Detection of CRISPR-Cas9-Mediated Mutations Using a Carbon Nanotube-Modified Electrochemical Genosensor

 ,
,  ,
,  ,
,  , and
, and 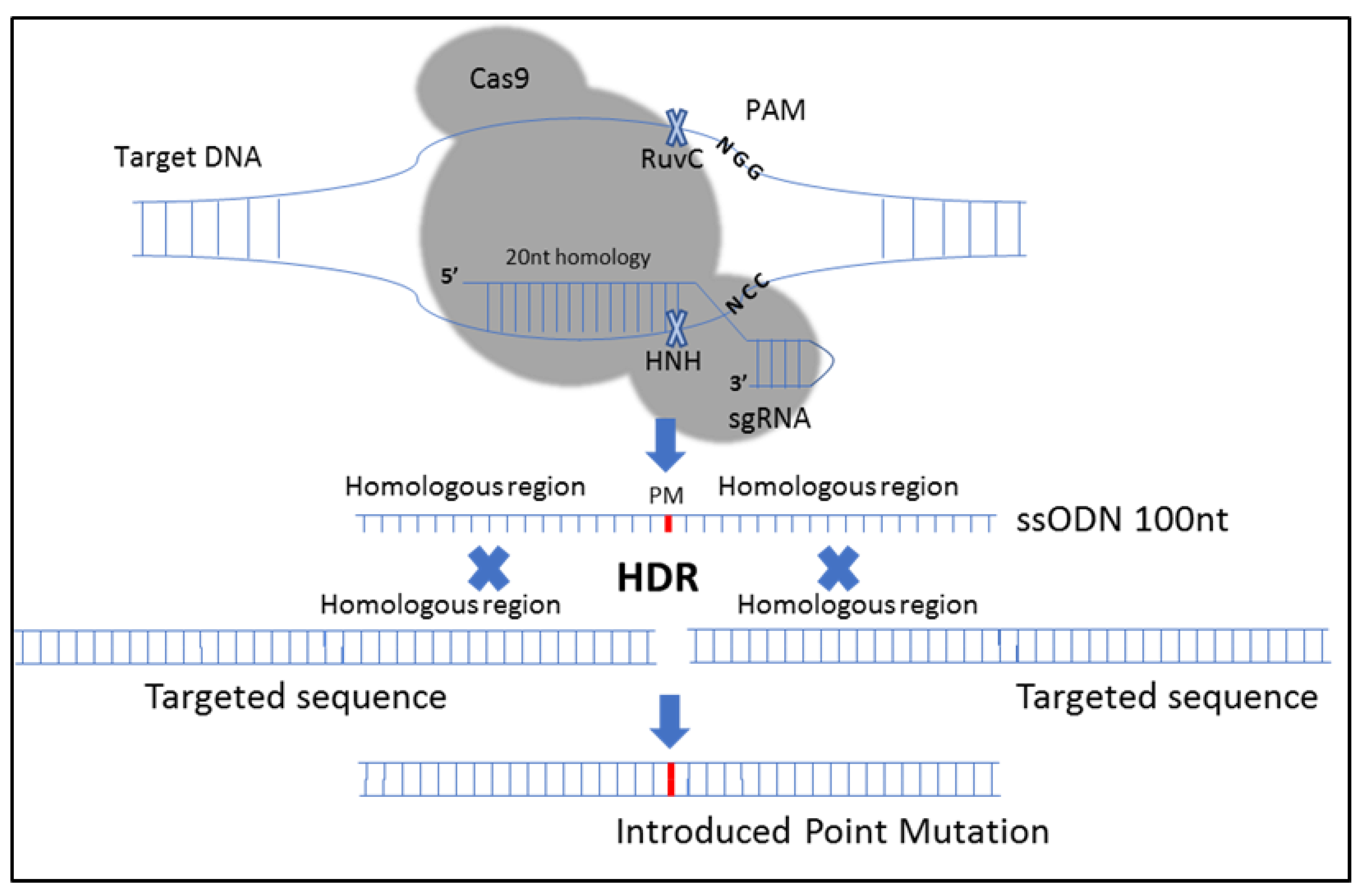{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Apparatus
2.2. Reagents and Solutions
- S-331 genome wild-type (WT) probe: 5′-NH2-C6-CICtAAITICTCTIIAIaIIT-3′
- Synthetic WT target: 5′-ACCTCTCCAGAGCACTTAGCG-3′
- S-331 genome mutant-type (MT) probe: 5′-NH2-C6-CICCAAITICTCTIIAICIIT-3′
- Synthetic MT target: 5′-ACCGCTCCAGAGCACTTGGCG-3′
- Synthetic non-complementary sequence: 5′-GGCAGCGGTGACTATGGCACC-3′
- CRISPR-Cas9-edited S-331 gene PCR amplicon (underlined bases refer to the introduced point mutation region via CRISPR/Cas9 system): 5′-TTAGGGCGATTGGGCCCTCTAGATGCATGCTCGAGCGGCCGCCAGTGTGATGGATATCTGCAGAATTCGCCCTTGGAACTGGGTCAAAGGCCTCTGGGAAGATAGAGCTTTGGTCTTCTTGGATTTGCTGGTTTGTTTTCATTTTTGAGACAATCTTGGCTGACCTAGAACTCACTATGTAGACCAGGCTGGCCTCAACTCTTCAGAAGAGATCCGCCTGTCTCTTCCTCCCTAGGGTCAGGATCAAAGGCATAGACCACCACAACTGGCTTTTTGCTTATCTTTGGATCTTTGCTAGCTCAGAGGAGTCCACCGAGAAAGGCCCTACAGGGCAGCCACAAGCAAGGGTCCAGCCTCAGACCCAGATGACAGCACCAAAGCAGACACAGACCCCGGATCGGCTGCCTGAGCCACCAGAAGTCCAAATGCTGCCGCGTATCCAGCCACAGGCACTGCAGATCCAGACCCAGCCAAAGCTGCTTTGGCTGGGTCTGAGGCAGGCACAGACACAGACCGCTCCAGAGCACTTGGCGCCCCAGCAGGATGTCCTGGAG-3′
