Formation and Stability of Pea Proteins Nanoparticles Using Ethanol-Induced Desolvation


Abstract
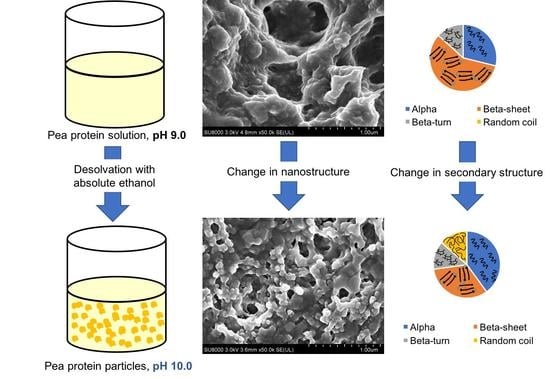
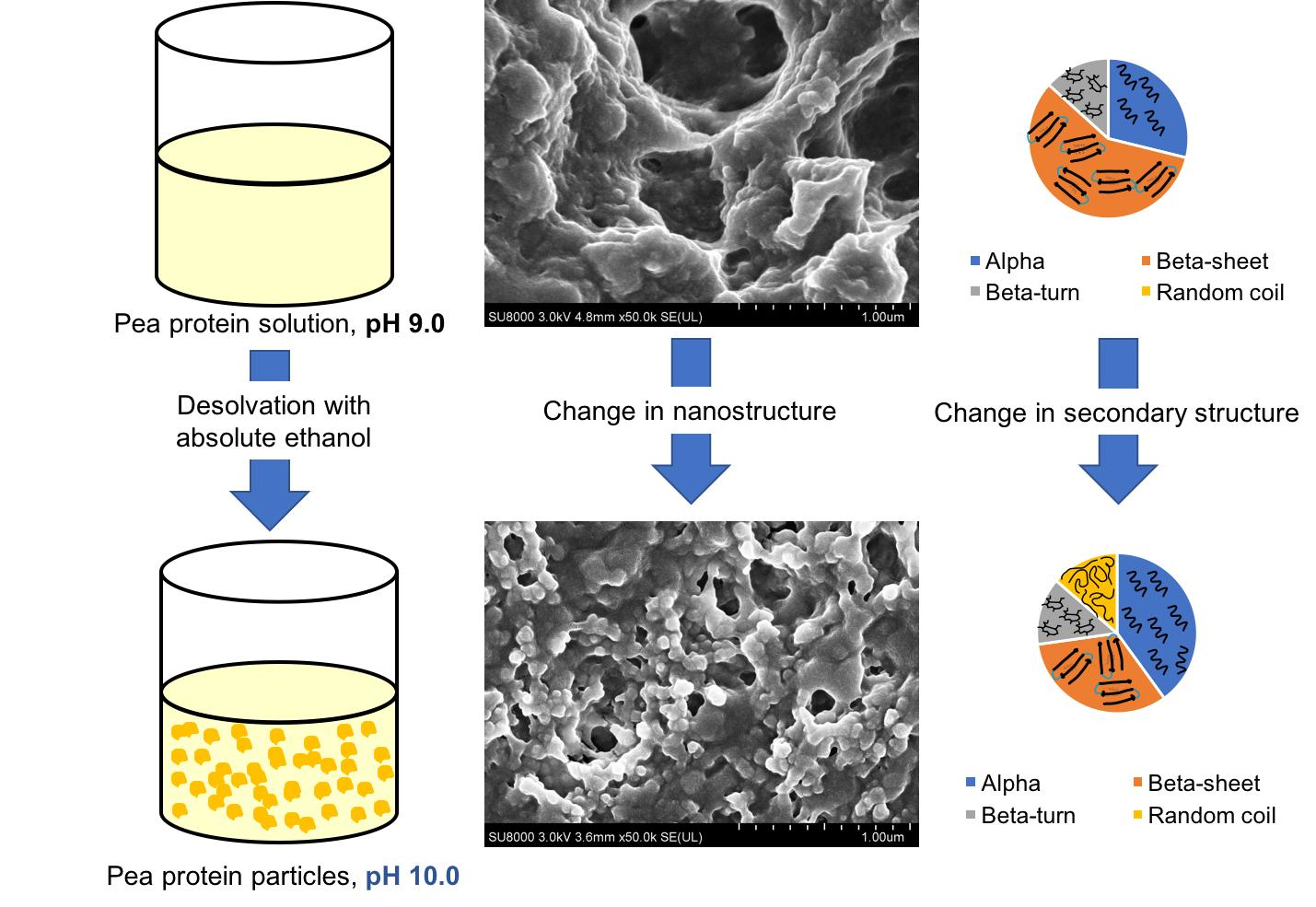{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Protein Solution
2.3. Determination of Protein Concentration
2.4. Synthesis of Pea Protein Nanoparticles (PPN)
2.5. Particle Size Distribution and Surface Charge
2.6. Secondary Structure Using Attenuated Total Internal Reflectance—Fourier Transform Infrared Spectroscopy (ATR-FTIR)
2.7. Particle Morphology
2.8. Re-Dispersibility of Pea Protein Nanoparticles
2.9. Accelerated Gravitational Sedimentation of Pea Protein Nanoparticles
2.10. Statistical Analysis
3. Results
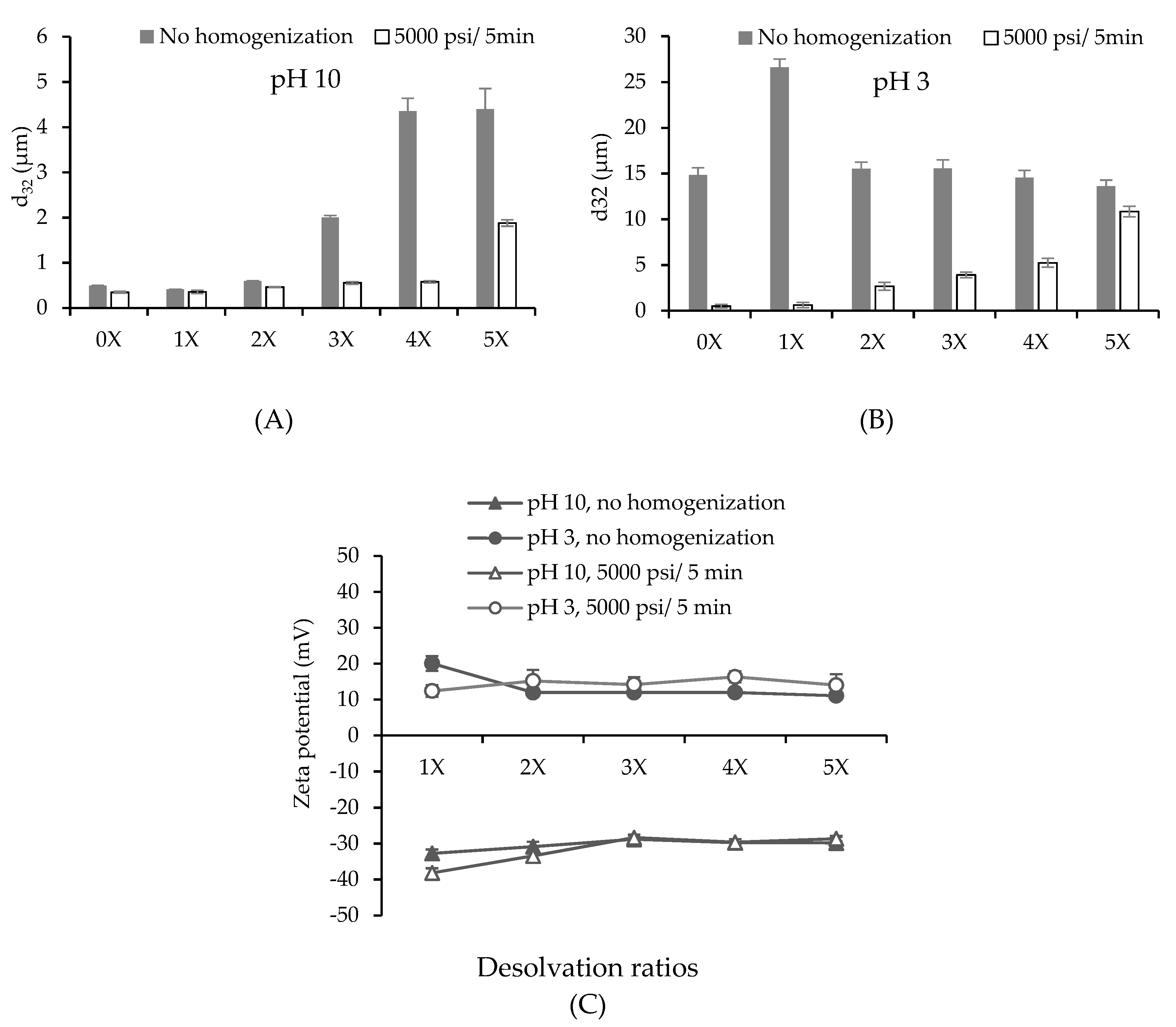
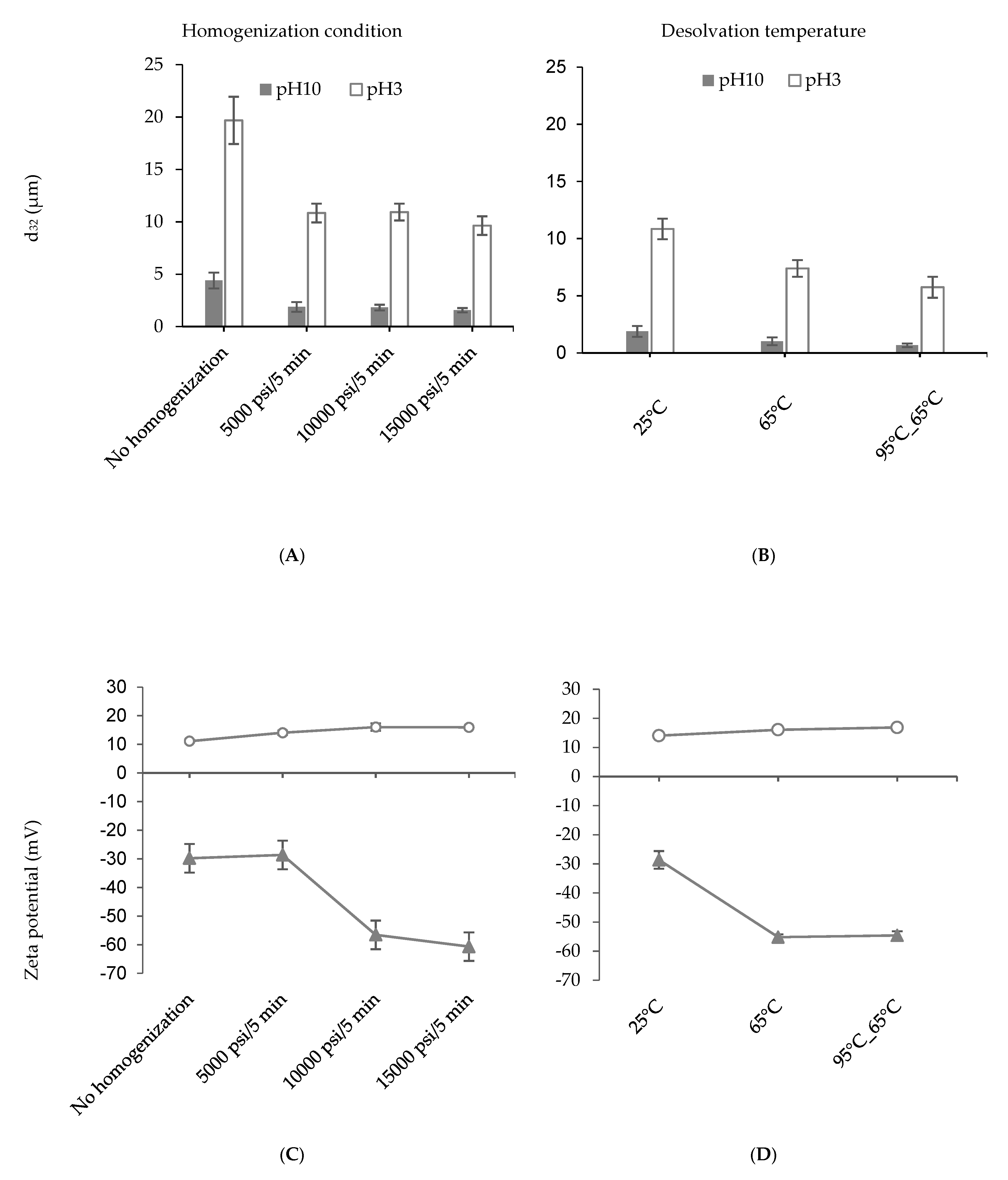
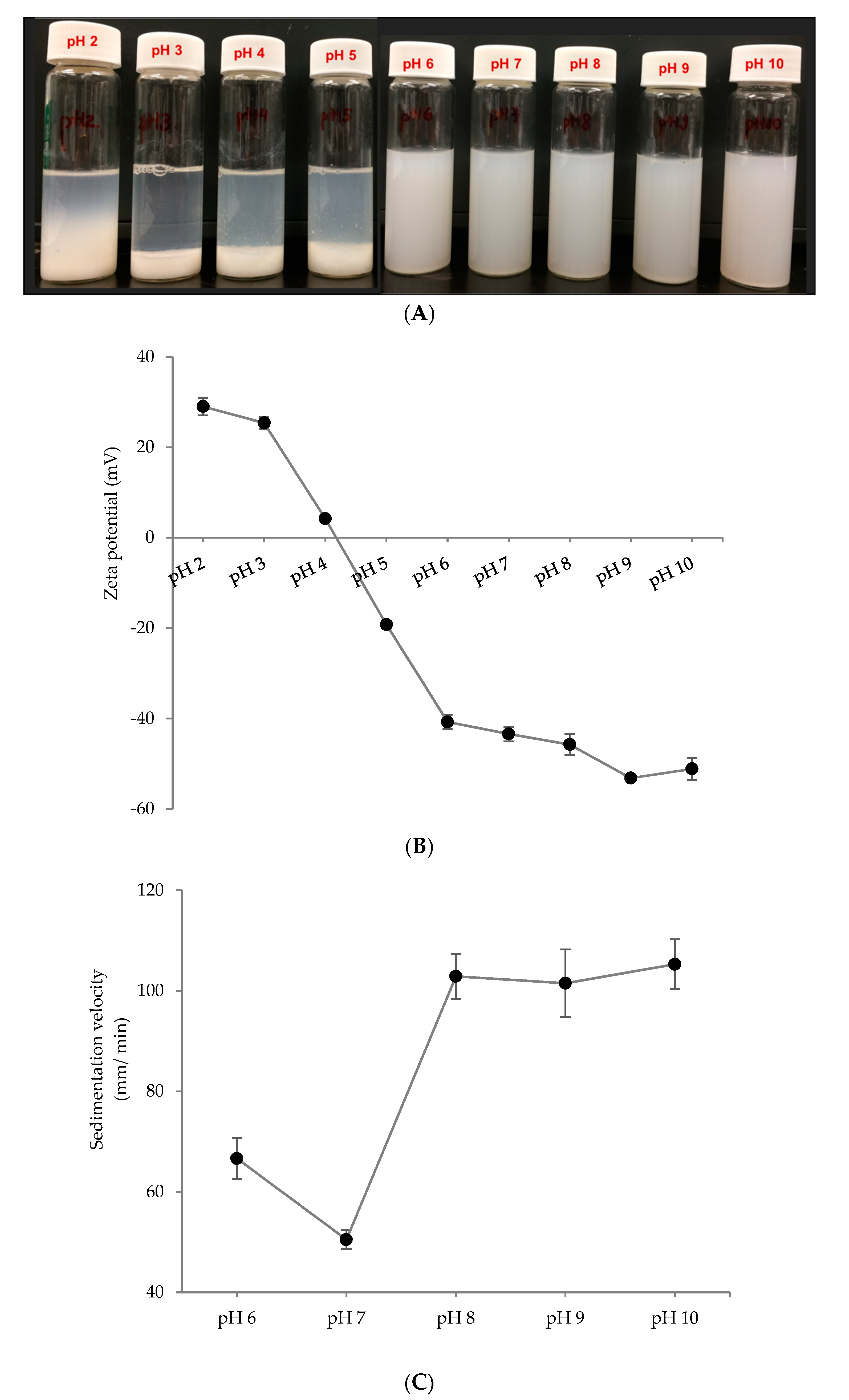
3.1. Nanoparticle Synthesis and Characterization
3.2. Particle Microstructure
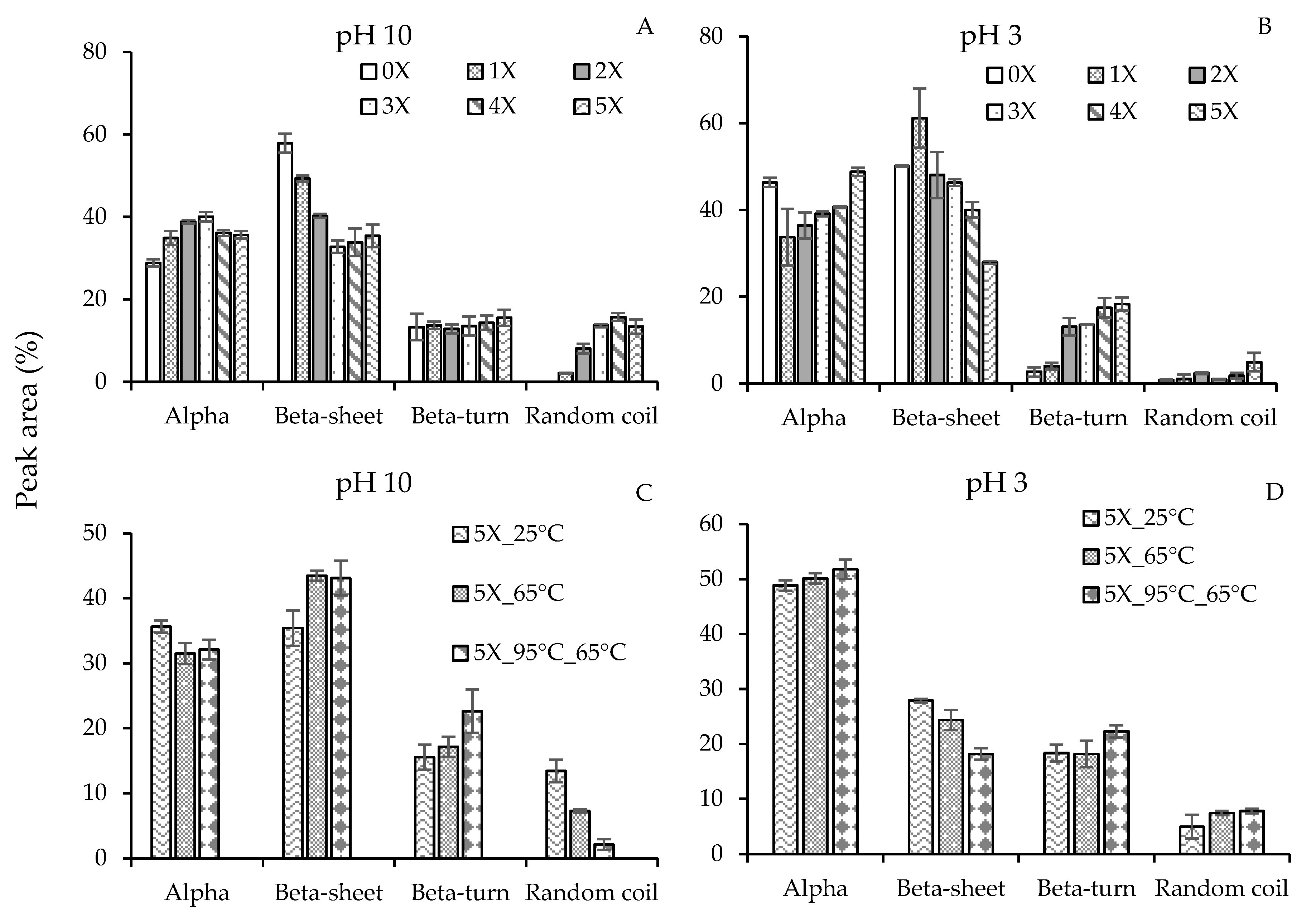
3.3. Conformational Changes in the Secondary Structures of Pea Protein Nanoparticles
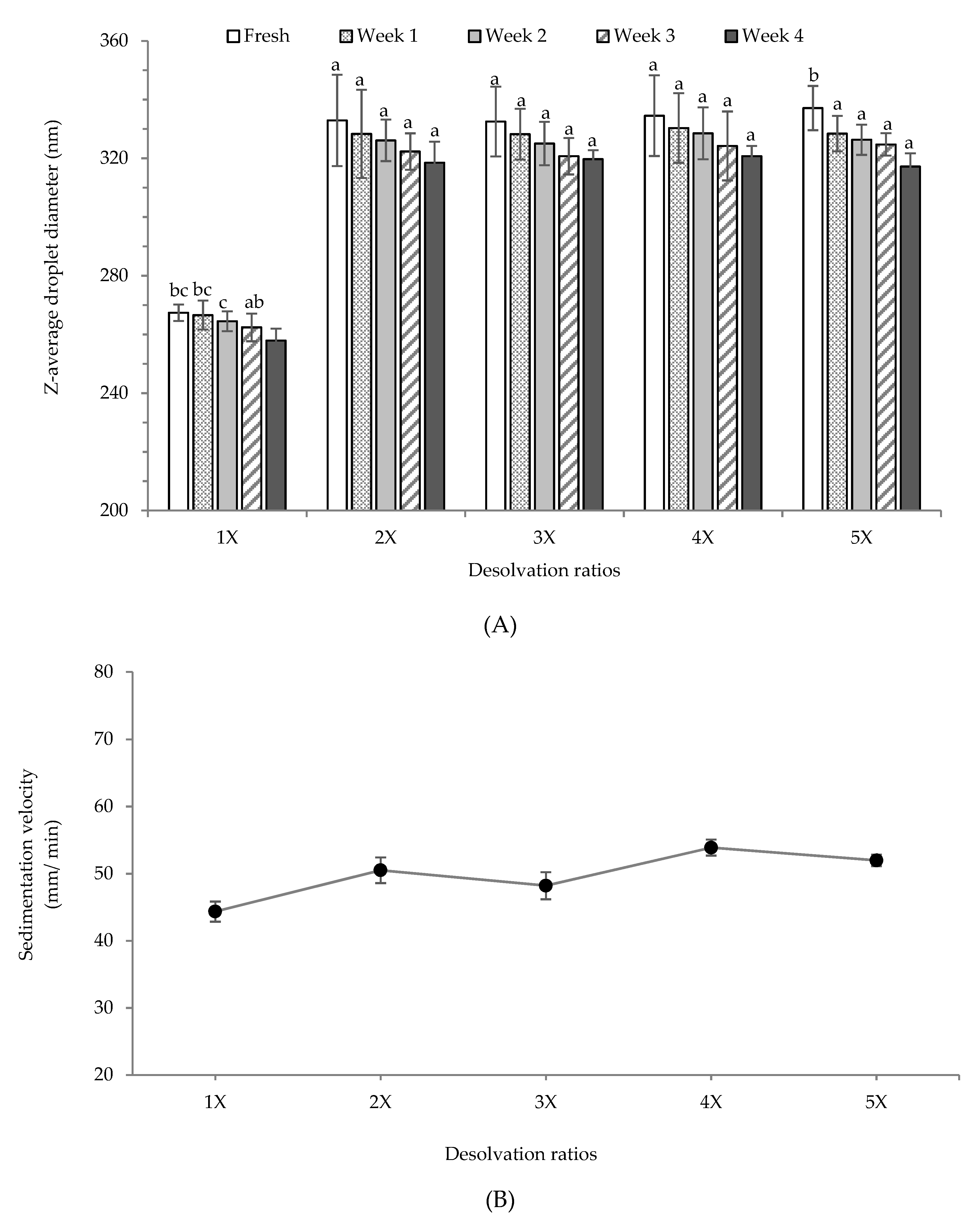
3.4. Storage Stability of Pea Protein Particles
