A Review on Brittle Fracture Nanomechanics by All-Atom Simulations



Abstract
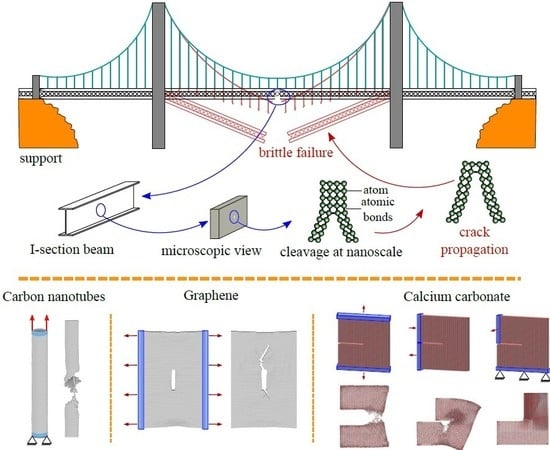
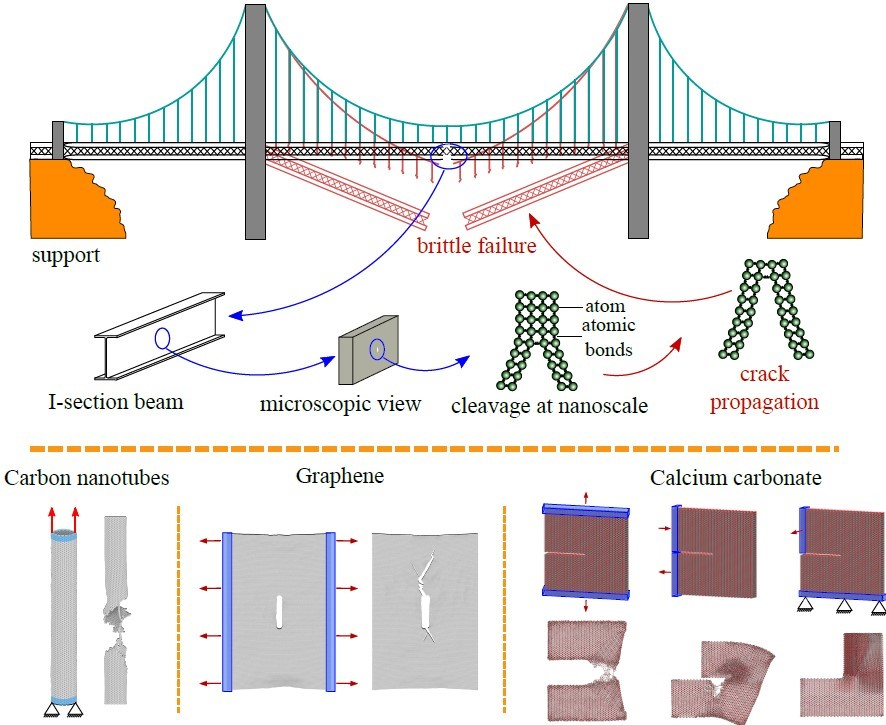
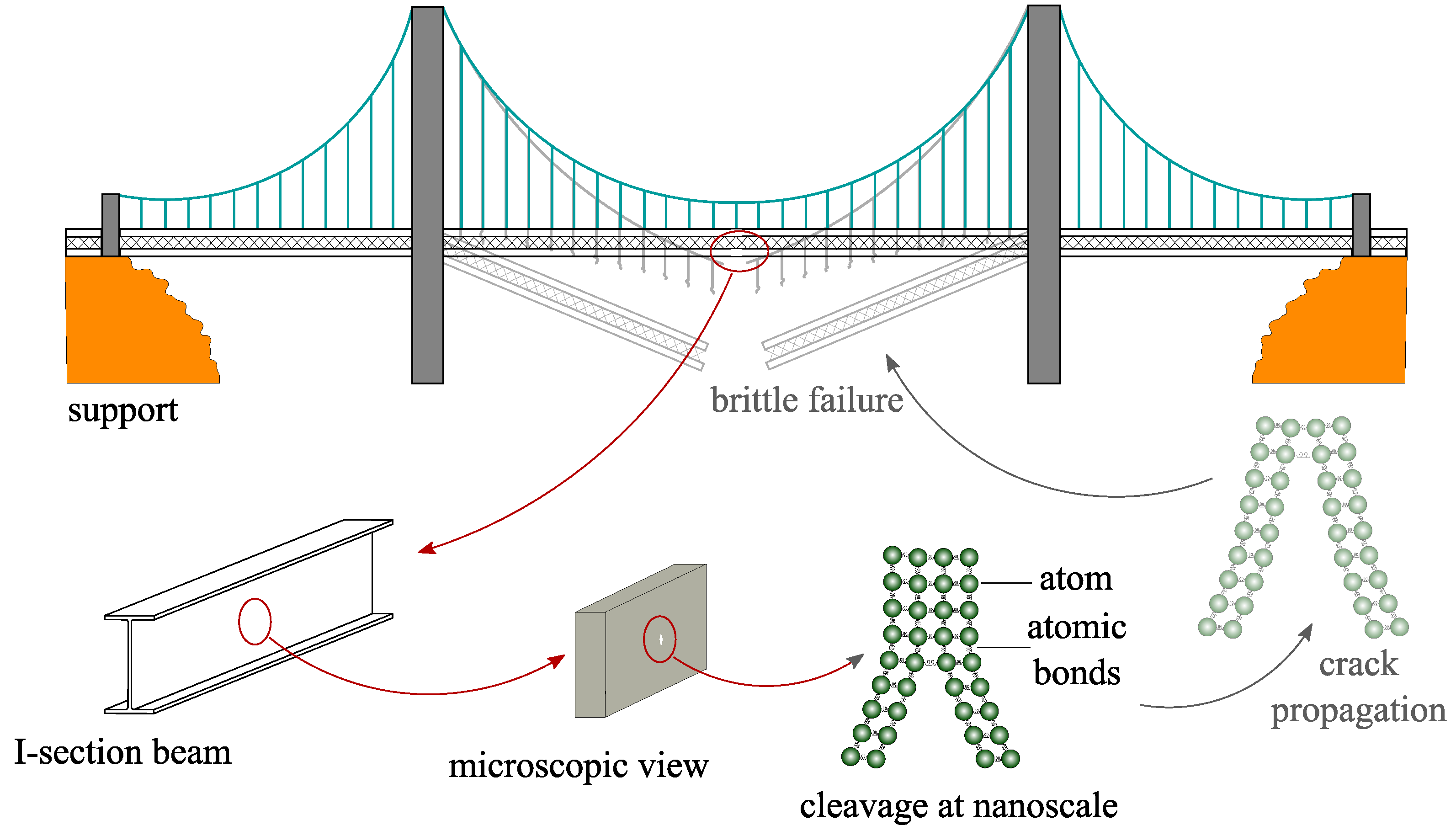
1. Introduction
2. Principles of MD Simulation
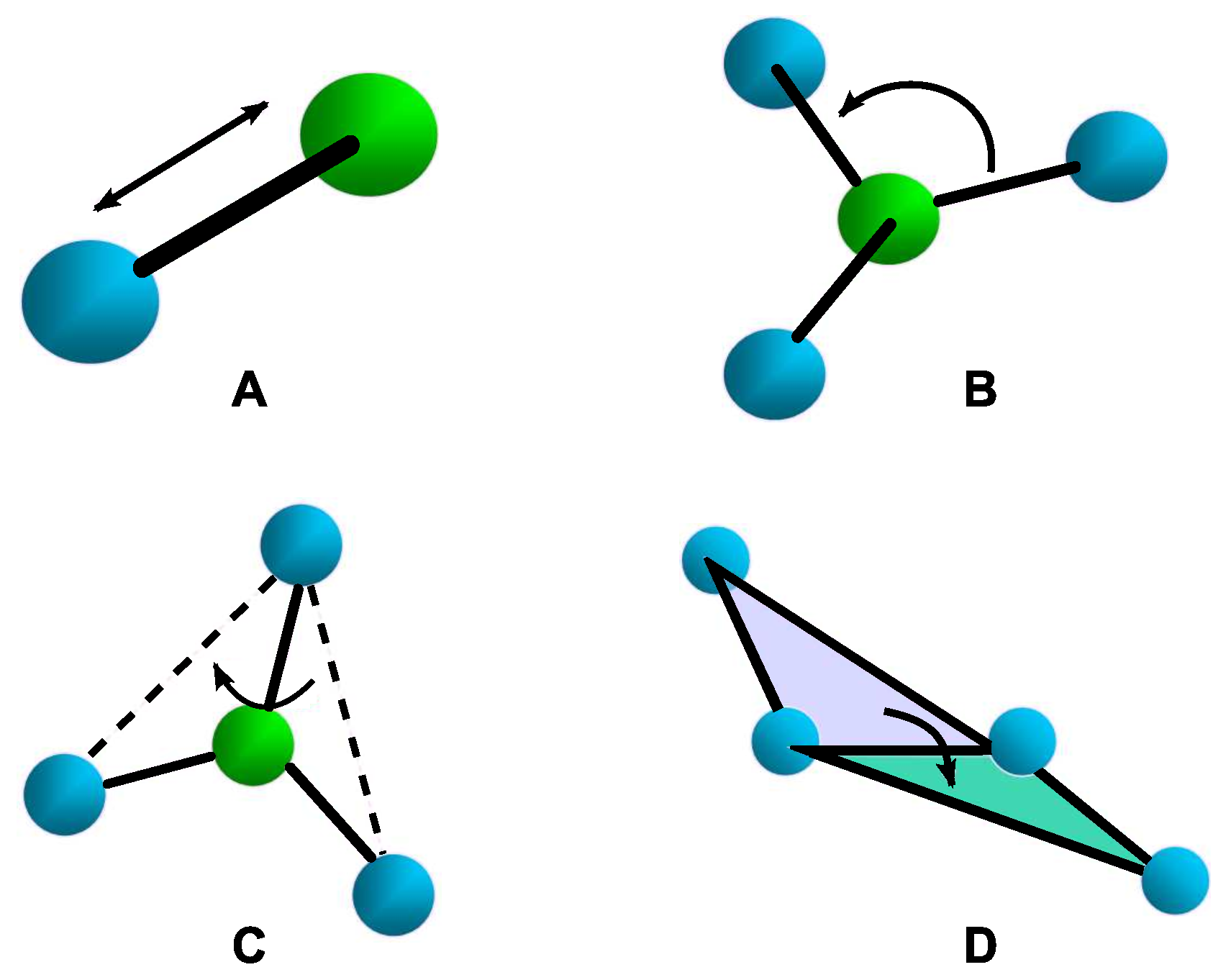
2.1. Bonded Interactions
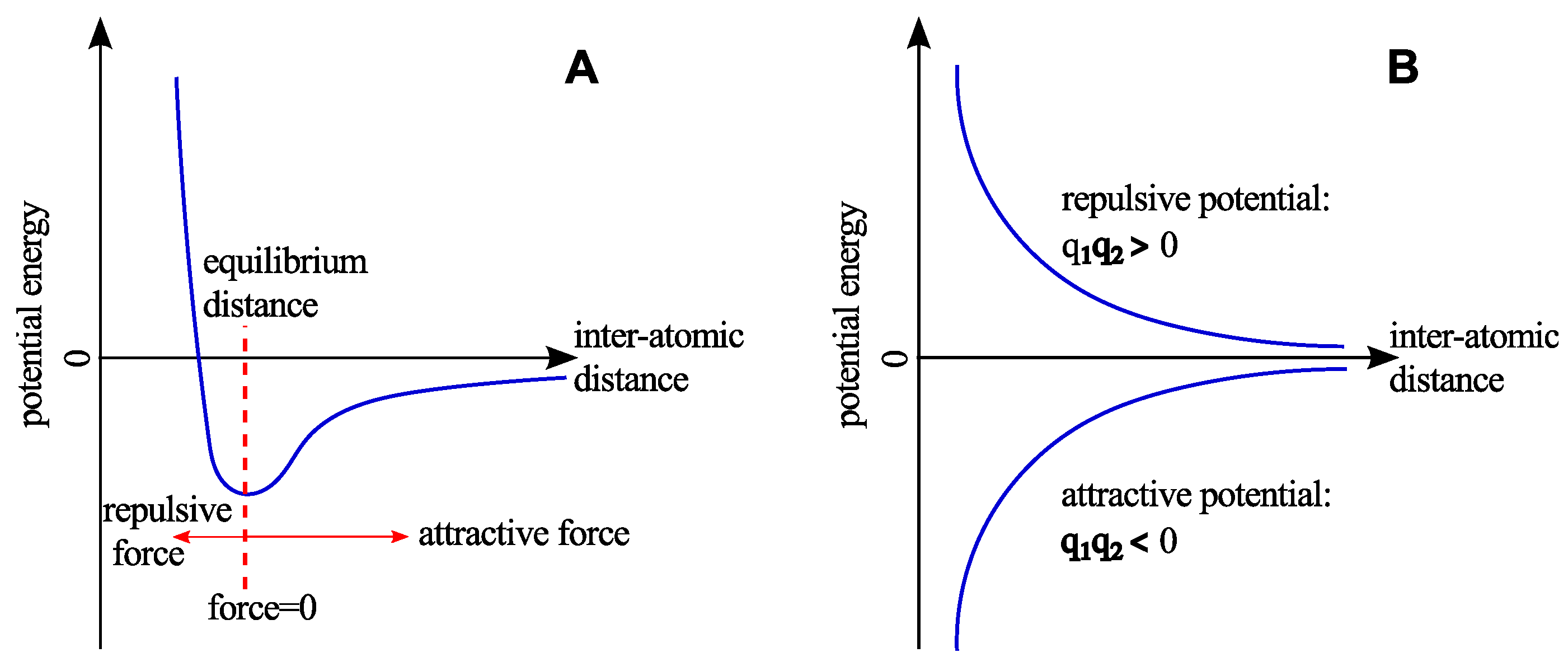
2.2. Non-Bonded Interactions
2.3. EAM Potentials for Metallic Atoms Interaction
2.4. Other Force-Fields
2.5. Numerical Solution of the Equations of Motion
2.6. Advantages and Limitations of MD Simulation of Fracture
