anyway. Therefore, we ruled out PDMS as reason and focused on other components of our setup e.g., the tubing. For bacterial MSCC, Tygon
® is a common material utilised for tubing. Although its status as biocompatible, there have been reports [
77] of negative influence on cell culture due to leachables. To control if these leachables have a negative effect on CHO cells, we exchanged the previously applied Tygon
® tubing against PTFE tubing. Another reason for exchanging Tygon
® against PTFE tubing was the lower gas permeability of PTFE: Many cell culture media are CO
2 buffered, thus losing the adjusted CO
2 concentration might lead to a pH shift and thereby to non-optimal growth conditions. Using an additional CO
2 incubation chamber for long-term MSCC also helped to guarantee stable CO
2 atmosphere in the chip’s surrounding.
Changing the tubing of the device led to cellular survival during MSCC but growth still stopped after 24 to 48 h and cells stayed in an unchanged status till the end of cultivation. Since cells did not demise, we concluded the problem not to be a technical one but rather assumed some limitations that prevented CHO cells from ongoing cell division. Looking at single-cell cloning procedures, growing isolated cells is also highly challenging when chemically defined media without any serum-percentage are applied. In MSCC all secreted factors were additionally diluted and washed out directly due to the constant perfusion of the device, which prohibited the accumulation of hormones and other beneficial factors. Without any single-cell medium available in the market, we decided to mix fresh medium with already conditioned medium from the main culture’s exponential growth phase to supply the isolated cells with essential signalling molecules and thus mislead them over their solitude inside our microfluidic device. This had already been successfully tested for various organisms such as bacteria [
78,
79]. With a ratio of 1:1, cells finally continued growing until the whole cultivation chamber was filled. However, generating the conditioned medium from the exponential growth phase seemed to be of utmost importance, as medium from the late stationary phase did not promote single-cell growth comparably but rather retarded it. For a more detailed explanation of the results of this case study, the reader is referred to Schmitz et al. [
63].
The realisation of MSCC for an organism not previously cultivated in microfluidic devices is associated with varying challenges which are individual for every new organism. For CHO suspension cells the unknown environment and materials turned out to be not only problematic but even toxic. Therefore, significant improvements could be made by adapting the tubing and guaranteeing a constant CO2 atmosphere during MSCC. Finally, supplementing perfusion medium with conditioned medium lead to stable growth. Initial non-growing behaviour of cells in minimal medium commonly can be tackled by introducing complex components into the medium. However, some complex compounds might alter cellular behaviour drastically like serum might trigger adherent growth in cell culture, compromising proper analysis. As a result, defined and reproducible cultivation cannot be performed.
4.2. Case Study II—Getting Cells to Grow Reproducibly
After MSCC setup and cultivation protocol have been successfully established for a new organism, getting the cells to grow in a reproducible way for quantitative single-cell studies still can be difficult to achieve. This can only be accomplished by using defined minimal medium, as a complex medium can slightly vary in composition and thus the cells’ behaviour might be affected concerning morphology or growth rate up to a complete growth arrest [
80]. However, for quantitative measurements it is important that the cells grow reproducibly to answer different research questions. Therefore, it must be investigated whether these irregularities have a technical or a biological origin. In most cases, a variety of parameters can affect the reproducibility of cellular growth. In the following, we present some cases that show which reasons may underlie non-reproducible growth.
