Micro-Mechanism of Interfacial Separation and Slippage of Graphene/Aluminum Nanolaminated Composites

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Model and Methodology
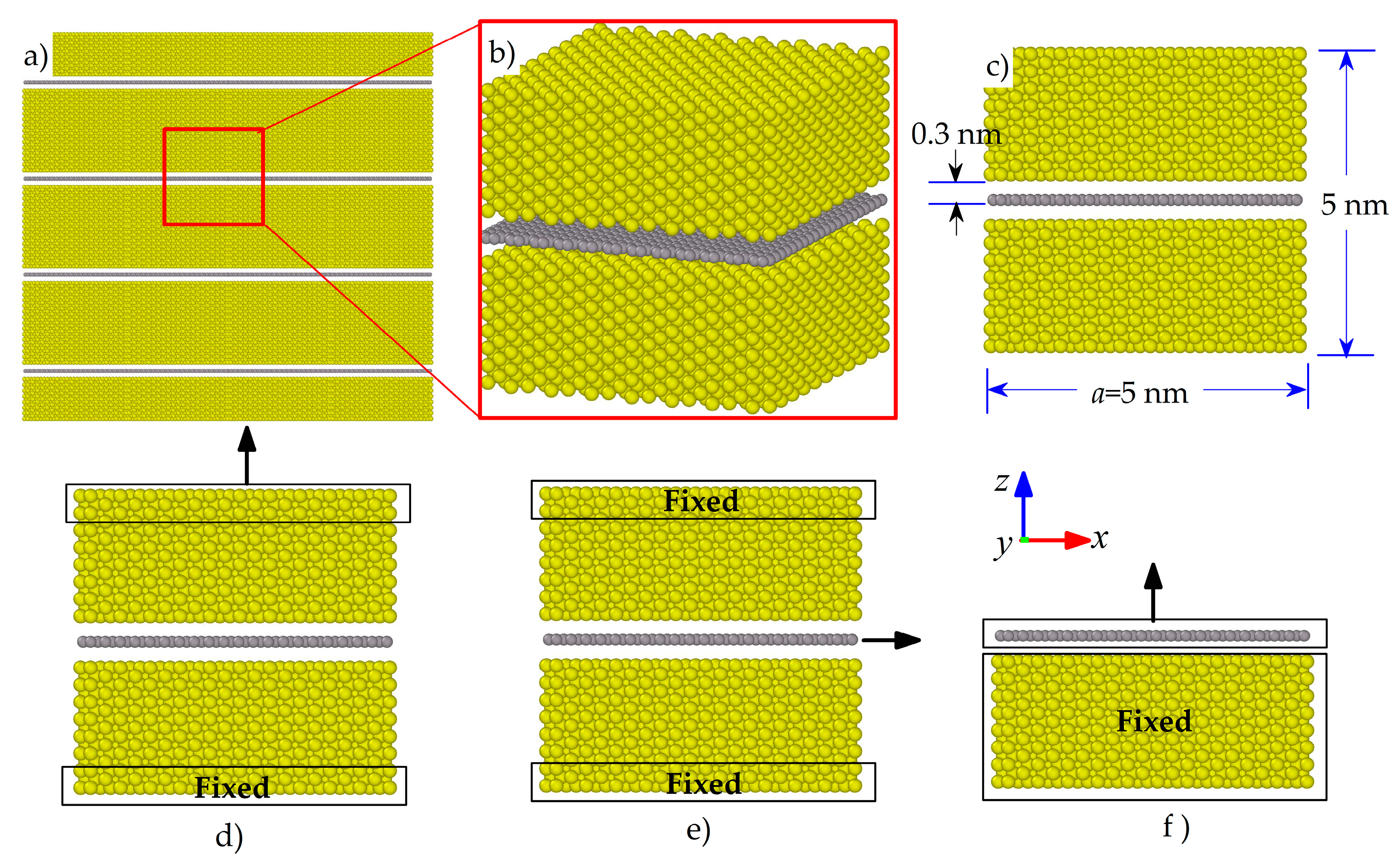