- Non complementary PCR amplicon (E. coli): 5′-AAAAGTGAAAGCGAACCGAATCTGTTAAATCAGCGAGTTGAGATCAAAAAATCTGACCTTGTTAACTATAATCCGATTGCGGAAAAGCACGTCAATGGGACGATGTCACTGGCTGAGCTTAGCGCGGCCGCGCTACAGTACAGCGATAACGTGGCGATGAATAAGCTGATTGCTCACGTTGGCGGCCCGGCTAGCGTCACCGCGTTCGCCCGACAGCTGGGAGACGAAACGTTCCGTCTCGACCGTACCGAGCCGACGTTAAACACCGCCATTCCGGGCGATCCGCGTGATACCACTTCACCTCGGGCAATGGCGCAAACTCTGCGGAATCTGACGCTGGGTAAAGCATTGGGCGACAGCCAACGGGCGCAGCTGGTGACATGGATGAAAGGCAATACCACCGGTGCAGCGAGCATTCAGGCTGGACTGCCTGCTTCCTGGGTTGTGGGGGATAAAACCGGCAGCGGTGACTATGGCACCACCAACGATATCGCGGTGATCTGGCCAAAAGATCGTGCGCCGCTGATTCTGGTCAC-3′
2.3. Methods
2.3.1. CRISPR-Cas9-Mediated Site-Directed Mutagenesis of the CIZ1 Gene
2.3.2. Quantitative Determination of Samples by Spectrophotometric Assay
2.3.3. Synthesis of CNTs
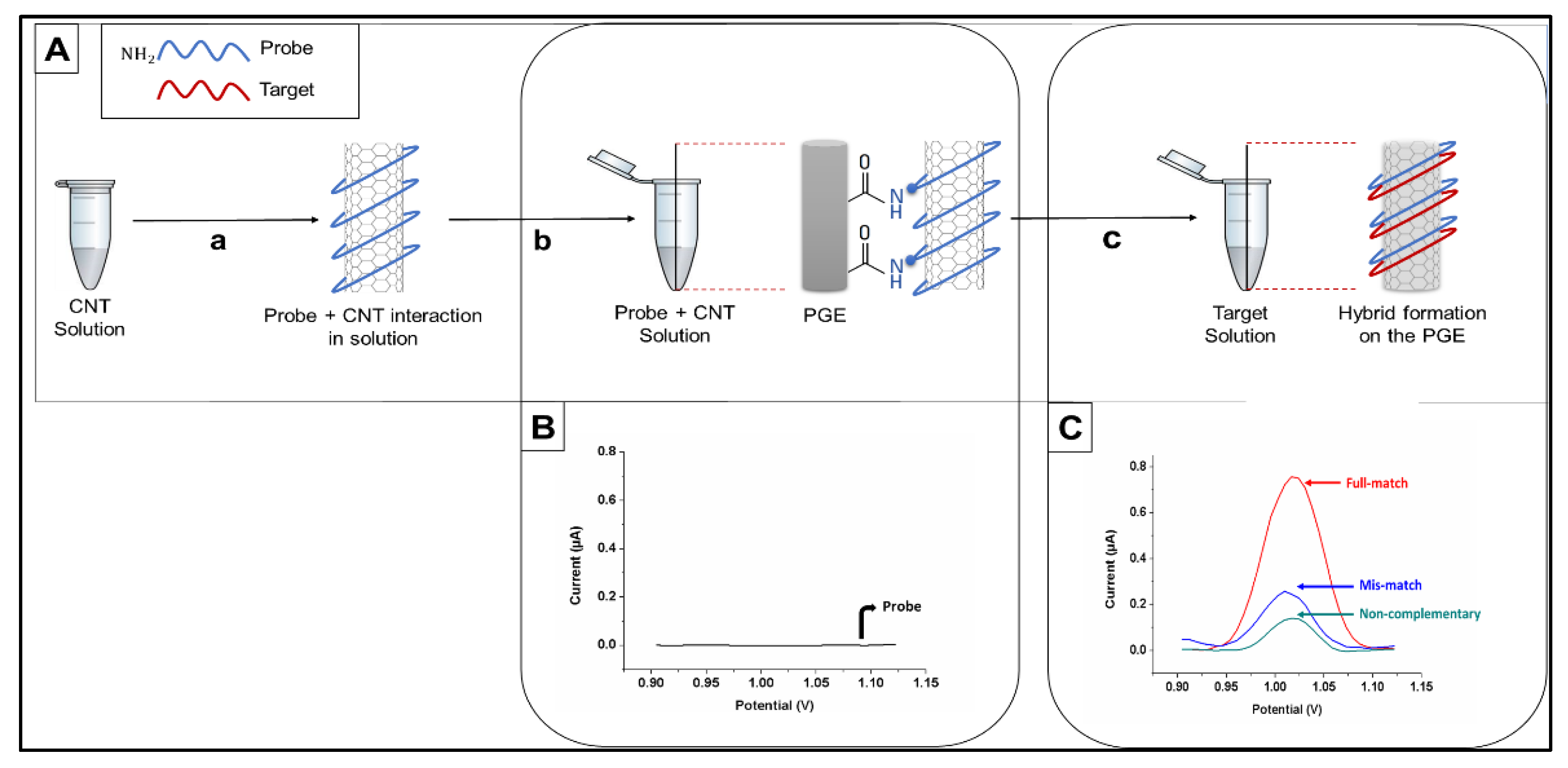
2.3.4. Electrode Modification and Probe Immobilization
2.3.5. Hybridization and Washing
2.3.6. Voltammetric Transduction
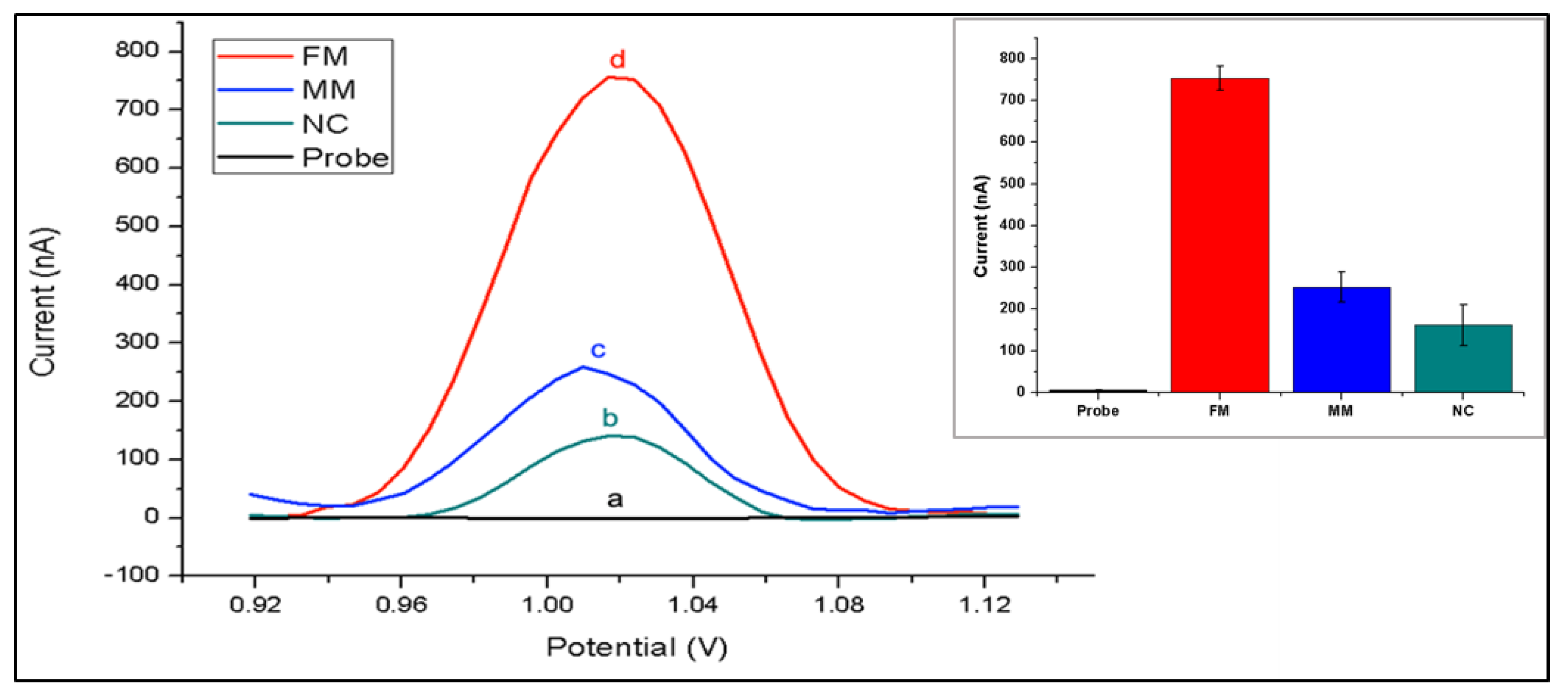
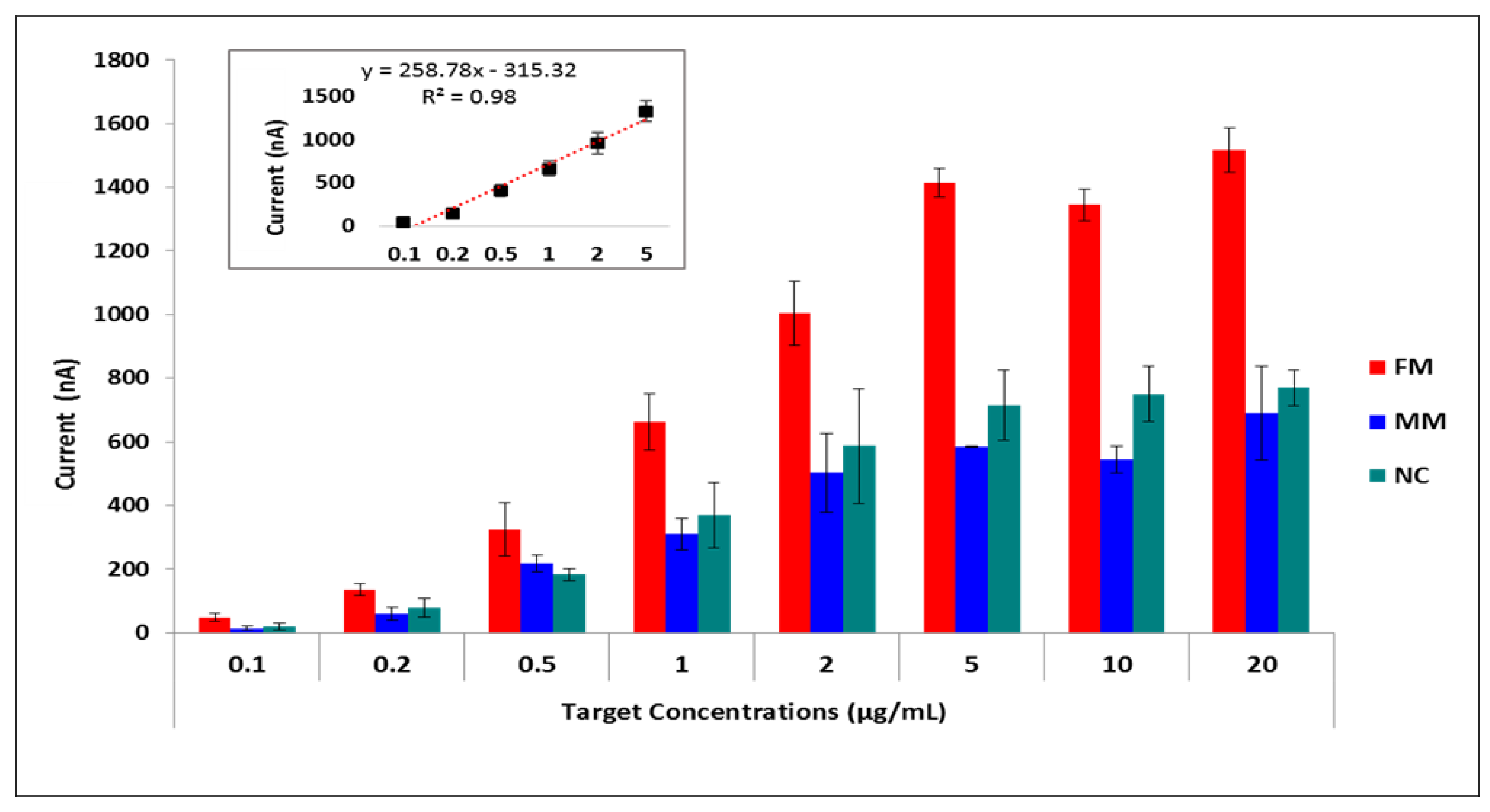
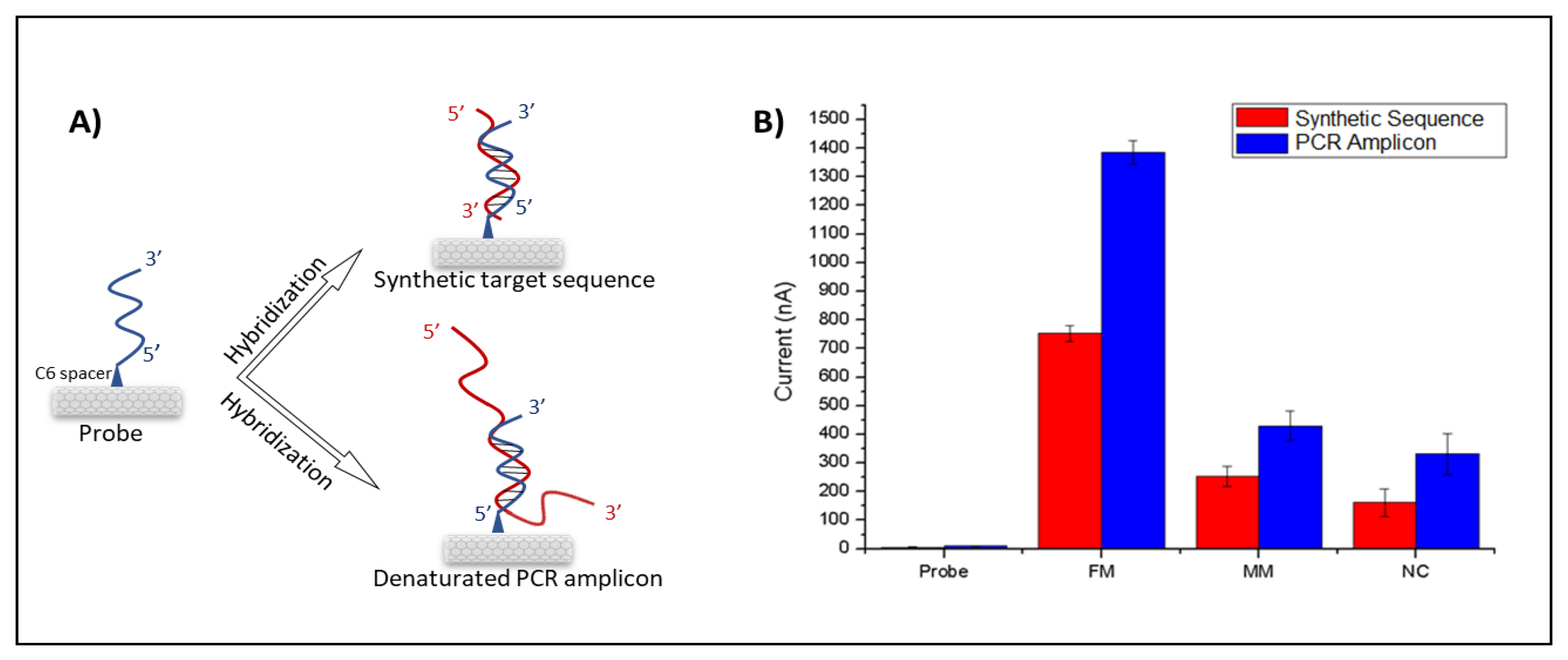
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, X.; Guo, Q.; Cui, D. Recent advances in nanotechnology applied to biosensors. Sensors 2009, 9, 1033–1053. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Yang, G.; Li, H.; Du, D.; Lin, Y. Electrochemical sensors and biosensors based on nanomaterials and nanostructures. Anal. Chem. 2015, 87, 230–249. [Google Scholar] [CrossRef]