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Joye, I.J.; McClements, D.J. Production of nanoparticles by anti-solvent precipitation for use in food systems. Trends Food Sci. Technol. 2013, 34, 109–123. [Google Scholar] [CrossRef]
- Teng, Z.; Luo, Y.; Wang, Q. Carboxymethyl chitosan–soy protein complex nanoparticles for the encapsulation and controlled release of vitamin D3. Food Chem. 2013, 141, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Gülseren, İ.; Fang, Y.; Corredig, M. Whey protein nanoparticles prepared with desolvation with ethanol: Characterization, thermal stability and interfacial behavior. Food Hydrocoll. 2012, 29, 258–264. [Google Scholar] [CrossRef]
- Hirota-Nakaoka, N.; Goto, Y. Alcohol-induced denaturation of β-lactoglobulin: A close correlation to the alcohol-induced α-helix formation of melittin. Bioorganic Med. Chem. 1999, 7, 67–73. [Google Scholar] [CrossRef]
- Nesterenko, A.; Alric, I.; Silvestre, F.; Durrieu, V. Vegetable proteins in microencapsulation: A review of recent interventions and their effectiveness. Ind. Crop. Prod. 2013, 42, 469–479. [Google Scholar] [CrossRef]
- Jenkins, J.A.; Breiteneder, H.; Mills, E.C. Evolutionary distance from human homologs reflects allergenicity of animal food proteins. J. Allergy Clin. Immunol. 2007, 120, 1399–1405. [Google Scholar] [CrossRef]