2.1. MD Models
2.2. MD Potentials
2.3. MD Simulation Method
3. Results and Discussion
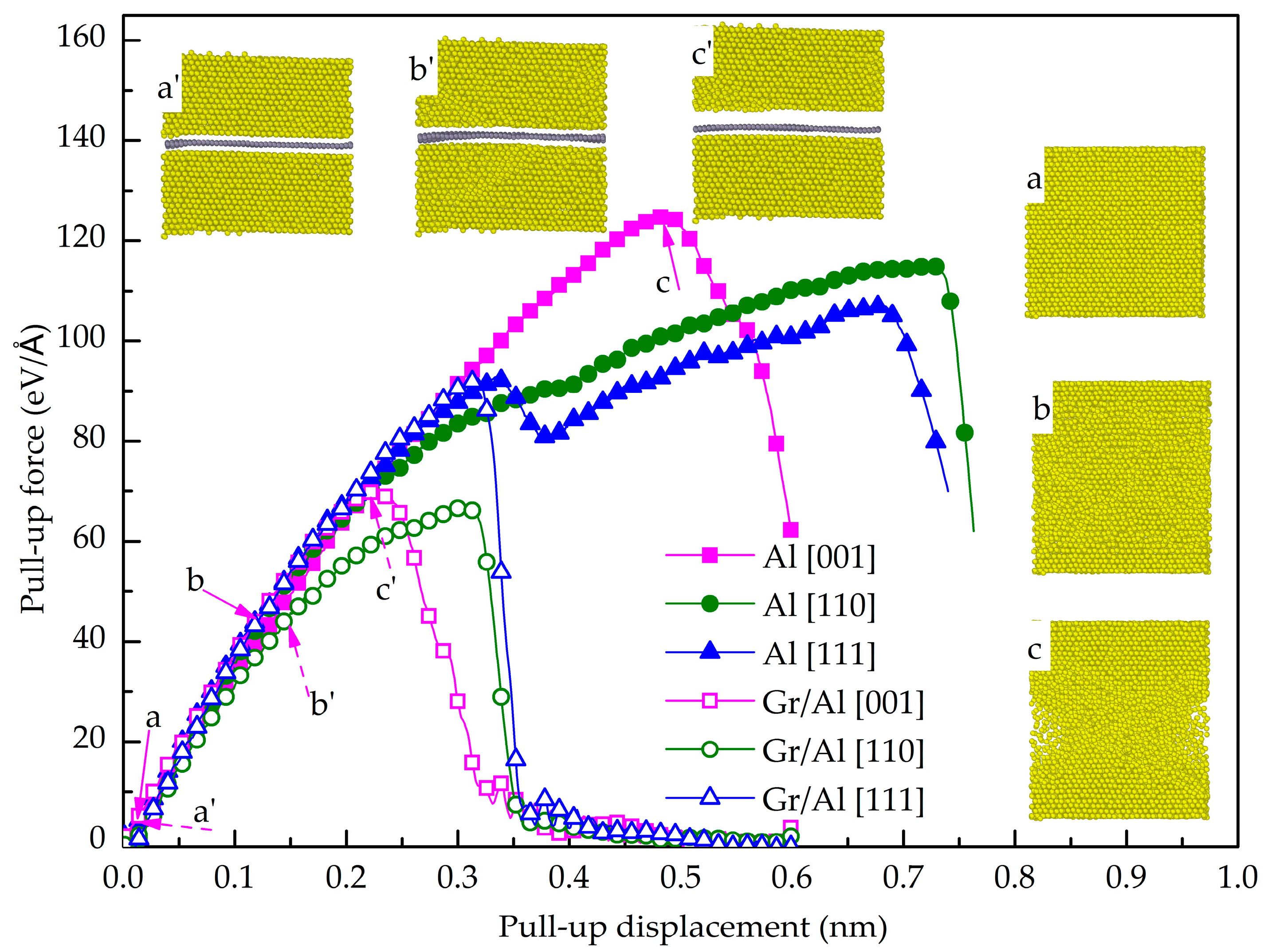