- Hu, C.; Yang, D.-P.; Zhu, F.; Jiang, F.; Shen, S.; Zhang, J. Enzyme-labeled Pt@ BSA nanocomposite as a facile electrochemical biosensing interface for sensitive glucose determination. ACS Appl. Mater. Interfaces 2014, 6, 4170–4178. [Google Scholar] [CrossRef]
- Sireesha, M.; Jagadeesh Babu, V.; Kranthi Kiran, A.S.; Ramakrishna, S. A review on carbon nanotubes in biosensor devices and their applications in medicine. Nanocomposites 2018, 4, 36–57. [Google Scholar] [CrossRef]
- Ozsoz, M.; Erdem, A.; Kerman, K.; Ozkan, D.; Tugrul, B.; Topcuoglu, N.; Ekren, H.; Taylan, M. Electrochemical genosensor based on colloidal gold nanoparticles for the detection of Factor V Leiden mutation using disposable pencil graphite electrodes. Anal. Chem. 2003, 75, 2181–2187. [Google Scholar] [CrossRef]
- Fan, C.; Plaxco, K.W.; Heeger, A.J. Electrochemical interrogation of conformational changes as a reagentless method for the sequence-specific detection of DNA. Proc. Natl. Acad. Sci. USA 2003, 100, 9134–9137. [Google Scholar] [CrossRef]
- Yan, F.; Erdem, A.; Meric, B.; Kerman, K.; Ozsoz, M.; Sadik, O.A. Electrochemical DNA biosensor for the detection of specific gene related to Microcystis species. Electrochem. Commun. 2001, 3, 224–228. [Google Scholar] [CrossRef]