- Li, H.; Zhu, K.; Zhou, H.; Peng, W. Effects of high hydrostatic pressure treatment on allergenicity and structural properties of soybean protein isolate for infant formula. Food Chem. 2012, 132, 808–814. [Google Scholar] [CrossRef]
- Koyoro, H.; Powers, J. Functional properties of pea globulin fractions. Cereal. Chem. 1987, 64, 97–101. [Google Scholar]
- Gueguen, J.; Barbot, J. Quantitative and qualitative variability of pea (Pisum sativum L.) protein composition. J. Sci. Food Agric. 1988, 42, 209–224. [Google Scholar] [CrossRef]
- Liu, S.; Elmer, C.; Low, N.; Nickerson, M. Effect of pH on the functional behaviour of pea protein isolate–gum Arabic complexes. Food Res. Int. 2010, 43, 489–495. [Google Scholar] [CrossRef]
- Shand, P.; Ya, H.; Pietrasik, Z.; Wanasundara, P. Physicochemical and textural properties of heat-induced pea protein isolate gels. Food Chem. 2007, 102, 1119–1130. [Google Scholar] [CrossRef]
- Choi, W.S.; Han, J.H. Physical and mechanical properties of pea-protein-based edible films. J. Food Sci. 2001, 66, 319–322. [Google Scholar] [CrossRef]
- Akintayo, E.; Oshodi, A.; Esuoso, K. Effects of NaCl, ionic strength and pH on the foaming and gelation of pigeon pea (Cajanus cajan) protein concentrates. Food Chem. 1999, 66, 51–56. [Google Scholar] [CrossRef]