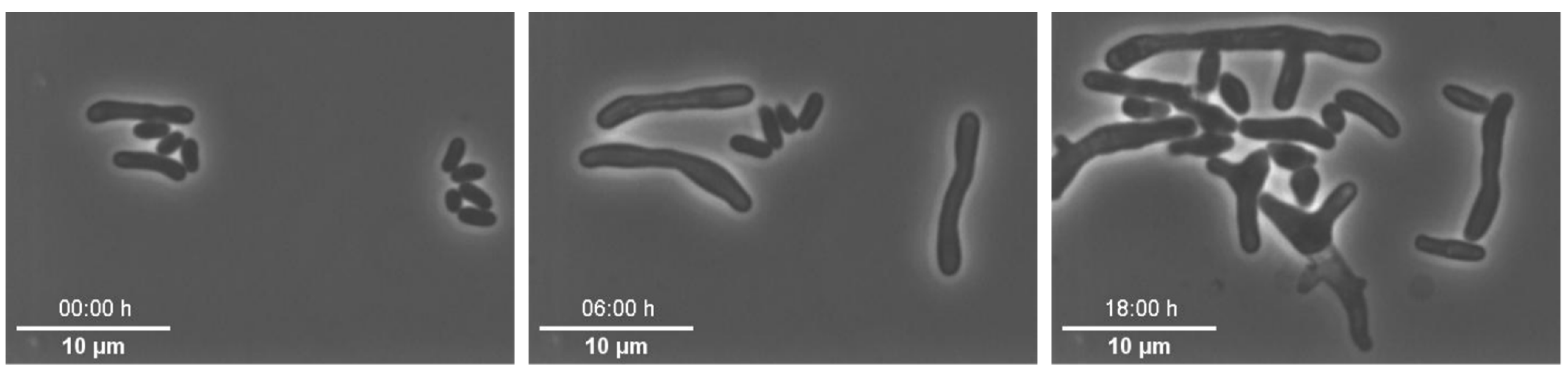
Example 1—increased lag phase of cells: In this series of experiments,
Corynebacterium glutamicum was cultivated under constant CGXII minimal medium [
27,
81] conditions at 30 °C in an open-box monolayer cultivation chamber (2D chamber) [
82]. After cell loading of our MSCC chip, we observed a distinct lag phase of all cells (see
Figure 5). As a first step, we examined the pre- and interculture and found that the growth phase of the main culture was a crucial factor for the initial lag phase on-chip. An increased lag-phase in the MSCC was observed when cells from the late exponential phase were applied for chip inoculation. We found that in precultures of an optical density (OD
600) above 0.5 the PCA is depleted from the medium and consequently the cells’ metabolism is changed [
27]. To prevent this, precultures were inoculated with an OD
600 between 0.05 and 0.1 and applied for chip loading when the OD
600 reached values between 0.2 and 0.4. We inoculated the MSCC device with an early exponential growth phase (OD
600~0.3) which resulted in reproducible growth of
C. glutamicum cells with no observable lag-phase.
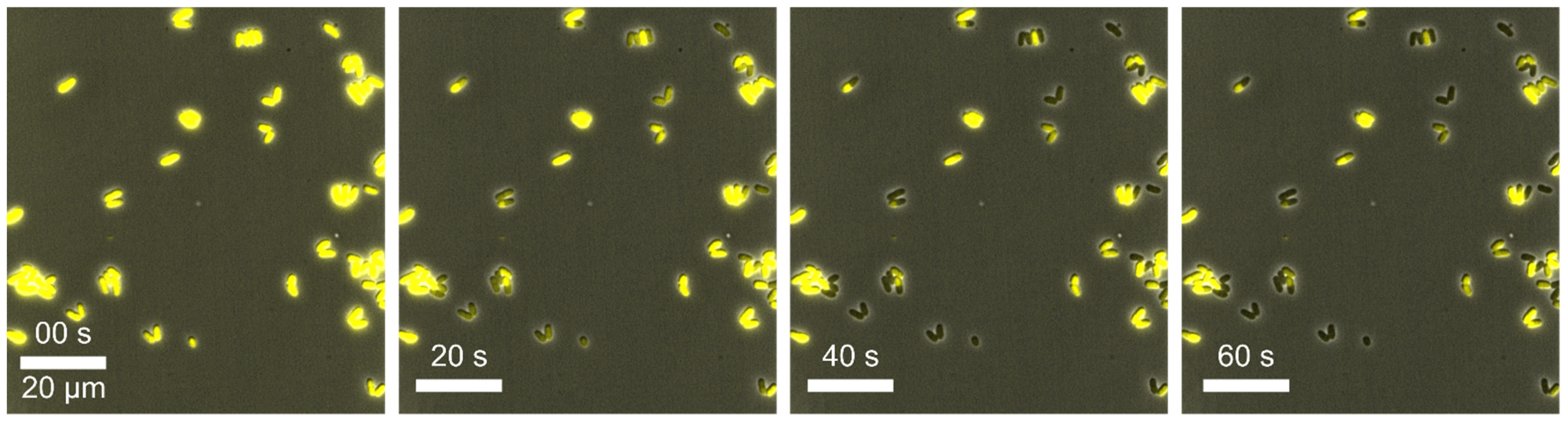
Example 2—stagnating growth of cells: To study the growth under optimal conditions, we cultivated
C. glutamicum in a 2D cultivation chamber system with constant perfusion of CGXII minimal medium at 30 °C. The minimal medium CGXII consists of six components: a base solution, CaCl
2, biotin, glucose, protocatechuic acid (PCA) and a trace elements solution. During this MSCC experiment, we observed just a few cell divisions (see
Figure 6), mostly two to three divisions at the beginning of the experiment, until growth completely stagnated. After excluding the microfluidic setup and its periphery as an error source, we checked the pre- and interculture to see if there were any problems during the shake flask cultivation that may affect the subsequent MSCC experiment. We found that the 3-(N-morpholino) propane sulfonic acid (MOPS) buffer in our CGXII medium was omitted during shake flask cultivation, since buffering medium in MSCC is not as relevant as in batch cultivations, due to the constant perfusion. As a result, there was a pH shift because of the lack of buffering during pre-cultivation seed train. After preparing the CGXII medium with MOPS buffer for shake flask cultivation, we unexpectedly still noticed decreased growth and finally growth arrest during MSCC experiments. Thus, we switched from the minimal medium CGXII to the complex medium BHI for MSCC and observed an optimal growth rate (µ ≈ 0.9 h
−1) [
62]. Based on the obtained results, we concluded that the decreased growth rate during the experiments was related to the minimal medium CGXII and its components. Various media components such as the base solution and glucose were systematically replaced with new stock solutions, but normal growth in CGXII medium during MSCC was not restored. Subsequently two more components of the applied medium were checked: the iron chelator PCA and the inserted biotin solution. We tested different stock solutions and found that using a different iron chelator such as citrate resulted in exponential growth for