- Kilic, T.; Topkaya, S.N.; Ariksoysal, D.O.; Ozsoz, M.; Ballar, P.; Erac, Y.; Gozen, O. Electrochemical based detection of microRNA, mir21 in breast cancer cells. Biosens. Bioelectron. 2012, 38, 195–201. [Google Scholar] [CrossRef]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Dai, Y.; Wu, Y.; Liu, G.; Gooding, J.J. CRISPR Mediated Biosensing Toward Understanding Cellular Biology and Point-of-Care Diagnosis. Angew. Chem. 2020. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef]
- Dai, Y.; Somoza, R.A.; Wang, L.; Welter, J.F.; Li, Y.; Caplan, A.I.; Liu, C.C. Exploring the Trans-Cleavage Activity of CRISPR-Cas12a (cpf1) for the Development of a Universal Electrochemical Biosensor. Angew. Chem. Int. Ed. 2019, 58, 17399–17405. [Google Scholar] [CrossRef]
- Hajian, R.; Balderston, S.; Tran, T.; DeBoer, T.; Etienne, J.; Sandhu, M.; Wauford, N.A.; Chung, J.-Y.; Nokes, J.; Athaiya, M. Detection of unamplified target genes via CRISPR–Cas9 immobilized on a graphene field-effect transistor. Nat. Biomed. Eng. 2019, 3, 427–437. [Google Scholar] [CrossRef]
- Xu, W.; Jin, T.; Dai, Y.; Liu, C.C. Surpassing the detection limit and accuracy of the electrochemical DNA sensor through the application of CRISPR Cas systems. Biosens. Bioelectron. 2020, 155, 112100. [Google Scholar] [CrossRef]