- Yerramilli, M.; Longmore, N.; Ghosh, S. Improved stabilization of nanoemulsions by partial replacement of sodium caseinate with pea protein isolate. Food Hydrocoll. 2017, 64, 99–111. [Google Scholar] [CrossRef]
- Irache, J.M.; Bergougnoux, L.; Ezpeleta, I.; Gueguen, J.; Orecchioni, A.-M. Optimization and in vitro stability of legumin nanoparticles obtained by a coacervation method. Int. J. Pharm. 1995, 126, 103–109. [Google Scholar] [CrossRef]
- Liu, F.; Chen, Z.; Tang, C. Microencapsulation properties of protein isolates from three selected Phaseolus legumes in comparison with soy protein isolate. Lwt-Food Sci. Technol. 2014, 55, 74–82. [Google Scholar] [CrossRef]
- Ko, S.; Gunasekaran, S. Preparation of sub-100-nm β-lactoglobulin (BLG) nanoparticles. J. Microencapsul. 2006, 23, 887–898. [Google Scholar] [CrossRef]
- Sadeghi, R.; Moosavi-Movahedi, A.; Emam-Jomeh, Z.; Kalbasi, A.; Razavi, S.; Karimi, M.; Kokini, J. The effect of different desolvating agents on BSA nanoparticle properties and encapsulation of curcumin. J. Nanoparticle Res. 2014, 16, 2565. [Google Scholar] [CrossRef]