C. glutamicum in MSCC. Therefore, the cause of our problem was a compromised PCA solution. A new PCA stock solution finally resulted in regular growth on-chip (µ ≈ 0.6 h
−1) [
82]. For a more detail explanation of the results of this case study, the reader is referred to Grünberger et al. [
22] and Täuber et al. [
62].
Example 3—prolonged division time of cells: For the analysis of single-cell events, we cultivated
C. glutamicum with constant CGXII medium supply at 30 °C in a 1D cultivation chamber design, modified mother machines, with a length of 20 µm and a width of 0.8 µm, which is open to the supply channel on both sides. Here, we observed an increase in doubling time (see
Figure 7) compared to cells which were cultivated in 2D cultivation chamber systems. Typically,
C. glutamicum divides in a V-shape, but under spatial restrictions, the cells no longer form this V-shape and divide along their main axis. The additional mechanical stress during division increased the doubling time. Similar observations have been reported by Yang et al. [
83] and Dusny et al. [
84] when cells are cultivated in agarose-pads. Therefore, we developed wider 1D cultivation chambers with a width between 1.5 and 2 µm, in which the cells finally can divide in their preferred V-shape again. With the adapted cultivation chamber design, we observed V-shaped division of our cells as well as optimal doubling times of t
D = 70 min.
Example 4—altered morphology of cells: In this set of experiments, we cultivated
C. glutamicum under constant CGXII medium supply, but with varying pH values at 30 °C to investigate its pH stress response [
82]. For each experiment, pH was adjusted to the desired pH value between pH 6 and 8. During all of these experiments we observed altered cellular morphology and non-uniform cell growth (see
Figure 8), which was not expectable based on previous experiments. First, we tested the individual medium components of the CGXII medium to determine if they were the reason for the inconsistent growth behaviour, but normal growth could not be restored. As we cultivated
C. glutamicum in the CGXII medium without pH adjustment, optimal growth was restored. Based on this result, we suspected that the cause of the altered morphology and growth rates must be related to the pH adjustment procedure. Therefore, we tested different approaches to exclude impurities. We prepared medium and adjusted the pH using different pH meters to rule out instrumental failure as a source of error but altered growth morphology remained unchanged. Afterwards, the pipettes used to transfer the pH adjusting agent into the medium were examined in more detail. Different pipettes were used: plastic single-use pipette tips, plastic Pasteur pipettes and glass Pasteur pipettes. After adjusting the pH value with plastic Pasteur pipettes and single-use tips, reduced growth and altered morphology still were observed. However, utilizing glass Pasteur pipettes resulted in expected growth and morphology. Thus, we suspect that leachable substances were released from the plastic pipettes as well as single-use pipettes. Consequently, we decided to make future pH adjustments with glass pipettes only. For a more detailed explanation of the results of this case study, the reader is referred to Täuber et al. [
82].