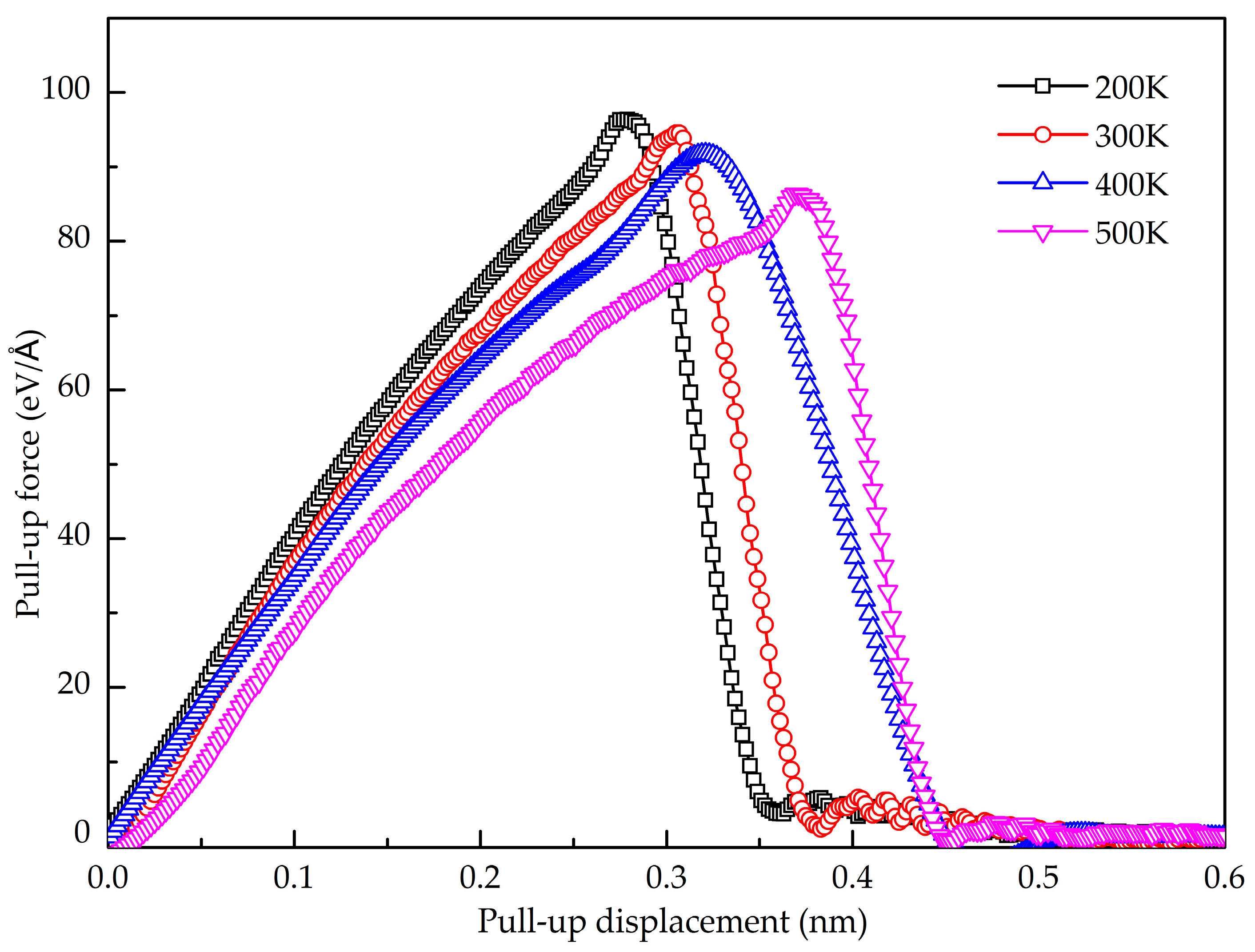
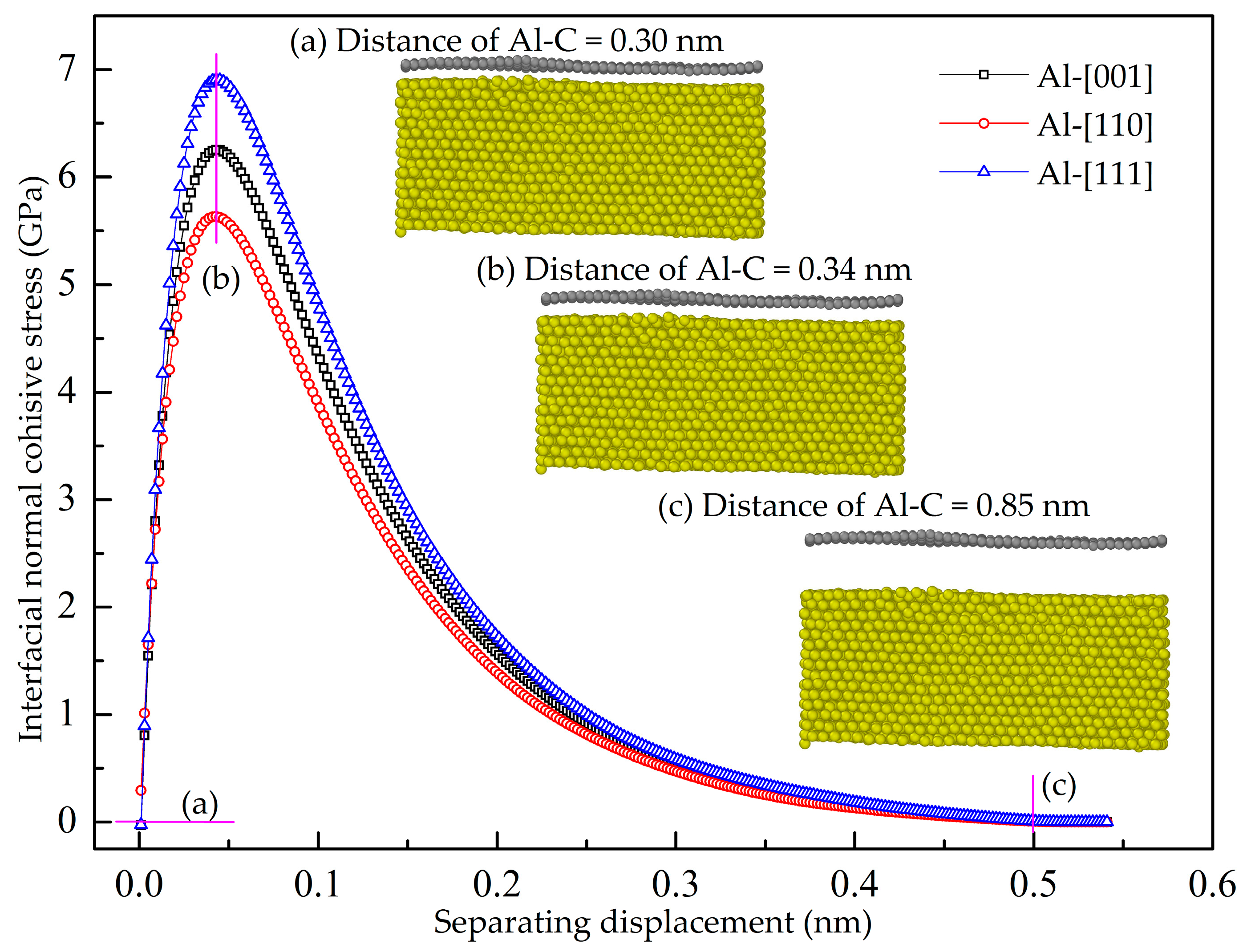