- Joung, J.; Ladha, A.; Saito, M.; Kim, N.-G.; Woolley, A.E.; Segel, M.; Barretto, R.P.; Ranu, A.; Macrae, R.K.; Faure, G. Detection of SARS-CoV-2 with SHERLOCK one-pot testing. N. Engl. J. Med. 2020, 383, 1492–1494. [Google Scholar] [CrossRef]
- Li, Y.; Li, S.; Wang, J.; Liu, G. CRISPR/Cas systems towards next-generation biosensing. Trends Biotechnol. 2019, 37, 730–743. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A• T to G• C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Sansbury, B.M.; Hewes, A.M.; Kmiec, E.B. Understanding the diversity of genetic outcomes from CRISPR-Cas generated homology-directed repair. Commun. Biol. 2019, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Arbab, M.; Shen, M.W.; Mok, B.; Wilson, C.; Matuszek, Ż.; Cassa, C.A.; Liu, D.R. Determinants of Base Editing Outcomes from Target Library Analysis and Machine Learning. Cell 2020. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Kara, P.; Ozkan, D.; Erdem, A.; Kerman, K.; Pehlivan, S.; Ozkinay, F.; Unuvar, D.; Itirli, G.; Ozsoz, M. Detection of achondroplasia G380R mutation from PCR amplicons by using inosine modified carbon electrodes based on electrochemical DNA chip technology. Clin. Chim. Acta 2003, 336, 57–64. [Google Scholar] [CrossRef]