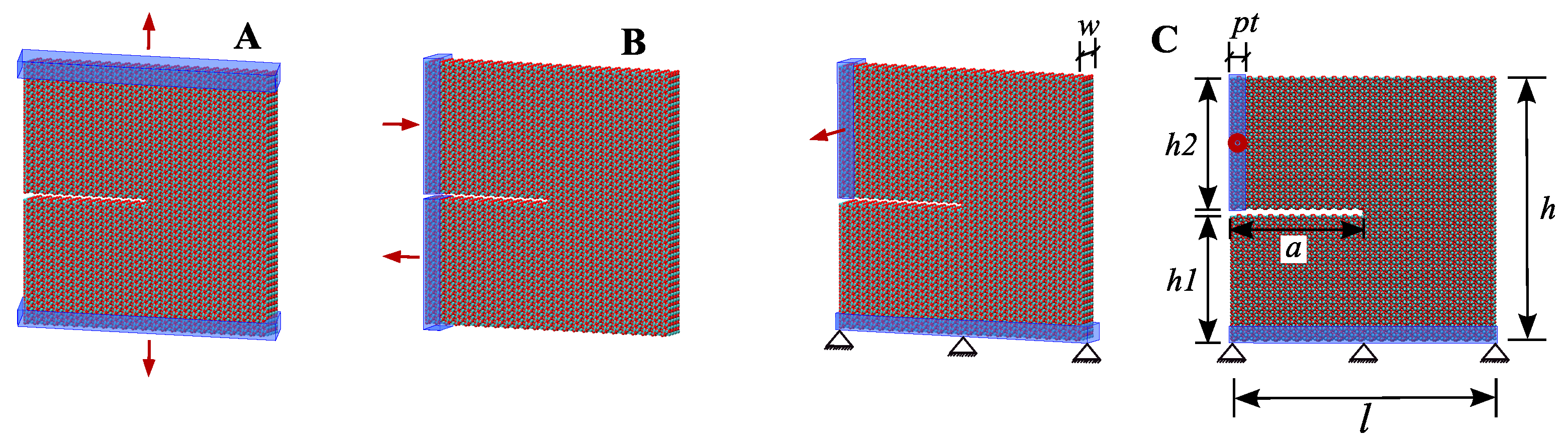
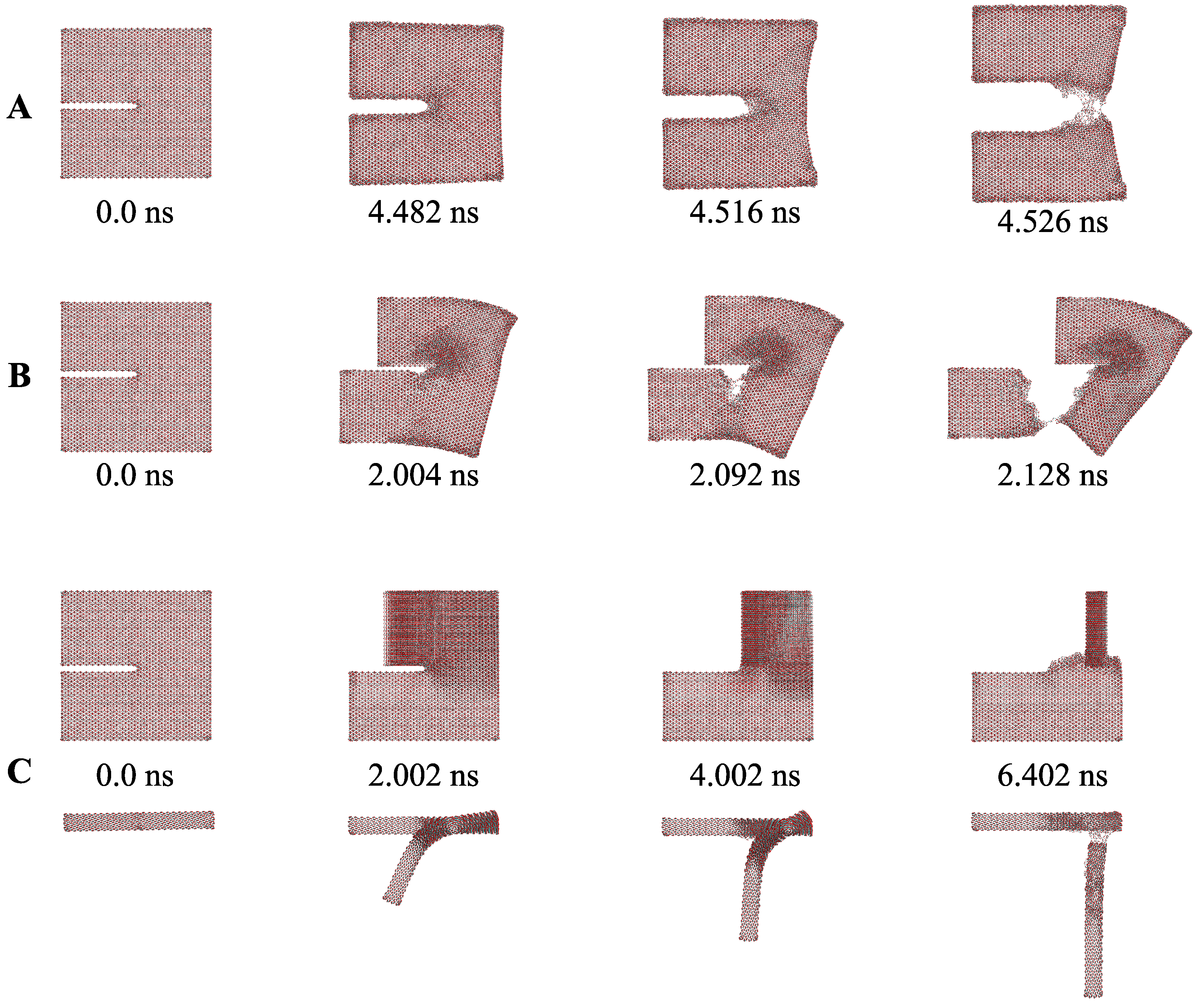
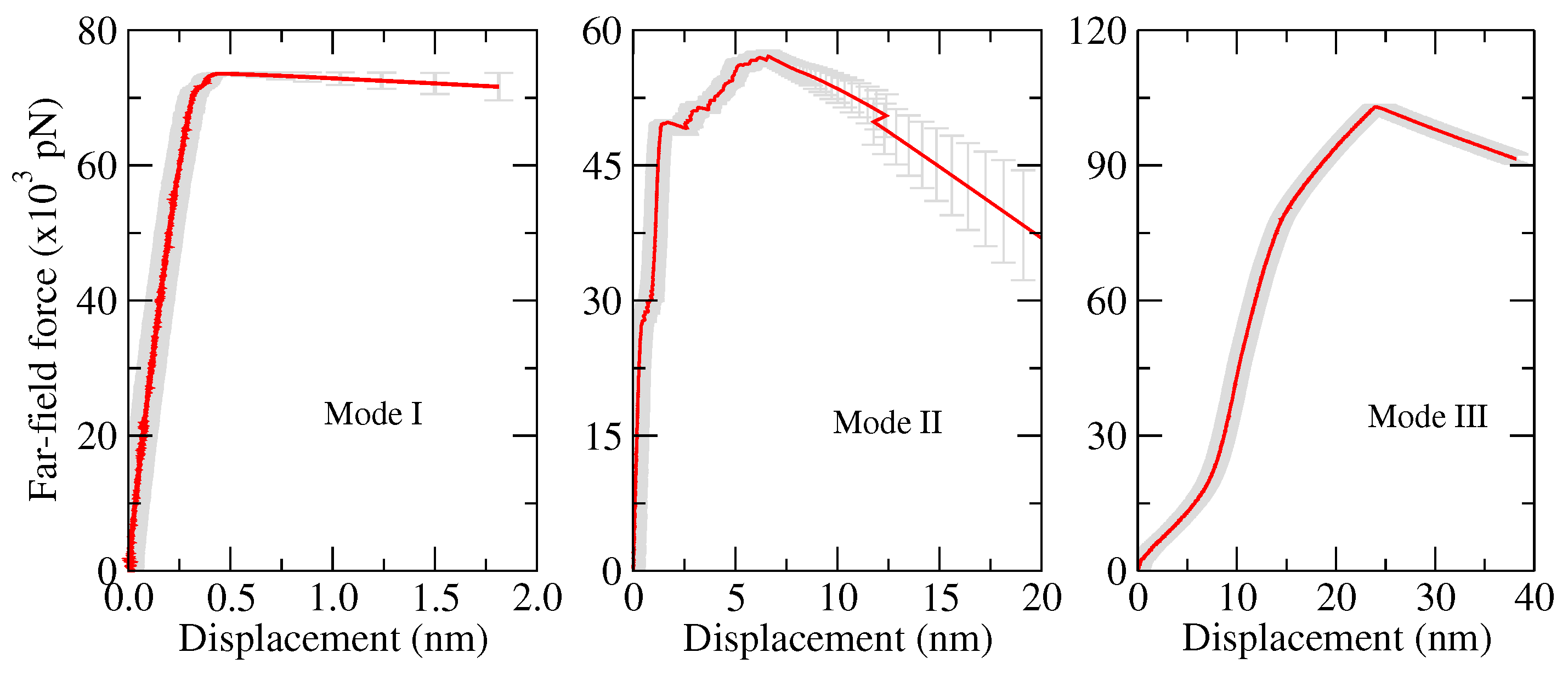
3. Fracture Mechanical Properties
- Opening mode (Mode I): tensile stresses are applied perpendicular to the crack plane.
- Shearing mode (Mode II): shear stresses act along the crack plane and normal to the crack front.
- Tearing mode (Mode III): shear stresses act along the crack plane and parallel to the crack front.
3.1. Griffith’s Criteria
3.2. Crack-Tip Opening Displacement (CTOD) Approach
3.3. J-Integral
3.4. Other Criteria
3.5. Milestones in MD Simulations to Study Brittle Fracture