- Dufour, E.; Robert, P.; Renard, D.; Llamas, G. Investigation of β-lactoglobulin gelation in water/ethanol solutions. Int. Dairy J. 1998, 8, 87–93. [Google Scholar] [CrossRef]
- Ohnishi, S.T.; Barr, J.K. A simplified method of quantitating protein using the biuret and phenol reagents. Anal. Biochem. 1978, 86, 193–200. [Google Scholar] [CrossRef]
- Kauppinen, J.K.; Moffatt, D.J.; Mantsch, H.H.; Cameron, D.G. Fourier self-deconvolution: A method for resolving intrinsically overlapped bands. Appl. Spectrosc. 1981, 35, 271–276. [Google Scholar] [CrossRef]
- Teng, Z.; Luo, Y.; Wang, Q. Nanoparticles synthesized from soy protein: Preparation, characterization, and application for nutraceutical encapsulation. J. Agric. Food Chem. 2012, 60, 2712–2720. [Google Scholar] [CrossRef] [PubMed]
- Subirade, M.; Gueguen, J.; Schwenke, K.D. Effect of dissociation and conformational changes on the surface behavior of pea legumin. J. Colloid Interface Sci. 1992, 152, 442–454. [Google Scholar] [CrossRef]
- Magdassi, S. Surface Activity of Proteins: Chemical and Physicochemical Modifications; CRC Press: Boca Raton, FL, USA, 1996. [Google Scholar]
- Donsì, F.; Senatore, B.; Huang, Q.; Ferrari, G. Development of novel pea protein-based nanoemulsions for delivery of nutraceuticals. J. Agric. Food Chem. 2010, 58, 10653–10660. [Google Scholar] [CrossRef] [PubMed]