- Ozkan, D.; Erdem, A.; Kara, P.; Kerman, K.; Meric, B.; Hassmann, J.; Ozsoz, M. Allele-specific genotype detection of factor V Leiden mutation from polymerase chain reaction amplicons based on label-free electrochemical genosensor. Anal. Chem. 2002, 74, 5931–5936. [Google Scholar] [CrossRef]
- Napier, M.E.; Loomis, C.R.; Sistare, M.F.; Kim, J.; Eckhardt, A.E.; Thorp, H.H. Probing biomolecule recognition with electron transfer: Electrochemical sensors for DNA hybridization. Bioconjug. Chem. 1997, 8, 906–913. [Google Scholar] [CrossRef]
- Kilic, T.; Erdem, A.; Erac, Y.; Seydibeyoglu, M.O.; Okur, S.; Ozsoz, M. Electrochemical Detection of a Cancer Biomarker mir-21 in Cell Lysates Using Graphene Modified Sensors. Electroanalysis 2015, 27, 317–326. [Google Scholar] [CrossRef]
- Sauer, P.; Muller, M.; Kang, J. Quantitation of DNA. Qiagen News 1998, 2, 23–26. [Google Scholar]
- Yardimci, A.I.; Yılmaz, S.; Selamet, Y. The effects of catalyst pretreatment, growth atmosphere and temperature on carbon nanotube synthesis using Co–Mo/MgO catalyst. Diam. Relat. Mater. 2015, 60, 81–86. [Google Scholar] [CrossRef]
- Rashidi, A.; Akbarnejad, M.; Khodadadi, A.; Mortazavi, Y.; Ahmadpourd, A. Single-wall carbon nanotubes synthesized using organic additives to Co–Mo catalysts supported on nanoporous MgO. Nanotechnology 2007, 18, 315605. [Google Scholar] [CrossRef]