4. Brittle Materials—Advances in Fracture Study at Nanoscale
4.1. Carbon Nanotube
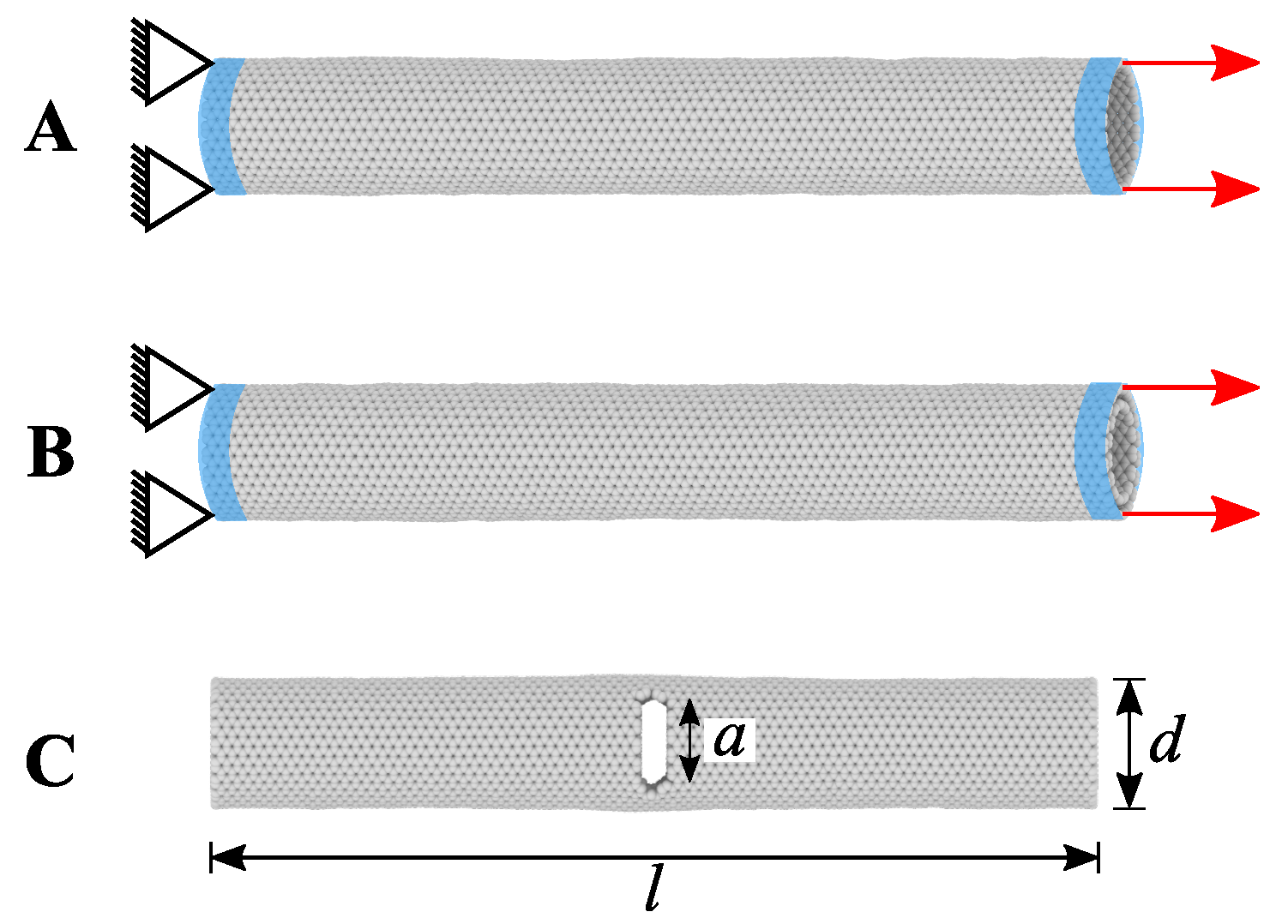
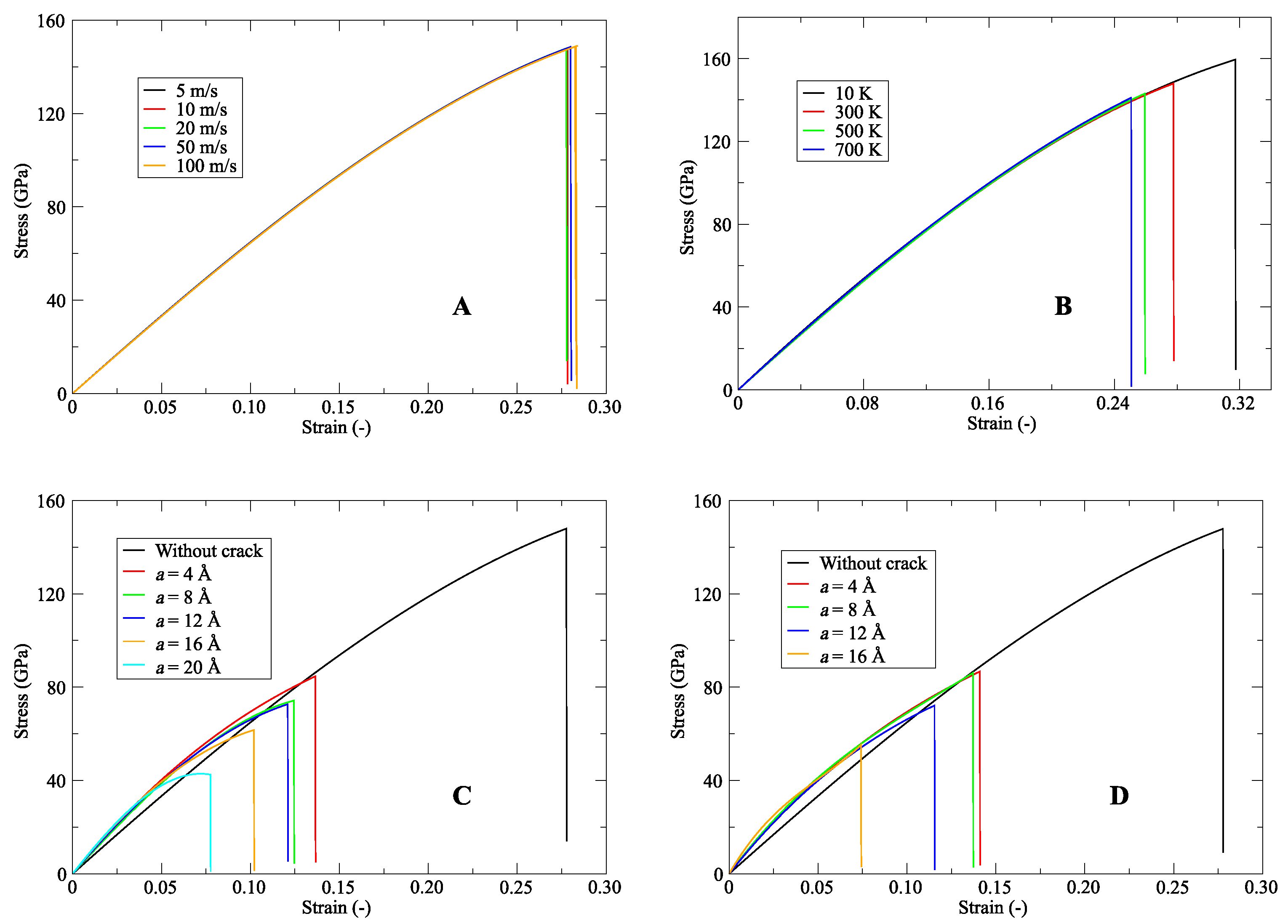
4.2. Literature Review of Carbon Nanotubes: Past and Recent Advances
- ■
- the scope of collapse strains of defect-free CNTs was in the range of 10 to 16%,
- ■
- a decrease in the strength was obtained in connection with defect occurrence,
- ■
- fracture strain showed strong dependency on the inflection threshold in the potential, and
- ■
- chirality seemed to have just a moderate influence on CNTs strength.