- Kakran, M.; Sahoo, N.G.; Tan, I.-L.; Li, L. Preparation of nanoparticles of poorly water-soluble antioxidant curcumin by antisolvent precipitation methods. J. Nanoparticle Res. 2012, 14, 757. [Google Scholar] [CrossRef]
- Chen, F.; Li, B.; Tang, C. Nanocomplexation between curcumin and soy protein isolate: Influence on curcumin stability/bioaccessibility and in vitro protein digestibility. J. Agric. Food Chem 2015, 63, 3559–3569. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-J.; Jeon, S.-Y.; Yeo, S.-D. Recrystallization of a pharmaceutical compound using liquid and supercritical antisolvents. Ind. Eng. Chem. Res. 2006, 45, 2287–2293. [Google Scholar] [CrossRef]
- Singh, S.M.; Cabello-Villegas, J.; Hutchings, R.L.; Mallela, K.M. Role of partial protein unfolding in alcohol-induced protein aggregation. Proteins: Struct. Funct. Bioinform. 2010, 78, 2625–2637. [Google Scholar] [CrossRef] [PubMed]
- Davidov-Pardo, G.; Joye, I.J.; McClements, D.J. Encapsulation of resveratrol in biopolymer particles produced using liquid antisolvent precipitation. Part 1: Preparation and characterization. Food Hydrocoll. 2015, 45, 309–316. [Google Scholar] [CrossRef]