3.1. Normal Interfacial Behavior by Pull-Up Simulation
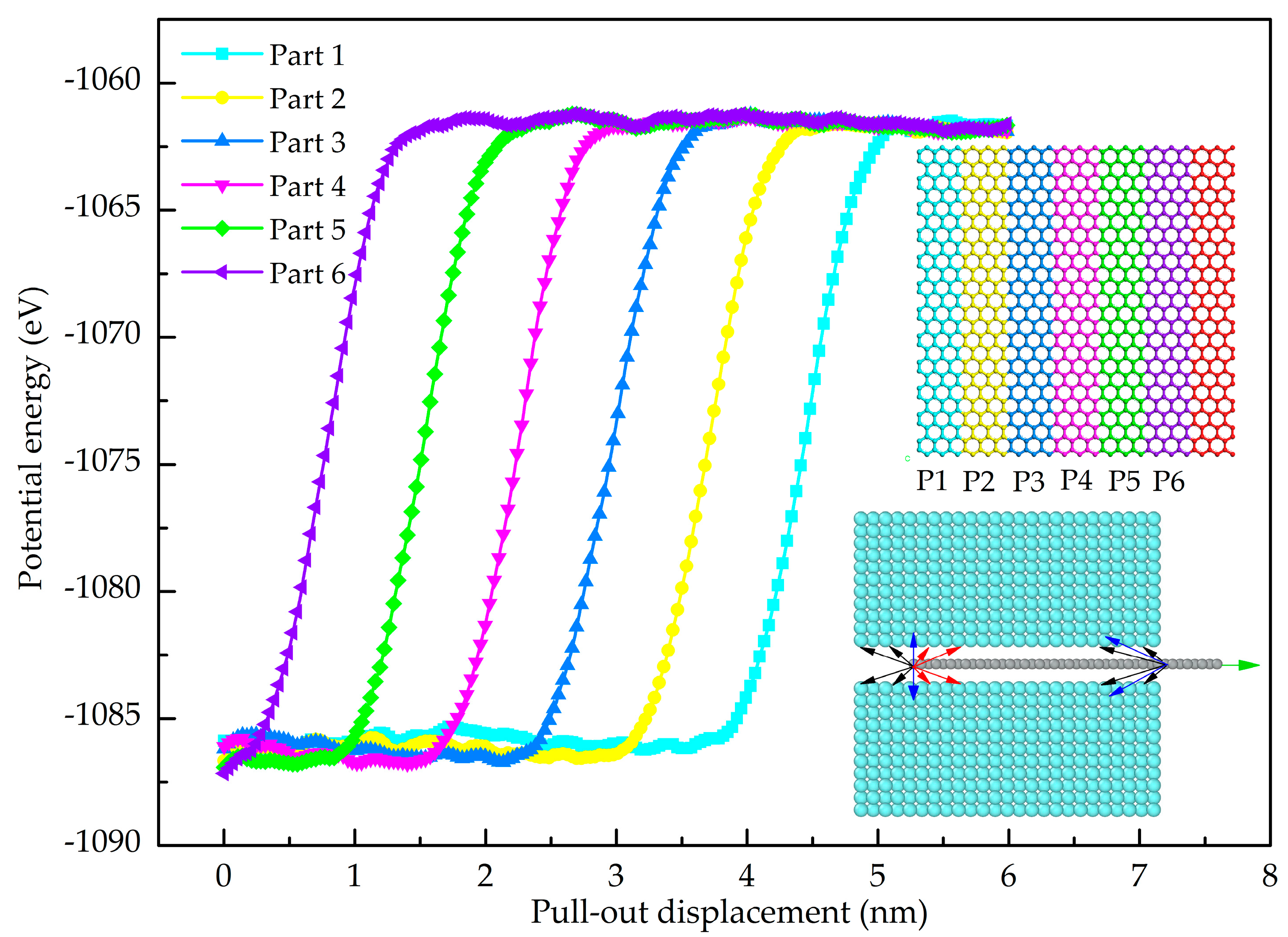
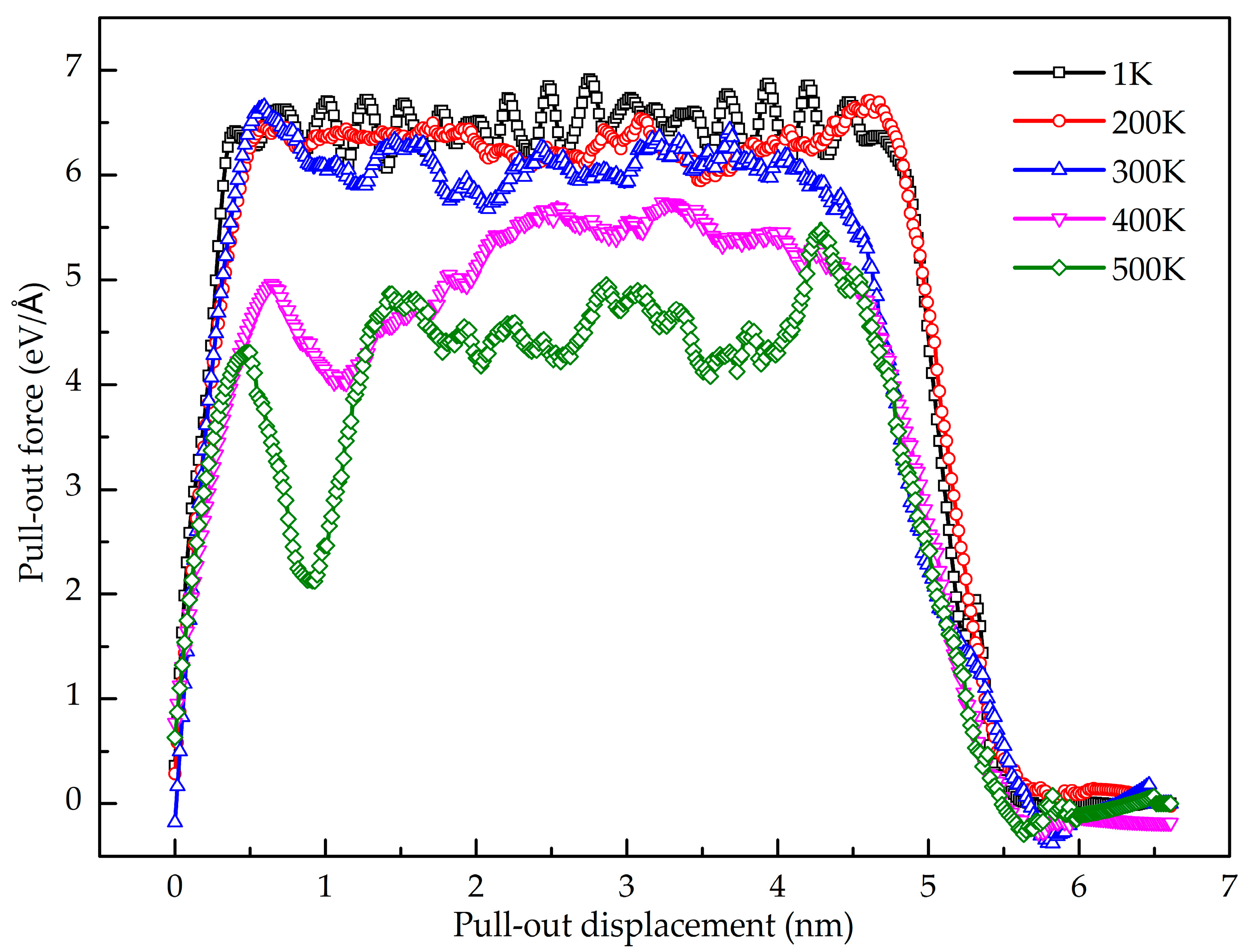