Example 5—no growth on-chip: In this experiment, we cultivated
C. glutamicum with BHI (complex) medium supply at 30 °C in perfusion to analyse the colony growth rate. However, no growth was observed in the MSCC device (see
Figure 9). To check whether the origin of the absence of growth was medium related, we cultivated our cells in CGXII minimal medium but were not able to detect any growth as well. Therefore, we assumed that this problem had its origin in a technical step prior to the MSCC experiment. We suspected the cleaning procedure of our PDMS chips and glass slides as critical step. We usually clean both parts, PDMS chip and glass slide, with isopropanol, thus it could be possible that isopropanol residues remained in the channel after cleaning, which will be toxic to the cells and most likely affect the growth. Therefore, we adapted our cleaning procedure in two different ways. First, we tried to wash the PDMS chip with ultrapure water after cleaning it with isopropanol to remove potential isopropanol residues. Second, we stored the PDMS chip overnight after cleaning with isopropanol before bonding the PDMS chip and the glass slide, which allowed the isopropanol residue to evaporate from the PDMS chip. With the new cleaning procedure, the cells grew optimally in MSCC experiments, therefore, both methods turned out to be successful in removing isopropanol residues.
Example 6—cell death: In this experiment,
C. glutamicum DM1800 pSenLysTK [
85] was cultivated in CGXII medium at 30 °C under constant conditions in 2D cultivation chambers. This strain on the one hand produces intracellular L-lysine, which, on the other hand, triggers the production of a yellow fluorescent protein (YFP) under the control of the lysine sensor pSenLys [
85]. This YFP molecule can be examined by fluorescence microscopy so that lysine production behaviour is quantifiable. During the MSCC experiments, we observed a steadily decrease in the fluorescence signal over the cultivation time (see
Figure 10) as well as a decrease in growth resulting finally in cell death for most of the analysed cells. We assumed that the decrease in fluorescence signal could be related to photobleaching. In this process, the fluorophore can decay, releasing harmful products like reactive oxygen species and hence lead to cell death [
86]. Based on this assumption, we investigated the fluorescence settings in more detail. We performed several cultivations with different fluorescence settings for the excitation light intensity and exposure time and found optimal parameters for our bacterial strain. The settings of the excitation intensities highly depend on the sensitivity and stability of the fluorophore in use as well as the sensitivity of the organisms. These parameters can be easily adjusted in live-cell imaging, where photobleaching directly represents the response to the corresponding excitation intensity. For our strain, an exposure time in the lower millisecond range and a low light intensity of less than 10% of the maximum intensity proved to be optimal [
87]. Using the correct fluorescence settings, no impairments of the fluorescence signal or growth was observed.
To sum up, achieving reproducible growth during MSCC is a very challenging task, as every single step, from microfluidic design over biological preparation to the microscope settings has to be considered as a potential source of influence on cellular growth and morphology. Based on our experience, the reason for altered growth often originates before the experimental step of live-cell imaging and the MSCC. Neglecting the importance of a proper seed train and properly stored chemicals or media already hampers the cultivation before it even started. Likewise, careless chip cleaning or adjusting the medium’s pH value with single-use plastic pipettes can show an effect which unfortunately is not noticeable until growth rates are determined after MSCC. During the actual experiment, cellular growth might be unexpectedly altered by spatial restriction or too intense light exposure. Because of the variety of sources of error, troubleshooting must start again from the beginning for each challenge that arises and does not follow a pre-set order, as it was the case in Case study I, and workflows must be followed in detail.