- Tsumura, K.; Enatsu, M.; Kuramori, K.; Morita, S.; Kugimiya, W.; Kuwada, M.; Shimura, Y.; Hasumi, H. Conformational change in a single molecular species, β3, of β-conglycinin in acidic ethanol solution. Biosci. Biotechnol. Biochem. 2001, 65, 292–297. [Google Scholar] [CrossRef]
- Thomas, P.D.; Dill, K.A. Local and nonlocal interactions in globular proteins and mechanisms of alcohol denaturation. Protein Sci. 1993, 2, 2050–2065. [Google Scholar] [CrossRef] [PubMed]
- Herskovits, T.T.; Gadegbeku, B.; Jaillet, H. On the structural stability and solvent denaturation of proteins I. Denaturation by the alcohols and glycols. J. Biol. Chem. 1970, 245, 2588–2598. [Google Scholar]
- Clark, D.C.; Smith, L.J. Influence of alcohol-containing spreading solvents on the secondary structssure of proteins: A circular dichroism investigation. J. Agric. Food Chem. 1989, 37, 627–633. [Google Scholar] [CrossRef]
- Marcelino, A.M.C.; Gierasch, L.M. Roles of β-turns in protein folding: From peptide models to protein engineering. Biopolym. Orig. Res. Biomol. 