- Ozkan-Ariksoysal, D.; Kayran, Y.U.; Yilmaz, F.F.; Ciucu, A.A.; David, I.G.; David, V.; Hosgor-Limoncu, M.; Ozsoz, M. DNA-wrapped multi-walled carbon nanotube modified electrochemical biosensor for the detection of Escherichia coli from real samples. Talanta 2017, 166, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Paquet, D.; Kwart, D.; Chen, A.; Sproul, A.; Jacob, S.; Teo, S.; Olsen, K.M.; Gregg, A.; Noggle, S.; Tessier-Lavigne, M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 2016, 533, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Copeland, N.A.; Sercombe, H.E.; Wilson, R.H.; Coverley, D. Cyclin-A–CDK2-mediated phosphorylation of CIZ1 blocks replisome formation and initiation of mammalian DNA replication. J. Cell Sci. 2015, 128, 1518–1527. [Google Scholar] [CrossRef]
- Copeland, N.A.; Sercombe, H.E.; Ainscough, J.F.; Coverley, D. Ciz1 cooperates with cyclin-A–CDK2 to activate mammalian DNA replication in vitro. J. Cell Sci. 2010, 123, 1108–1115. [Google Scholar] [CrossRef]
- Kara, P.; Cavusoglu, C.; Cavdar, S.; Ozsoz, M. Direct electrochemical genosensing for multiple point mutation detection of Mycobacterium tuberculosis during the development of rifampin resistance. Biosens. Bioelectron. 