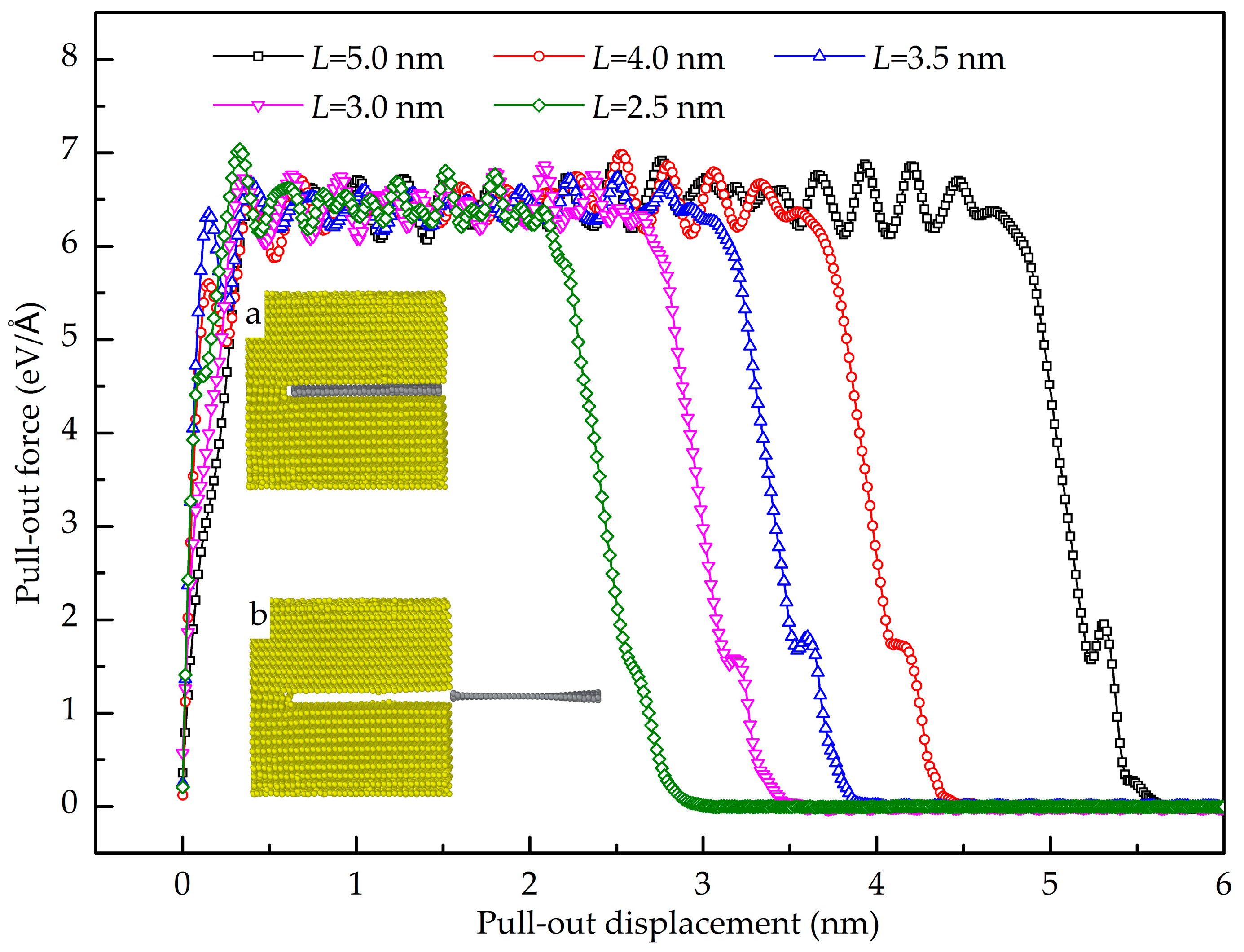
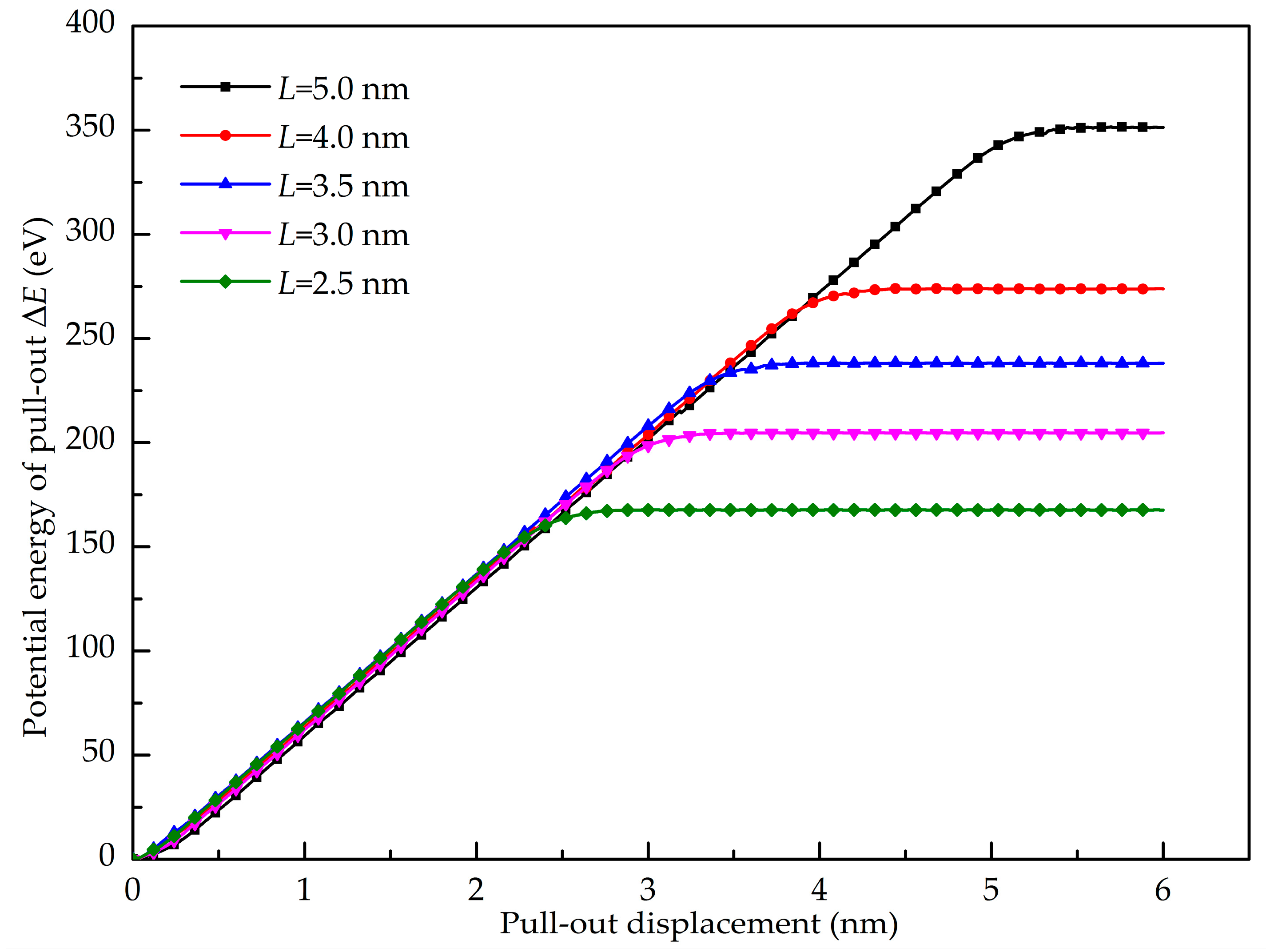