4.3. Graphene
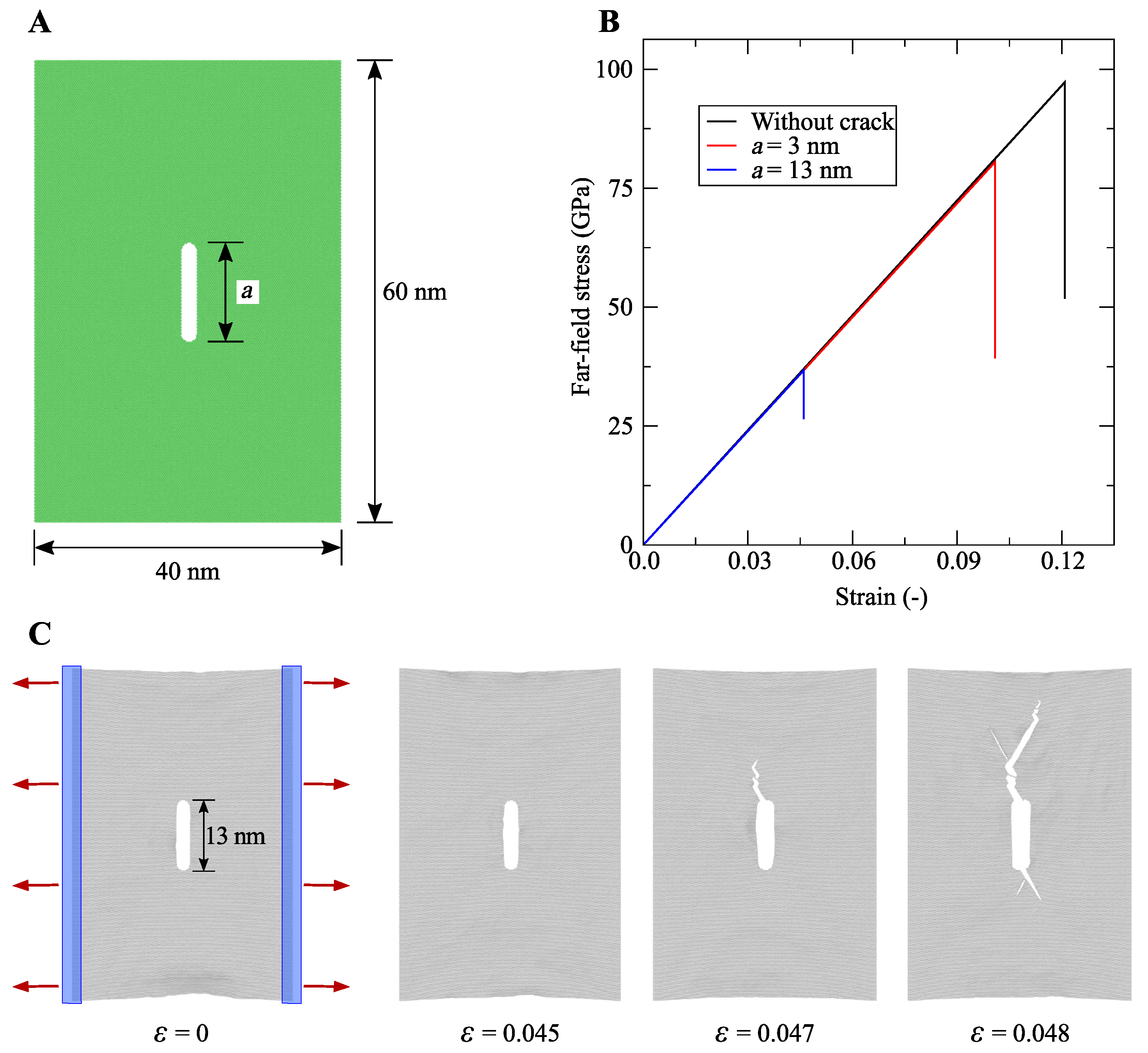
4.4. Literature Review of Graphene: Past and Recent Advances
4.5. Silicon Carbide
4.6. Literature Review of Silicon Carbide: Past and Recent Advances
4.7. Amorphous Silica
4.8. Literature Review of Amorphous Silica: Past and Recent Advances
4.9. Calcium Carbonate
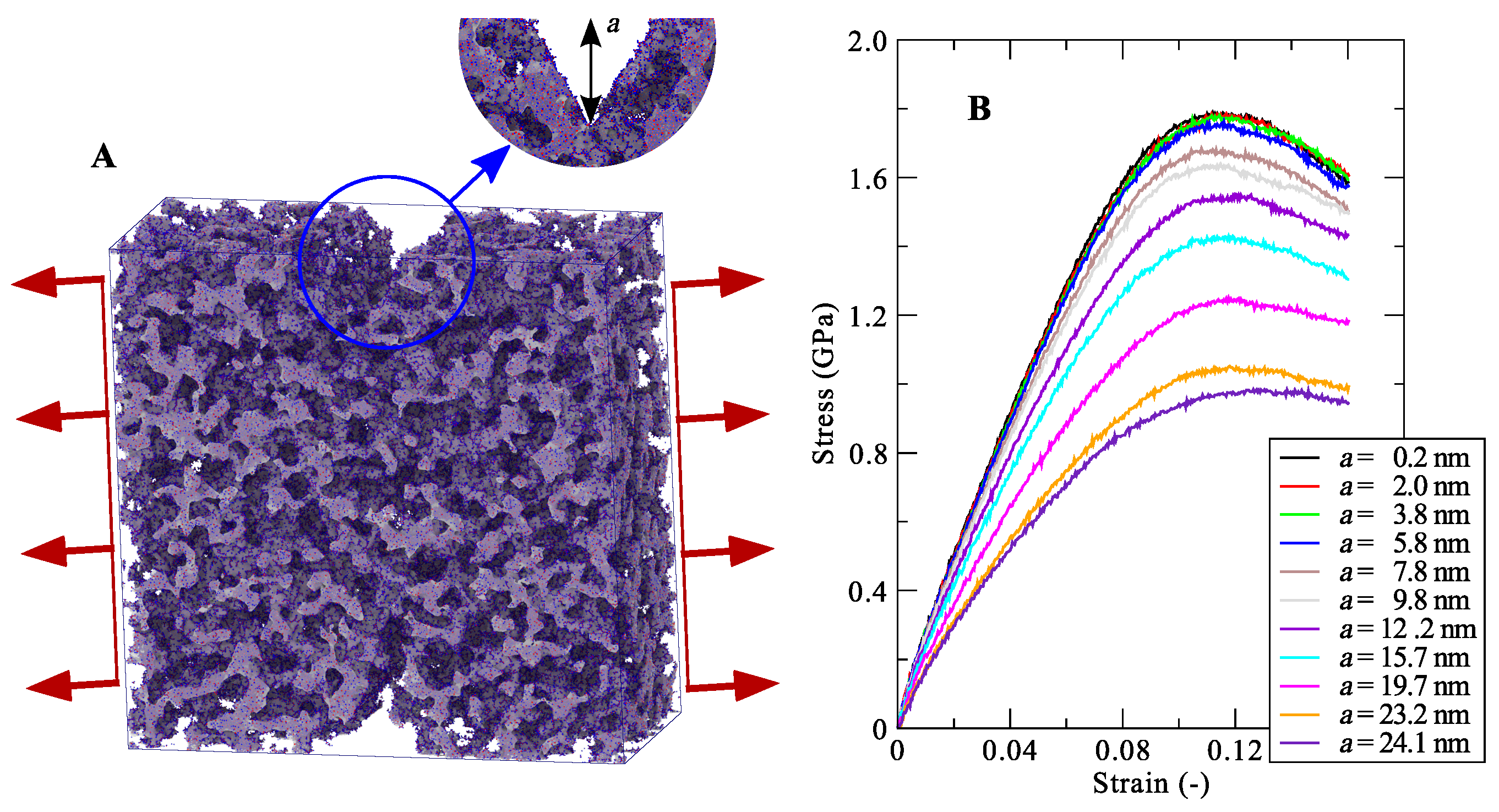
4.10. Silica Aerogel
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wallin, K. Small Specimen Fracture Toughness Characterization-State of the Art and Beyond. In Proceedings of the ICF 9, Sydney, Australia, 14 October 1997; Karihaloo, B.L., Mai, Y.-W., Ripley, M.I., Ritchie, R.O., Eds.; Pergamon: Oxford, UK, 1997. [Google Scholar]
- Zerbst, U.; Mädler, K.; Hintze, H. Fracture mechanics in railway applications—An overview. Eng. Fract. Mech. 2005, 72, 163–194. [Google Scholar] [CrossRef]
- Taylor, D. Fracture Mechanics in Biology and Medicine. Procedia Struct. Integr. 2016, 2, 42–49. [Google Scholar] [CrossRef][Green Version]
- Council, N.R. Rock Fractures and Fluid Flow: Contemporary Understanding and Applications; National Academies Press: Washington, DC, USA, 1996. [Google Scholar]
- Delale, F.; Erdogan, F. The crack problem for a nonhomogeneous plane. J. Appl. Mech. 1983, 50, 609–614. [Google Scholar] [CrossRef]
- Gol’Dstein, R.; Salganik, R. Brittle fracture of solids with arbitrary cracks. Int. J. Fract. 1974, 10, 507–523. [Google Scholar] [CrossRef]
- Müller, J.J.; Bitzek, E.; Janisch, R.; ul Hassan, H.; Hartmaier, A. Fracture ab initio: A force-based scaling law for atomistically informed continuum models. J. Mater. Res. 2018, 33, 3750–3761. [Google Scholar] [CrossRef]
- Hicks, J. Welded Joint Design; Industrial Press Inc.: New York, NY, USA, 1999. [Google Scholar]
- Berto, F.; Marsavina, L.; Ayatollahi, M.R.; Panin, S.V.; Tserpes, K.I. Brittle or quasi-brittle fracture of engineering materials 2016. Adv. Mater. Sci. Eng. 2016, 2016, 7094298. [Google Scholar] [CrossRef]
- Valintine, A.P.; Hull, D. Effect of temperature on the brittle fracture of polycrystalline tungsten. J. Less Common Met. 1969, 17, 353–361. [Google Scholar] [CrossRef]
- Maccagno, T.; Knott, J. The low temperature brittle fracture behaviour of steel in mixed modes I and II. Eng. Fract. Mech. 1991, 38, 111–128. [Google Scholar] [CrossRef]
- Scibetta, M. Mixed mode brittle fracture and fatigue in support to structural integrity assessment of laminar flaws. Procedia Mater. Sci. 2014, 3, 847–854. [Google Scholar] [CrossRef][Green Version]
- Rafii-Tabar, H. Nanoscopic modelling of the adhesion, indentation and fracture characteristics of metallic systems via molecular dynamics simulations. In Mesoscopic Dynamics of Fracture; Springer: Berlin, Heidelberg, Germany, 1998; pp. 36–48. [Google Scholar]
- Abraham, F.F.; Broughton, J.Q.; Bernstein, N.; Kaxiras, E. Spanning the continuum to quantum length scales in a dynamic simulation of brittle fracture. Europhys. Lett. 1998, 44, 783–787. [Google Scholar] [CrossRef]
- Ashurst, W.T.; Hoover, W.G. Microscopic fracture studies in the two-dimensional triangular lattice. Phys. Rev. B 1976, 14, 1465. [Google Scholar] [CrossRef]
- Bitzek, E.; Kermode, J.R.; Gumbsch, P. Atomistic aspects of fracture. Int. J. Fract. 2015, 191, 13–30. [Google Scholar] [CrossRef]
- Patil, S.P.; Rege, A.; Itskov, M.; Markert, B. Fracture of silica aerogels: An all-atom simulation study. J. Non-Cryst. Solids 2018, 498, 125–129. [Google Scholar] [CrossRef]
- Swadener, J.; Baskes, M.; Nastasi, M. Molecular dynamics simulation of brittle fracture in silicon. Phys. Rev. Lett. 2002, 89, 085503. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, H.; Kalia, R.K.; Nakano, A.; Vashishta, P.; Branicio, P.S.; Shimojo, F. Brittle dynamic fracture of crystalline cubic silicon carbide (3C-SiC) via molecular dynamics simulation. J. Appl. Phys. 2005, 98, 103524. [Google Scholar] [CrossRef]
- Karimi, M.; Roarty, T.; Kaplan, T. Molecular dynamics simulations of crack propagation in Ni with defects. Modell. Simul. Mater. Sci. Eng. 2006, 14, 1409. [Google Scholar] [CrossRef]
- Kumar, S.; Li, X.; Haque, A.; Gao, H. Is stress concentration relevant for nanocrystalline metals? Nano Lett. 2011, 11, 2510–2516. [Google Scholar] [CrossRef]
- Cheng, S.H.; Sun, C. Size-dependent fracture toughness of nanoscale structures: Crack-tip stress approach in molecular dynamics. J. Nanomech. Micromech. 2013, 4, A4014001. [Google Scholar] [CrossRef]
- Zhang, P.; Ma, L.; Fan, F.; Zeng, Z.; Peng, C.; Loya, P.E.; Liu, Z.; Gong, Y.; Zhang, J.; Zhang, X.; et al. Fracture toughness of graphene. Nat. Commun. 2014, 5, 3782. [Google Scholar] [CrossRef]
- Dewapriya, M.; Rajapakse, R.; Phani, A. Atomistic and continuum modelling of temperature-dependent fracture of graphene. Int. J. Fract. 2014, 187, 199–212. [Google Scholar] [CrossRef]
- Yin, H.; Qi, H.J.; Fan, F.; Zhu, T.; Wang, B.; Wei, Y. Griffith criterion for brittle fracture in graphene. Nano Lett. 2015, 15, 1918–1924. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.P.; Heider, Y.; Padilla, C.A.H.; Cruz-Chú, E.R.; Markert, B. A comparative molecular dynamics-phase- field modeling approach to brittle fracture. Comput. Methods Appl. Mech. Eng. 2016, 312, 117–129. [Google Scholar]
- Cruz-Chu, E.R.; Xiao, S.; Patil, S.P.; Gkagkas, K.; Gräter, F. Organic Filling Mitigates Flaw-Sensitivity of Nanoscale Aragonite. ACS Biomater. Sci. Eng. 2017, 3, 260–268. [Google Scholar] [CrossRef]
- Ferdous, S.F.; Adnan, A. Mode-I Fracture Toughness Prediction of Diamond at the Nanoscale. J. Nanomech. Micromech. 2017, 7, 04017010. [Google Scholar]
- Rapaport, D.C.; Rapaport, D.C.R. The Art of Molecular Dynamics Simulation; Cambridge University Press: Cambridge, England, UK, 2004. [Google Scholar]
- Daw, M.S.; Baskes, M.I. Embedded-atom method: Derivation and application to impurities, surfaces, and other defects in metals. Phys. Rev. B 1984, 29, 6443. [Google Scholar]
- Baskes, M. Modified embedded-atom potentials for cubic materials and impurities. Phys. Rev. B 1992, 46, 2727. [Google Scholar] [CrossRef] [PubMed]
- Foiles, S.M. Embedded-atom and related methods for modeling metallic systems. MRS Bull. 1996, 21, 24–28. [Google Scholar]
- Vashishta, P.; Kalia, R.K.; Rino, J.P.; Ebbsjö, I. Interaction potential for SiO2: A molecular-dynamics study of structural correlations. Phys. Rev. B 1990, 41, 12197–12209. [Google Scholar] [CrossRef] [PubMed]
- Vashishta, P.; Kalia, R.K.; Nakano, A.; Li, W.; Ebbsjö, I. Molecular dynamics methods and large-scale simulations of amorphous materials. Amor. Insul. Semiconduct. 1996, 33, 151–213. [Google Scholar]