2008, 89, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Davidov-Pardo, G.; Joye, I.J.; McClements, D.J. Food-grade protein-based nanoparticles and microparticles for bioactive delivery: Fabrication, characterization, and utilization. In Advances in protein chemistry and structural biology; Elsevier: Amsterdam, The Netherlands, 2015; Volume 98, pp. 293–325. [Google Scholar]
- Selling, G.W.; Hamaker, S.A.; Sessa, D.J. Effect of solvent and Temperature on secondary and tertiary structure of zein by circular dichroism. Cereal Chem. 2007, 84, 265–270. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doan, C.D.; Ghosh, S. Formation and Stability of Pea Proteins Nanoparticles Using Ethanol-Induced Desolvation. Nanomaterials 2019, 9, 949. https://doi.org/10.3390/nano9070949
Doan CD, Ghosh S. Formation and Stability of Pea Proteins Nanoparticles Using Ethanol-Induced Desolvation. Nanomaterials. 2019; 9(7):949. https://doi.org/10.3390/nano9070949
Chicago/Turabian StyleDoan, Chi Diem, and Supratim Ghosh. 2019. "Formation and Stability of Pea Proteins Nanoparticles Using Ethanol-Induced Desolvation" Nanomaterials 9, no. 7: 949. https://doi.org/10.3390/nano9070949
APA StyleDoan, C. D., & Ghosh, S. (2019). Formation and Stability of Pea Proteins Nanoparticles Using Ethanol-Induced Desolvation. Nanomaterials, 9(7), 949. https://doi.org/10.3390/nano9070949