2009, 24, 1796–1800. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kivrak, E.; Pauzaite, T.; Copeland, N.A.; Hardy, J.G.; Kara, P.; Firlak, M.; Yardimci, A.I.; Yilmaz, S.; Palaz, F.; Ozsoz, M. Detection of CRISPR-Cas9-Mediated Mutations Using a Carbon Nanotube-Modified Electrochemical Genosensor. Biosensors 2021, 11, 17. https://doi.org/10.3390/bios11010017
Kivrak E, Pauzaite T, Copeland NA, Hardy JG, Kara P, Firlak M, Yardimci AI, Yilmaz S, Palaz F, Ozsoz M. Detection of CRISPR-Cas9-Mediated Mutations Using a Carbon Nanotube-Modified Electrochemical Genosensor. Biosensors. 2021; 11(1):17. https://doi.org/10.3390/bios11010017
Chicago/Turabian StyleKivrak, Ezgi, Tekle Pauzaite, Nikki A. Copeland, John G. Hardy, Pinar Kara, Melike Firlak, Atike I. Yardimci, Selahattin Yilmaz, Fahreddin Palaz, and Mehmet Ozsoz. 2021. "Detection of CRISPR-Cas9-Mediated Mutations Using a Carbon Nanotube-Modified Electrochemical Genosensor" Biosensors 11, no. 1: 17. https://doi.org/10.3390/bios11010017
APA StyleKivrak, E., Pauzaite, T., Copeland, N. A., Hardy, J. G., Kara, P., Firlak, M., Yardimci, A. I., Yilmaz, S., Palaz, F., & Ozsoz, M. (2021). Detection of CRISPR-Cas9-Mediated Mutations Using a Carbon Nanotube-Modified Electrochemical Genosensor. Biosensors, 11(1), 17. https://doi.org/10.3390/bios11010017