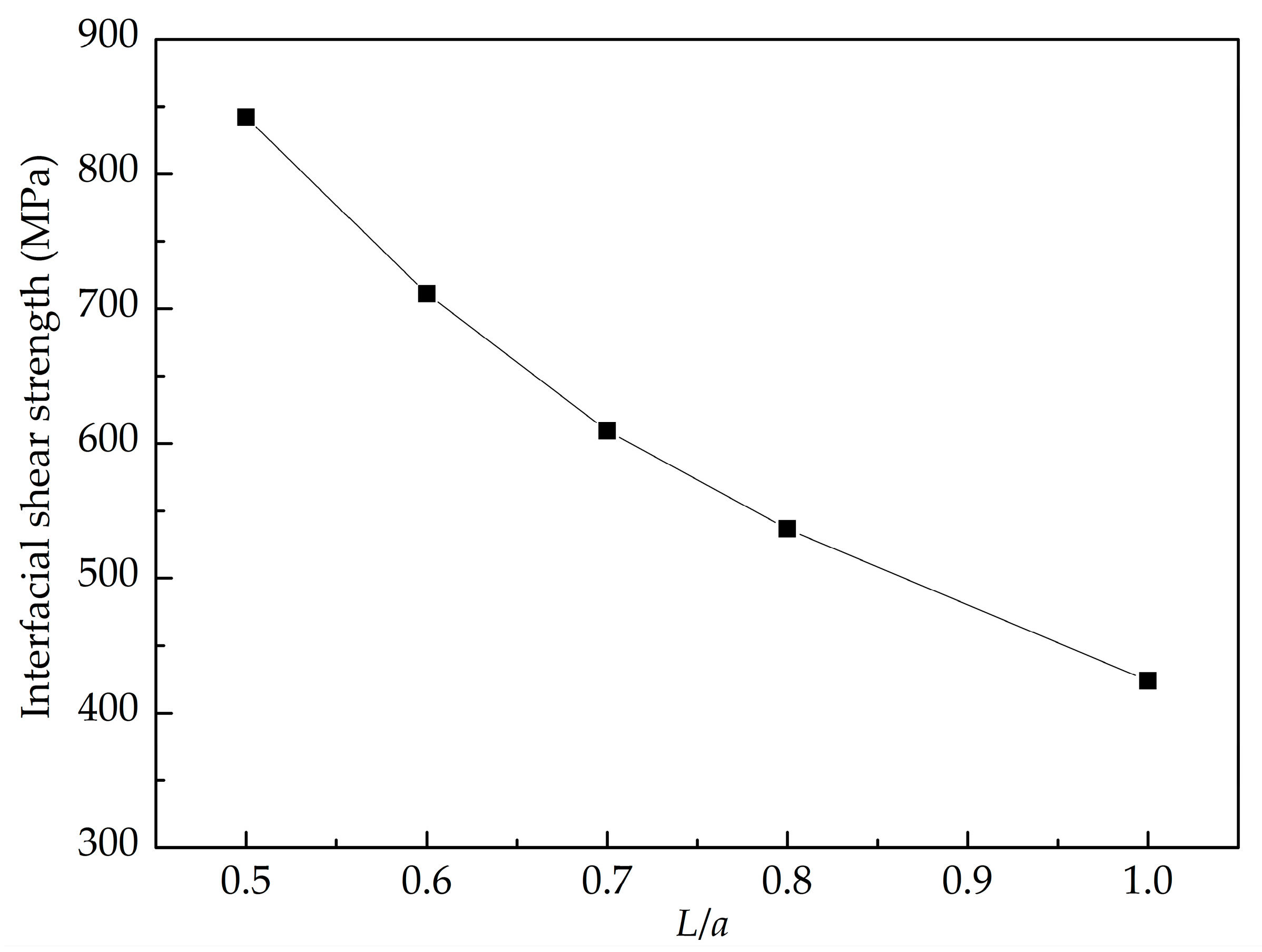
3.2. Shear Interfacial Properties by Pull-Out Simulations
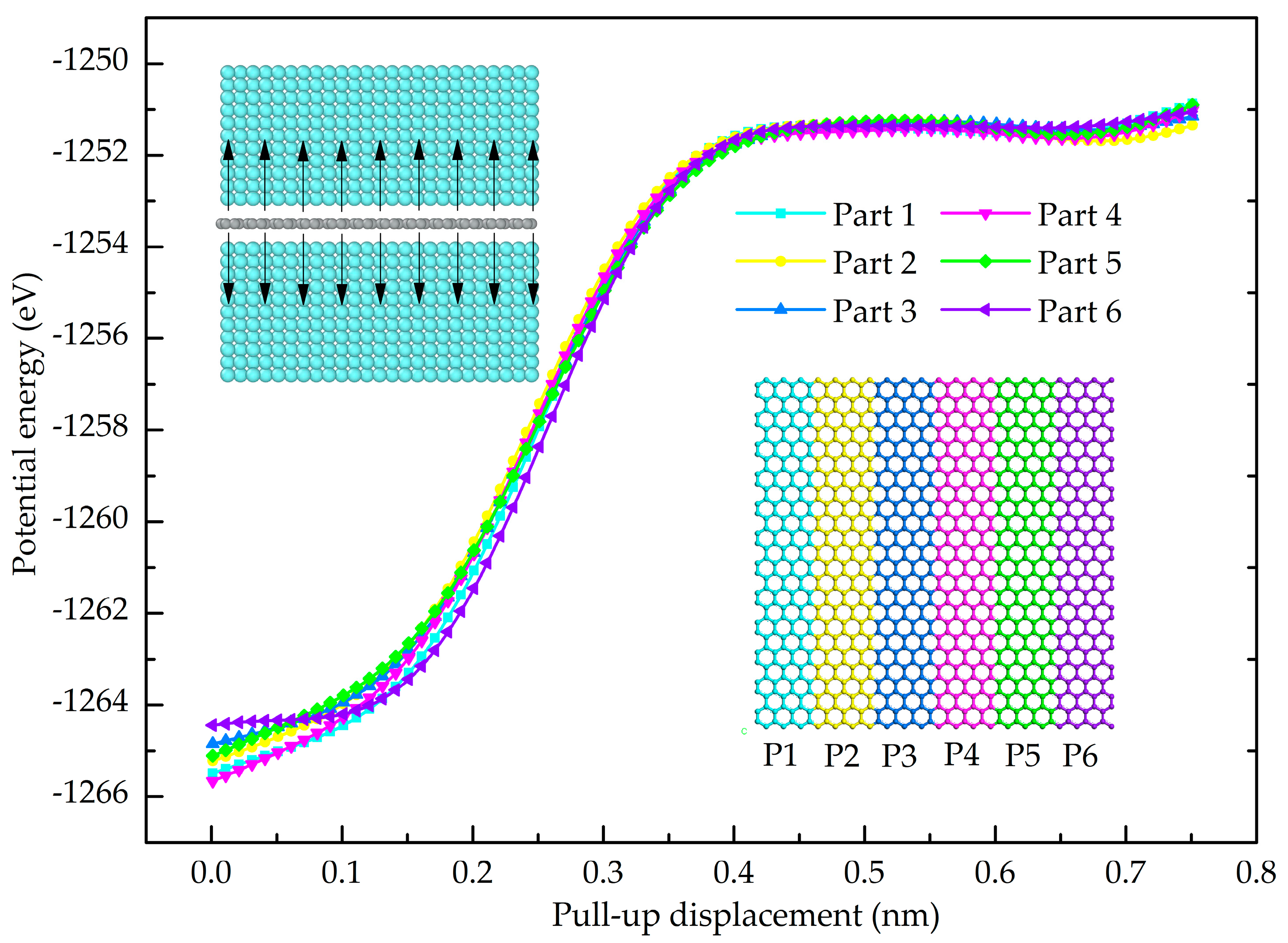