- Patil, S.P.; Rege, A.; Sagardas; Itskov, M.; Markert, B. Mechanics of Nanostructured Porous Silica Aerogel Resulting from Molecular Dynamics Simulations. J. Phys. Chem. B 2017, 121, 5660–5668. [Google Scholar]
- Stuart, S.J.; Tutein, A.B.; Harrison, J.A. A reactive potential for hydrocarbons with intermolecular interactions. J. Chem. Phys. 2000, 112, 6472–6486. [Google Scholar] [CrossRef]
- Brenner, D.W.; Shenderova, O.A.; Harrison, J.A.; Stuart, S.J.; Ni, B.; Sinnott, S.B. A second-generation reactive empirical bond order (REBO) potential energy expression for hydrocarbons. J. Phys. Condens. Matter 2002, 14, 783. [Google Scholar] [CrossRef]
- Belytschko, T.; Xiao, S.; Schatz, G.; Ruoff, R. Atomistic simulations of nanotube fracture. Phys. Rev. B 2002, 65, 235430. [Google Scholar] [CrossRef]
- Zhao, H.; Min, K.; Aluru, N. Size and chirality dependent elastic properties of graphene nanoribbons under uniaxial tension. Nano Lett. 2009, 9, 3012–3015. [Google Scholar] [CrossRef] [PubMed]
- Dewapriya, M.; Rajapakse, R. Molecular dynamics simulations and continuum modeling of temperature and strain rate dependent fracture strength of graphene with vacancy defects. J. Appl. Mech. 2014, 81, 081010. [Google Scholar] [CrossRef]
- Hockney, R.; Eastwood., J. Computer Simulation Using Particles; A. Hilger: Philadelphia, PA, USA, 1988. [Google Scholar]
- Verlet, L. Computer “Experiments” on Classical Fluids. I. Thermodynamical Properties of Lennard-Jones Molecules. Phys. Rev. 1967, 159, 98–103. [Google Scholar] [CrossRef]
- Anderson, T.L. Fracture Mechanics: Fundamentals and Applications, 3rd ed.; CRC: Boca Raton, FL, USA, 2005. [Google Scholar]
- Broberg, K.B. Cracks and Fracture; Academic Press: San Diego, CA, USA, 1999. [Google Scholar]
- Zhu, X.K.; Joyce, J.A. Review of fracture toughness (G, K, J, CTOD, CTOA) testing and standardization. Eng. Fract. Mech. 2012, 85, 1–46. [Google Scholar] [CrossRef]
- Sun, C.T.; Jin, Z.H. Fracture Mechanics; Academic Press: Boston, MA, USA, 2012. [Google Scholar]
- Griffith, A.A. VI. The phenomena of rupture and flow in solids. Phil. Trans. R. Soc. Lond. A 1921, 221, 163–198. [Google Scholar] [CrossRef]
- Perez, N. Fracture Mechanics; Kluwer Academic Publisher Group: Boston, MA, USA, 2004. [Google Scholar]
- Irwin, G.R. Analysis of stresses and strains near the end of a crack traversing a plate. J. Appl. Mech. 1957, 24, 361–364. [Google Scholar]
- Adnan, A.; Sun, C. Evolution of nanoscale defects to planar cracks in a brittle solid. J. Mech. Phys. Solids 2010, 58, 983–1000. [Google Scholar] [CrossRef]
- Sakib, A.N.; Adnan, A. On the size-dependent critical stress intensity factor of confined brittle nanofilms. Eng. Fract. Mech. 2012, 86, 13–22. [Google Scholar] [CrossRef]
- Sakib, A.N.; De, N.N.; Adnan, A. Shear fracture of confined NaCl nanofilms. Comput. Mater. Sci. 2013, 68, 271–279. [Google Scholar] [CrossRef]
- Kravchenko, S.G.; Kravchenko, O.G.; Sun, C. A two-parameter fracture mechanics model for fatigue crack growth in brittle materials. Eng. Fract. Mech. 2014, 119, 132–147. [Google Scholar] [CrossRef]
- Shimada, T.; Ouchi, K.; Chihara, Y.; Kitamura, T. Breakdown of continuum fracture mechanics at the nanoscale. Sci. Rep. 2015, 5, 8596. [Google Scholar] [CrossRef] [PubMed]
- Wells, A. Application of fracture mechanics at and beyond general yielding. Br. Weld. J. 1963, 10, 563–570. [Google Scholar]
- Dugdale, D.S. Yielding of steel sheets containing slits. J. Mech. Phys. Solids 1960, 8, 100–104. [Google Scholar] [CrossRef]
- Xia, X.X.; Wang, W.H.; Greer, A.L. Plastic zone at crack tip: A nanolab for formation and study of metallic glassy nanostructures. J. Mater. Res. 2009, 24, 2986–2992. [Google Scholar] [CrossRef]
- Kelling, A.; Mangipudi, K.R.; Knorr, I.; Liese, T.; Krebs, H.U.; Volkert, C.A. Investigating fracture of nanoscale metal–ceramic multilayers in the transmission electron microscope. Scr. Mater. 2016, 115, 42–45. [Google Scholar] [CrossRef]
- Matsumoto, R.; Nakagaki, M.; Nakatani, A.; Kitagawa, H. Molecular-dynamics study on crack growth behavior relevant to crystal nucleation in amorphous metal. Comput. Model. Eng. Sci. 2005, 9, 75. [Google Scholar]
- Rice, J.R. A path independent integral and the approximate analysis of strain concentration by notches and cracks. J. Appl. Mech. 1968, 35, 379–386. [Google Scholar] [CrossRef]
- Cao, C.; Mukherjee, S.; Howe, J.Y.; Perovic, D.D.; Sun, Y.; Singh, C.V.; Filleter, T. Nonlinear fracture toughness measurement and crack propagation resistance of functionalized graphene multilayers. Sci. Adv. 2018, 4, eaao7202. [Google Scholar] [CrossRef]
- Jin, Y.; Yuan, F. Atomistic simulations of J-integral in 2D graphene nanosystems. J. Nanosci. Nanotechnol. 2005, 5, 2099–2107. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, K.; Nakatani, A.; Sugiyama, Y.; Kitagawa, H. Molecular dynamics study on mechanical properties and fracture in amorphous metal. AIAA J. 2000, 38, 695–701. [Google Scholar] [CrossRef]
- Khare, R.; Mielke, S.L.; Paci, J.T.; Zhang, S.; Ballarini, R.; Schatz, G.C.; Belytschko, T. Coupled quantum mechanical/molecular mechanical modeling of the fracture of defective carbon nanotubes and graphene sheets. Phys. Rev. B 2007, 75, 075412. [Google Scholar] [CrossRef]
- Neuber, H. Theory of Notch Stresses; Splinger: Berlin, Germany, 1958. [Google Scholar]
- Novozhilov, V. On a necessary and sufficient criterion for brittle strength. J. Appl. Math. Mech. 1969, 33, 201–210. [Google Scholar] [CrossRef]
- Rivlin, R.; Thomas, A.G. Rupture of rubber. I. Characteristic energy for tearing. J. Polym. Sci. Part A Polym. Chem. 1953, 10, 291–318. [Google Scholar] [CrossRef]
- Cotterell, B. The past, present, and future of fracture mechanics. Eng. Fract. Mech. 2002, 69, 533–553. [Google Scholar] [CrossRef]
- Kalthoff, J.; Shockey, D. Instability of cracks under impulse loads. J. Appl. Phys. 1977, 48, 986–993. [Google Scholar] [CrossRef]
- Decelis, B.; Argon, A.S.; Yip, S. Molecular dynamics simulation of crack tip processes in alpha-iron and copper. J. Appl. Phys. 1983, 54, 4864–4878. [Google Scholar] [CrossRef]
- Smith, R.W.; Was, G.S. Application of molecular dynamics to the study of hydrogen embrittlement in Ni-Cr-Fe alloys. Phys. Rev. B 1989, 40, 10322. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, T.; Tang, Q. Brittle and ductile fracture at the atomistic crack tip in copper crystals. Scr. Metall. Mater. 1995, 33, 267–274. [Google Scholar] [CrossRef][Green Version]