4.3. Case Study III—Growing Cells without Carbon Source
Performing successful MSCC experiments include the execution of negative control experiments, as for any study in microbiology and applied biotechnology. These experiments are necessary to validate well-designed scientific experiments and findings. As described before, quantitative growth studies rely on the use of minimal and defined medium allowing to draw conclusive interpretation on the obtained MSCC data. After the selection and design of the desired medium, one typical control experiment is the cultivation of cells without the main carbon source. In the past, these experiments revealed significant scientific surprises and led to the development of adjusted microfluidic cultivation media. Here, we will demonstrate the importance on the example of C. glutamicum WT as well as Escherichia coli MG1655 K12.
C. glutamicum MSCC experiments are typically performed with the well-established CGXII minimal medium (see Case study II). Analysing microcolony growth with multiple replicates repeatably resulted in growth rates around µ ≈ 0.15 h
−1, when glucose was omitted within the CGXII medium (negative control experiments). Thus, we concluded that the microbial cells must gain their demanded carbon from a yet unknown source [
27]. In successive steps, PDMS chips were washed with n-Pentane to remove any monomer residuals that were in suspicion to be metabolised by the organism. In the next step, tubing was exchanged to rule out leakage of carbon-based material into the experimental setup. After exclusion of these factors, we were convinced that the residual growth must be based on a carbon source that had to be part of the medium composition. After a careful review of the CGXII compounds, only MOPS as a buffer compound and PCA as iron chelator have been identified as compounds that contain carbon elements. Since pH conditions are constant in perfusion cultivations, MOPS was omitted from the cultivation medium. The experiments still revealed a remaining growth rate of approximately µ ≈ 0.15 h
−1 and thus we could conclude that MOPS was not responsible for residual growth. Removing the iron chelator PCA from the cultivation medium finally resulted in zero growth [
27]. However, simply removing the iron chelator for further limitation studies to analyse carbon source limitation was not a solution, since its primary function cannot be compensated by other medium ingredients. On the first instance, negative control experiments were successful. Unfortunately,
C. glutamicum was not able to grow on a main carbon source (here glucose) when PCA was omitted due to its function as iron chelator. After repetitive experimental trial and error, we identified citrate as an alternative iron chelator, that was not metabolised as carbon source at CGXII medium composition at the given environmental conditions (unpublished data).
In a similar study,
E. coli cells were cultivated under limiting condition in M9 medium [
88]. Here, cellular growth was determined at different carbon source concentrations ranging from pM to mM. Under limiting conditions, decreased growth rates up to the point of zero growth were expected. Again, at limiting concentrations and without any carbon source, significant cellular growth was detectable (unpublished data). Biased from our previous experience with
C. glutamicum, our first approach was to check whether the applied minimal medium exhibits any potential secondary carbon sources like the iron chelator ethylenediaminetetraacetic acid (EDTA). Any experiments altering the iron chelator molecule and its concentration resulted in residual growth at zero and limiting carbon conditions, thus we concluded that not the medium compounds per se lead to the remaining growth within MSCC. In the next step, we logically checked all base chemicals for manufacturer related contaminants that could be metabolised by the cells. Here, no notable elements have been found. Next, we examined the containers and bottles in which the various stock solutions were prepared and stored. The preparation of new stock solutions resulted in the expected non-growth at low carbon conditions. In the following, we separated workflows in cleaning procedures and the preparation of stock solution and medium preparation of medium containing no or only small amounts of carbon source. As a result, quantitative reproducible experiments at low carbon concentrations were obtained.
Using growth medium with its standard component concentrations for MSCC of just a few cells always results in excessive supply of nutrients. Therefore, even reducing e.g., carbon source inside the medium drastically will still allow full-speed growth of the loaded cells, although cells cultivated in bigger scales might reach carbon limitations already after a few hours of cultivation. Operating MSCC in perfusion mode makes it even more difficult to achieve real limiting conditions. As we had to find out, even medium components like PCA, that are not sufficient to promote steady growth in shake flasks or bioreactors because of their low concentration, enable constant growth in a MSCC. Likewise, minimal contaminations of the applied bottles during medium preparation might result in decreased but still detectable growth. Thus, not only the choice of medium is crucial but also its preparation for MSCC has to be even more careful than for conventional cultivation approaches.

 ,
, 



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}