- Swiler, T.P.; Simmons, J.H.; Wright, A.C. Molecular dynamics study of brittle fracture in silica glass and cristobalite. J. Non-Cryst. Solids 1995, 182, 68–77. [Google Scholar] [CrossRef]
- Abraham, F.F.; Brodbeck, D.; Rudge, W.E.; Xu, X. A molecular dynamics investigation of rapid fracture mechanics. J. Mech. Phys. Solids 1997, 45, 1595–1619. [Google Scholar] [CrossRef]
- Holland, D.; Marder, M. Ideal brittle fracture of silicon studied with molecular dynamics. Phys. Rev. Lett. 1998, 80, 746. [Google Scholar] [CrossRef]
- Latapie, A.; Farkas, D. Molecular dynamics investigation of the fracture behavior of nanocrystalline α-Fe. Phys. Rev. B 2004, 69, 134110. [Google Scholar] [CrossRef]
- Mielke, S.L.; Troya, D.; Zhang, S.; Li, J.L.; Xiao, S.; Car, R.; Ruoff, R.S.; Schatz, G.C.; Belytschko, T. The role of vacancy defects and holes in the fracture of carbon nanotubes. Chem. Phys. Lett. 2004, 390, 413–420. [Google Scholar] [CrossRef]
- Zhou, X.; Zimmerman, J.; Reedy, E., Jr.; Moody, N. Molecular dynamics simulation based cohesive surface representation of mixed mode fracture. Mech. Mater. 2008, 40, 832–845. [Google Scholar] [CrossRef]
- Kang, K.; Cai, W. Size and temperature effects on the fracture mechanisms of silicon nanowires: Molecular dynamics simulations. Int. J. Plast. 2010, 26, 1387–1401. [Google Scholar] [CrossRef]
- Terdalkar, S.S.; Huang, S.; Yuan, H.; Rencis, J.J.; Zhu, T.; Zhang, S. Nanoscale fracture in graphene. Chem. Phys. Lett. 2010, 494, 218–222. [Google Scholar] [CrossRef]
- Zhang, B.; Mei, L.; Xiao, H. Nanofracture in graphene under complex mechanical stresses. Appl. Phys. Lett. 2012, 101, 121915. [Google Scholar] [CrossRef]
- Tang, D.M.; Ren, C.L.; Wang, M.S.; Wei, X.; Kawamoto, N.; Liu, C.; Bando, Y.; Mitome, M.; Fukata, N.; Golberg, D. Mechanical properties of Si nanowires as revealed by in situ transmission electron microscopy and molecular dynamics simulations. Nano Lett. 2012, 12, 1898–1904. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, Y.; Lin, L.; He, Y.; Huang, H. Fracture Strain of SiC Nanowires and Direct Evidence of Electron-Beam Induced Amorphisation in the Strained Nanowires. Small 2015, 11, 1672–1676. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiang, S.; Zhu, X.; Zhao, Y. Mechanisms of crack propagation in nanoscale single crystal, bicrystal and tricrystal nickels based on molecular dynamics simulation. Results Phys. 2017, 7, 1722–1733. [Google Scholar] [CrossRef]
- Sumigawa, T.; Shimada, T.; Tanaka, S.; Unno, H.; Ozaki, N.; Ashida, S.; Kitamura, T. Griffith Criterion for Nanoscale Stress Singularity in Brittle Silicon. ACS Nano 2017, 11, 6271–6276. [Google Scholar] [CrossRef]
- Bao, H.; Huang, Y.; Yang, Z.; Sun, Y.; Bai, Y.; Miao, Y.; Chu, P.K.; Xu, K.; Ma, F. Molecular Dynamics Simulation of Nanocrack Propagation in Single-Layer MoS2 Nanosheets. J. Phys. Chem. C 2018, 122, 1351–1360. [Google Scholar] [CrossRef]
- Iijima, S. Helical microtubules of graphitic carbon. Nature 1991, 354, 56. [Google Scholar] [CrossRef]
- Thostenson, E.T.; Ren, Z.; Chou, T.W. Advances in the science and technology of carbon nanotubes and their composites: A review. Compos. Sci. Technol. 2001, 61, 1899–1912. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Haile, J.; Johnston, I.; Mallinckrodt, A.J.; McKay, S. Molecular dynamics simulation: Elementary methods. Comput. Phys. 1993, 7. [Google Scholar] [CrossRef]
- Wei, C.; Cho, K.; Srivastava, D. Tensile strength of carbon nanotubes under realistic temperature and strain rate. Phys. Rev. B 2003, 67, 115407. [Google Scholar] [CrossRef]
- Yakobson, B.; Campbell, M.; Brabec, C.; Bernholc, J. High strain rate fracture and C-chain unraveling in carbon nanotubes. Comput. Mater. Sci. 1997, 8, 341–348. [Google Scholar] [CrossRef]
- Li, C.; Chou, T.W. A structural mechanics approach for the analysis of carbon nanotubes. Int. J. Solids Struct. 2003, 40, 2487–2499. [Google Scholar] [CrossRef]
- Liew, K.; He, X.; Wong, C. On the study of elastic and plastic properties of multi-walled carbon nanotubes under axial tension using molecular dynamics simulation. Acta Mater. 2004, 52, 2521–2527. [Google Scholar] [CrossRef]
- Buehler, M.J. Mesoscale modeling of mechanics of carbon nanotubes: Self-assembly, self-folding, and fracture. J. Mater. Res. 2006, 21, 2855–2869. [Google Scholar] [CrossRef]
- Han, Y.; Elliott, J. Molecular dynamics simulations of the elastic properties of polymer/carbon nanotube composites. Comput. Mater. Sci. 2007, 39, 315–323. [Google Scholar] [CrossRef]
- Nikiforov, I.; Zhang, D.B.; James, R.D.; Dumitrică, T. Wavelike rippling in multiwalled carbon nanotubes under pure bending. Appl. Phys. Lett. 2010, 96, 123107. [Google Scholar] [CrossRef]
- Neyts, E.C.; van Duin, A.C.; Bogaerts, A. Changing chirality during single-walled carbon nanotube growth: A reactive molecular dynamics/Monte Carlo study. J. Am. Chem. Soc. 2011, 133, 17225–17231. [Google Scholar] [CrossRef]
- Vijayaraghavan, V.; Wong, C. Temperature, defect and size effect on the elastic properties of imperfectly straight carbon nanotubes by using molecular dynamics simulation. Comput. Mater. Sci. 2013, 71, 184–191. [Google Scholar] [CrossRef]
- Li, M.; Kang, Z.; Li, R.; Meng, X.; Lu, Y. A molecular dynamics study on tensile strength and failure modes of carbon nanotube junctions. J. Phys. D Appl. Phys. 2013, 46, 495301. [Google Scholar] [CrossRef]
- Yang, L.; Greenfeld, I.; Wagner, H.D. Toughness of carbon nanotubes conforms to classic fracture mechanics. Sci. Adv. 2016, 2, e1500969. [Google Scholar] [CrossRef]
- Arash, B.; Wang, Q.; Varadan, V. Mechanical properties of carbon nanotube/polymer composites. Sci. Rep. 2014, 4, 6479. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.; Subramanian, N.; Koo, B.; Chattopadhyay, A. Multiscale damage analysis of carbon nanotube nanocomposite using a continuum damage mechanics approach. J. Compos. Mater. 2017, 51, 847–858. [Google Scholar] [CrossRef]
- Hossain, M.; Hao, T.; Silverman, B. Stillinger–Weber potential for elastic and fracture properties in graphene and carbon nanotubes. J. Phys. Condens. Matter 2018, 30, 055901. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, S.; Wang, Q.; Xing, M. Enhancement of fracture properties of polymer composites reinforced by carbon nanotubes: A molecular dynamics study. Carbon 2018, 129, 504–509. [Google Scholar] [CrossRef]
- Wang, S.; Gao, E.; Xu, Z. Interfacial failure boosts mechanical energy dissipation in carbon nanotube films under ballistic impact. Carbon 2019, 146, 139–146. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Ni, Z.; Bu, H.; Zou, M.; Yi, H.; Bi, K.; Chen, Y. Anisotropic mechanical properties of graphene sheets from molecular dynamics. Phys. B Condens. Matter 2010, 405, 1301–1306. [Google Scholar] [CrossRef]
- Zhao, H.; Aluru, N. Temperature and strain-rate dependent fracture strength of graphene. J. Appl. Phys. 2010, 108, 064321. [Google Scholar] [CrossRef]
- Pei, Q.X.; Zhang, Y.W.; Shenoy, V.B. Mechanical properties of methyl functionalized graphene: A molecular dynamics study. Nanotechnology 2010, 21, 115709. [Google Scholar] [CrossRef] [PubMed]
- Min, K.; Aluru, N. Mechanical properties of graphene under shear deformation. Appl. Phys. Lett. 2011, 98, 013113. [Google Scholar] [CrossRef]
- Wang, M.; Yan, C.; Ma, L.; Hu, N.; Chen, M. Effect of defects on fracture strength of graphene sheets. Comput. Mater. Sci. 2012, 54, 236–239. [Google Scholar] [CrossRef]
- Xu, M.; Tabarraei, A.; Paci, J.T.; Oswald, J.; Belytschko, T. A coupled quantum/continuum mechanics study of graphene fracture. Int. J. Fract. 2012, 173, 163–173. [Google Scholar] [CrossRef]
- Zhang, Y.; Pei, Q.; Wang, C. Mechanical properties of graphynes under tension: A molecular dynamics study. Appl. Phys. Lett. 2012, 101, 081909. [Google Scholar] [CrossRef]
- Yang, Y.; Xu, X. Mechanical properties of graphyne and its family—A molecular dynamics investigation. Comput. Mater. Sci. 2012, 61, 83–88. [Google Scholar] [CrossRef]
- Zhang, H.; Duan, Z.; Zhang, X.; Liu, C.; Zhang, J.; Zhao, J. Strength and fracture behavior of graphene grain boundaries: Effects of temperature, inflection, and symmetry from molecular dynamics. Phys. Chem. Chem. Phys. 2013, 15, 11794–11799. [Google Scholar] [CrossRef]
- Ansari, R.; Sahmani, S. Prediction of biaxial buckling behavior of single-layered graphene sheets based on nonlocal plate models and molecular dynamics simulations. Appl. Math. Modell. 2013, 37, 7338–7351. [Google Scholar] [CrossRef]
- Rajasekaran, G.; Parashar, A. Molecular dynamics study on the mechanical response and failure behaviour of graphene: Performance enhancement via 5–7–7–5 defects. RSC Adv. 2016, 6, 26361–26373. [Google Scholar] [CrossRef]
- Shekhawat, A.; Ritchie, R.O. Toughness and strength of nanocrystalline graphene. Nat. Commun. 2016, 7, 10546. [Google Scholar] [CrossRef]
- Qin, H.; Sun, Y.; Liu, J.Z.; Li, M.; Liu, Y. Negative Poisson’s ratio in rippled graphene. Nanoscale 2017, 9, 4135–4142. [Google Scholar] [CrossRef] [PubMed]
- Winczewski, S.; Shaheen, M.Y.; Rybicki, J. Interatomic potential suitable for the modeling of penta-graphene: Molecular statics/molecular dynamics studies. Carbon 2018, 126, 165–175. [Google Scholar] [CrossRef]
- Awasthi, A.P.; Lagoudas, D.C.; Hammerand, D.C. Modeling of graphene–polymer interfacial mechanical behavior using molecular dynamics. Model. Simul. Mater. Sci. Eng. 2008, 17, 015002. [Google Scholar] [CrossRef]
- Alian, A.; Dewapriya, M.; Meguid, S. Molecular dynamics study of the reinforcement effect of graphene in multilayered polymer nanocomposites. Mater. Des. 2017, 124, 47–57. [Google Scholar] [CrossRef]
- Patil, S.P. Nanoindentation of Graphene-Reinforced Silica Aerogel: A Molecular Dynamics Study. Molecules 2019, 24, 1336. [Google Scholar] [CrossRef] [PubMed]
- Hansen-Dörr, A.C.; Wilkens, L.; Croy, A.; Dianat, A.; Cuniberti, G.; Kästner, M. Combined molecular dynamics and phase-field modelling of crack propagation in defective graphene. Comput. Mater. Sci. 2019, 163, 117–126. [Google Scholar] [CrossRef]
- Zhang, R.; Li, H.; Zhang, Z.; Wang, Z.; Zhou, S.; Wang, Z.; Li, T.; Liu, J.; Fu, D. Graphene synthesis on SiC: Reduced graphitization temperature by C-cluster and Ar-ion implantation. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms. 2015, 356, 99–102. [Google Scholar] [CrossRef]
- Rountree, C.L.; Kalia, R.K.; Lidorikis, E.; Nakano, A.; Van Brutzel, L.; Vashishta, P. Atomistic aspects of crack propagation in brittle materials: Multimillion atom molecular dynamics simulations. Annu. Rev. Mater. Res. 2002, 32, 377–400. [Google Scholar] [CrossRef]
- Szlufarska, I.; Kalia, R.K.; Nakano, A.; Vashishta, P. Nanoindentation-induced amorphization in silicon carbide. Appl. Phys. Lett. 2004, 85, 378–380. [Google Scholar] [CrossRef]
- Kermode, J.; Albaret, T.; Sherman, D.; Bernstein, N.; Gumbsch, P.; Payne, M.; Csányi, G.; De Vita, A. Low-speed fracture instabilities in a brittle crystal. Nature 2008, 455, 1224. [Google Scholar] [CrossRef]
- Pan, H.; Si, X. Molecular dynamics simulations of diameter dependence tensile behavior of silicon carbide nanotubes. Phys. B Condens. Matter 2009, 404, 1809–1812. [Google Scholar] [CrossRef]
- Makeev, M.A.; Srivastava, D.; Menon, M. Silicon carbide nanowires under external loads: An atomistic simulation study. Phys. Rev. B 2006, 74, 165303. [Google Scholar] [CrossRef]
- Wang, J.; Lu, C.; Wang, Q.; Xiao, P.; Ke, F.; Bai, Y.; Shen, Y.; Liao, X.; Gao, H. Influence of microstructures on mechanical behaviours of SiC nanowires: A molecular dynamics study. Nanotechnology 2011, 23, 025703. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pizzagalli, L.; Godet, J.; Guénolé, J.; Brochard, S.; Holmstrom, E.; Nordlund, K.; Albaret, T. A new parametrization of the Stillinger–Weber potential for an improved description of defects and plasticity of silicon. J. Phys. Condens. Matter 2013, 25, 055801. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Luo, X.; Reuben, R.L. Molecular dynamics simulation model for the quantitative assessment of tool wear during single point diamond turning of cubic silicon carbide. Comput. Mater. Sci. 2012, 51, 402–408. [Google Scholar] [CrossRef]
- Xiao, G.; To, S.; Zhang, G. Molecular dynamics modelling of brittle–ductile cutting mode transition: Case study on silicon carbide. Int. J. Mach. Tools Manuf. 2015, 88, 214–222. [Google Scholar] [CrossRef]
- Chavoshi, S.Z.; Luo, X. Molecular dynamics simulation study of deformation mechanisms in 3C–SiC during nanometric cutting at elevated temperatures. Mater. Sci. Eng. A 2016, 654, 400–417. [Google Scholar] [CrossRef]
- Wang, J.; Fang, F.; Zhang, X. Nanometric cutting of silicon with an amorphous-crystalline layered structure: A molecular dynamics study. Nanoscale Res. Lett. 2017, 12, 41. [Google Scholar] [CrossRef]
- Liu, Y.; Li, B.; Kong, L. Atomistic insights on the nanoscale single grain scratching mechanism of silicon carbide ceramic based on molecular dynamics simulation. AIP Adv. 2018, 8, 035109. [Google Scholar] [CrossRef]
- Li, W.; Hahn, E.N.; Yao, X.; Germann, T.C.; Zhang, X. Shock induced damage and fracture in SiC at elevated temperature and high strain rate. Acta Mater. 2019, 167, 51–70. [Google Scholar] [CrossRef]
- Muralidharan, K.; Simmons, J.; Deymier, P.; Runge, K. Molecular dynamics studies of brittle fracture in vitreous silica: Review and recent progress. J. Non-Cryst. Solids 2005, 351, 1532–1542. [Google Scholar] [CrossRef]
- Ochoa, R.; Swiler, T.P.; Simmons, J.H. Molecular dynamics studies of brittle failure in silica: Effect of thermal vibrations. J. Non-Cryst. Solids 1991, 128, 57–68. [Google Scholar] [CrossRef]
- Nakano, A.; Kalia, R.K.; Vashishta, P. Growth of pore interfaces and roughness of fracture surfaces in porous silica: Million particle molecular-dynamics simulations. Phys. Rev. Lett. 1994, 73, 2336. [Google Scholar] [CrossRef] [PubMed]
- Van Brutzel, L.; Rountree, C.L.; Kalia, R.K.; Nakano, A.; Vashishta, P. Dynamic fracture mechanisms in nanostructured and amorphous silica glasses million-atom molecular dynamics simulations. Mater. Res. Soc. Symp. Proc. 2001, 703. [Google Scholar] [CrossRef]
- Pedone, A.; Malavasi, G.; Menziani, M.C.; Segre, U.; Cormack, A.N. Molecular dynamics studies of stress-strain behavior of silica glass under a tensile load. Chem. Mater. 2008, 20, 4356–4366. [Google Scholar] [CrossRef]
- Rountree, C.; Prades, S.; Bonamy, D.; Bouchaud, E.; Kalia, R.; Guillot, C. A unified study of crack propagation in amorphous silica: Using experiments and simulations. J. Alloys Compd. 2007, 434, 60–63. [Google Scholar] [CrossRef]
- Vashishta, P.; Kalia, R.K.; Nakano, A.; Rino, J.P. Interaction potential for silicon carbide: A molecular dynamics study of elastic constants and vibrational density of states for crystalline and amorphous silicon carbide. J. Appl. Phys. 2007, 101, 103515. [Google Scholar] [CrossRef]
- Van Beest, B.; Kramer, G.J.; Van Santen, R. Force fields for silicas and aluminophosphates based on ab initio calculations. Phys. Rev. Lett. 1990, 64, 1955. [Google Scholar] [CrossRef]
- Muralidharan, K.; Oh, K.D.; Deymier, P.; Runge, K.; Simmons, J. Molecular dynamics simulations of atomic-level brittle fracture mechanisms in amorphous silica. J. Mater. Sci. 2007, 42, 4159–4169. [Google Scholar] [CrossRef]
- Yuan, F.; Huang, L. Molecular dynamics simulation of amorphous silica under uniaxial tension: From bulk to nanowire. J. Non-Cryst. Solids 2012, 358, 3481–3487. [Google Scholar] [CrossRef]
- Nomura, K.i.; Chen, Y.C.; Weiqiang, W.; Kalia, R.K.; Nakano, A.; Vashishta, P.; Yang, L.H. Interaction and coalescence of nanovoids and dynamic fracture in silica glass: Multimillion-to-billion atom molecular dynamics simulations. J. Phys. D Appl. Phys. 2009, 42, 214011. [Google Scholar] [CrossRef]
- Castro-Marcano, F.; van Duin, A.C. Comparison of thermal and catalytic cracking of 1-heptene from ReaxFF reactive molecular dynamics simulations. Combust. Flame 2013, 160, 766–775. [Google Scholar] [CrossRef]
- Yuan, F.; Huang, L. Brittle to ductile transition in densified silica glass. Sci. Rep. 2014, 4, 5035. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.C.; Haque, B.Z.G.; Gillespie, J.W. Molecular dynamics simulations of the structure and mechanical properties of silica glass using ReaxFF. J. Mater. Sci. 2016, 51, 10139–10159. [Google Scholar] [CrossRef]
- Rimsza, J.M.; Jones, R.E.; Criscenti, L.J. Crack propagation in silica from reactive classical molecular dynamics simulations. J. Am. Ceram. Soc. 2018, 101, 1488–1499. [Google Scholar] [CrossRef]
- Irving, J.; Kirkwood, J.G. The statistical mechanical theory of transport processes. IV. The equations of hydrodynamics. J. Chem. Phys. 1950, 18, 817–829. [Google Scholar] [CrossRef]
- Chowdhury, S.C.; Wise, E.A.; Ganesh, R.; Gillespie, J.W., Jr. Effects of surface crack on the mechanical properties of Silica: A molecular dynamics simulation study. Eng. Fract. Mech. 2019, 207, 99–108. [Google Scholar] [CrossRef]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Xiao, S.; Edwards, S.A.; Gräter, F. A New Transferable Forcefield for Simulating the Mechanics of CaCO3 Crystals. J. Phys. Chem. C 2011, 115, 20067–20075. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Grubmüller, H.; Heymann, B.; Tavan, P. Ligand binding: Molecular mechanics calculation of the streptavidin-biotin rupture force. Science 1996, 271, 997–999. [Google Scholar] [CrossRef] [PubMed]
- Barthelat, F.; Espinoza, H.D. Elastic Properties of Nacre Aragonite Tablets. In Proceedings of the SEM Annual Conference and Exposition on Experimental and Applied Mechanics, Charlotte, NC, USA, 2–4 June 2003; p. 187. [Google Scholar]
- Barthelat, F.; Li, C.M.; Comi, C.; Espinosa, H.D. Mechanical Properties of Nacre Constituents and Their Impact on Mechanical Performance. J. Mater. Res. 2006, 21, 1977–1986. [Google Scholar] [CrossRef]
- Forbes, T.Z.; Radha, A.V.; Navrotsky, A. The Energetics of Nanophase Calcite. GeoChim. Cosmochim. Acta 2011, 75, 7893–7905. [Google Scholar] [CrossRef]
- Padilla, C.H.; Patil, S.; Heider, Y.; Markert, B. 3D modelling of brittle fracture using a joint all-atom and phase-field approach. GAMM-Mitteilungen 2017, 40, 91–101. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Authors and Year | MD System and Used Criteria | Brief Research Summary |
|---|---|---|
| Ashurst and Hoover (1976) [15] |
|
|
| Decelis et al. (1983) [70] |
|
|
| Smith et al. (1989) [71] |
|
|
| Zhang et al. (1995) [72] |
|
|
| Swiler et al. (1995) [73] |
|
|
| Abraham et al. (1997) [74] |
|
|
| Holland and Marder (1998) [75] |
|
|
| Latapie and Farkas (2004) [76] |
|
|
| Mielke et al. (2004) [77] |
|
|
| Zhou et al. (2008) [78] |
|
|
| Kang et al. (2010) [79] |
|
|
| Terdalkar et al. (2010) [80] |
|
|
| Zhang et al. (2012) [81] |
|
|
| Tang et al. (2012) [82] |
|
|
| Dewapriya et al. (2014) [24] |
|
|
| Wang et al. (2015) [83] |
|
|
| Patil et al. (2016) [26] |
|
|
| Zhang et al.(2017) [84] |
|
|
| Sumigawa et al. (2017) [85] |
|
|
| Bao et al. (2018) [86] |
|
|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patil, S.P.; Heider, Y. A Review on Brittle Fracture Nanomechanics by All-Atom Simulations. Nanomaterials 2019, 9, 1050. https://doi.org/10.3390/nano9071050
Patil SP, Heider Y. A Review on Brittle Fracture Nanomechanics by All-Atom Simulations. Nanomaterials. 2019; 9(7):1050. https://doi.org/10.3390/nano9071050
Chicago/Turabian StylePatil, Sandeep P., and Yousef Heider. 2019. "A Review on Brittle Fracture Nanomechanics by All-Atom Simulations" Nanomaterials 9, no. 7: 1050. https://doi.org/10.3390/nano9071050
APA StylePatil, S. P., & Heider, Y. (2019). A Review on Brittle Fracture Nanomechanics by All-Atom Simulations. Nanomaterials, 9(7), 1050. https://doi.org/10.3390/nano9071050