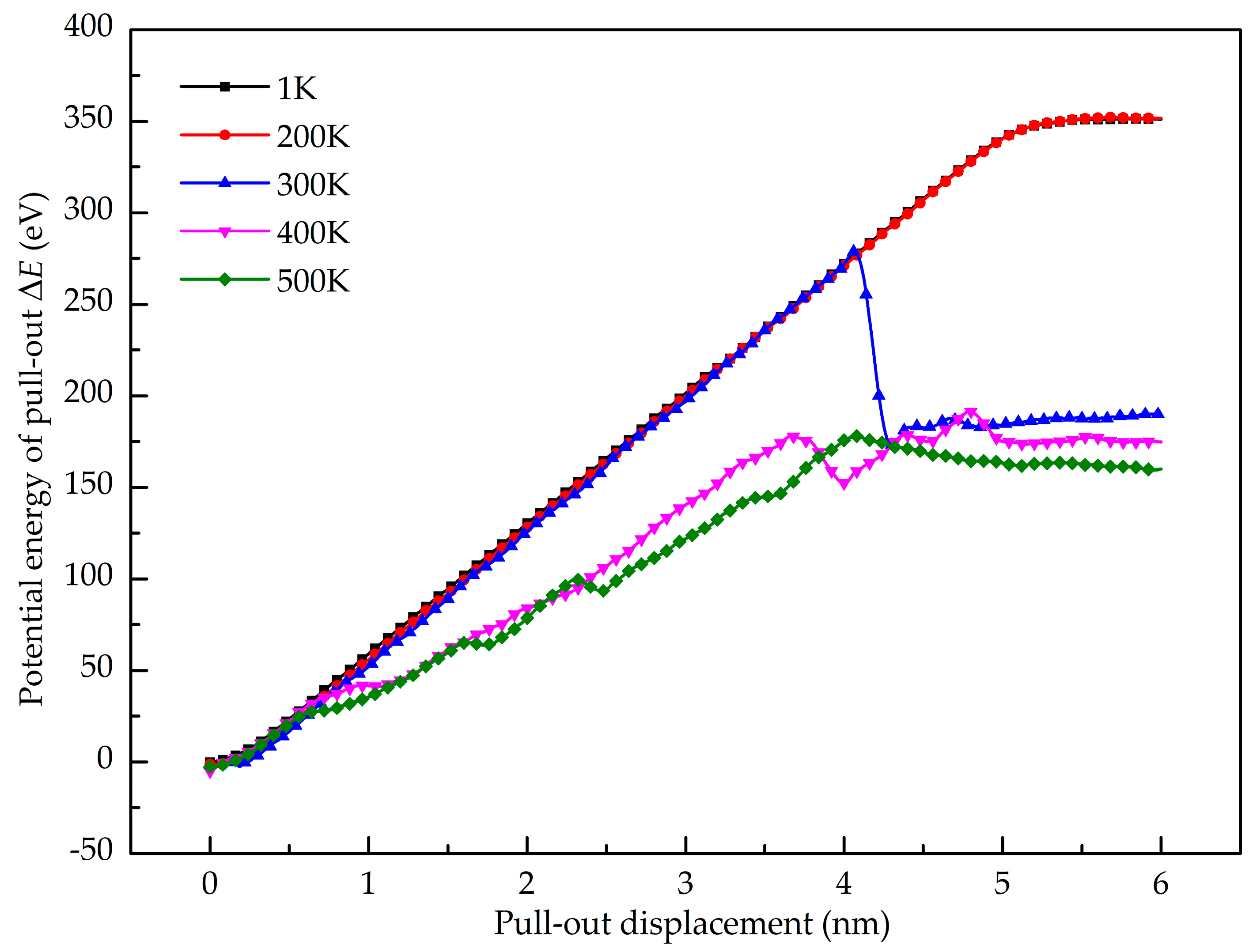
3.3. Cohesion Law of Gr/Al Interface
4. Conclusions
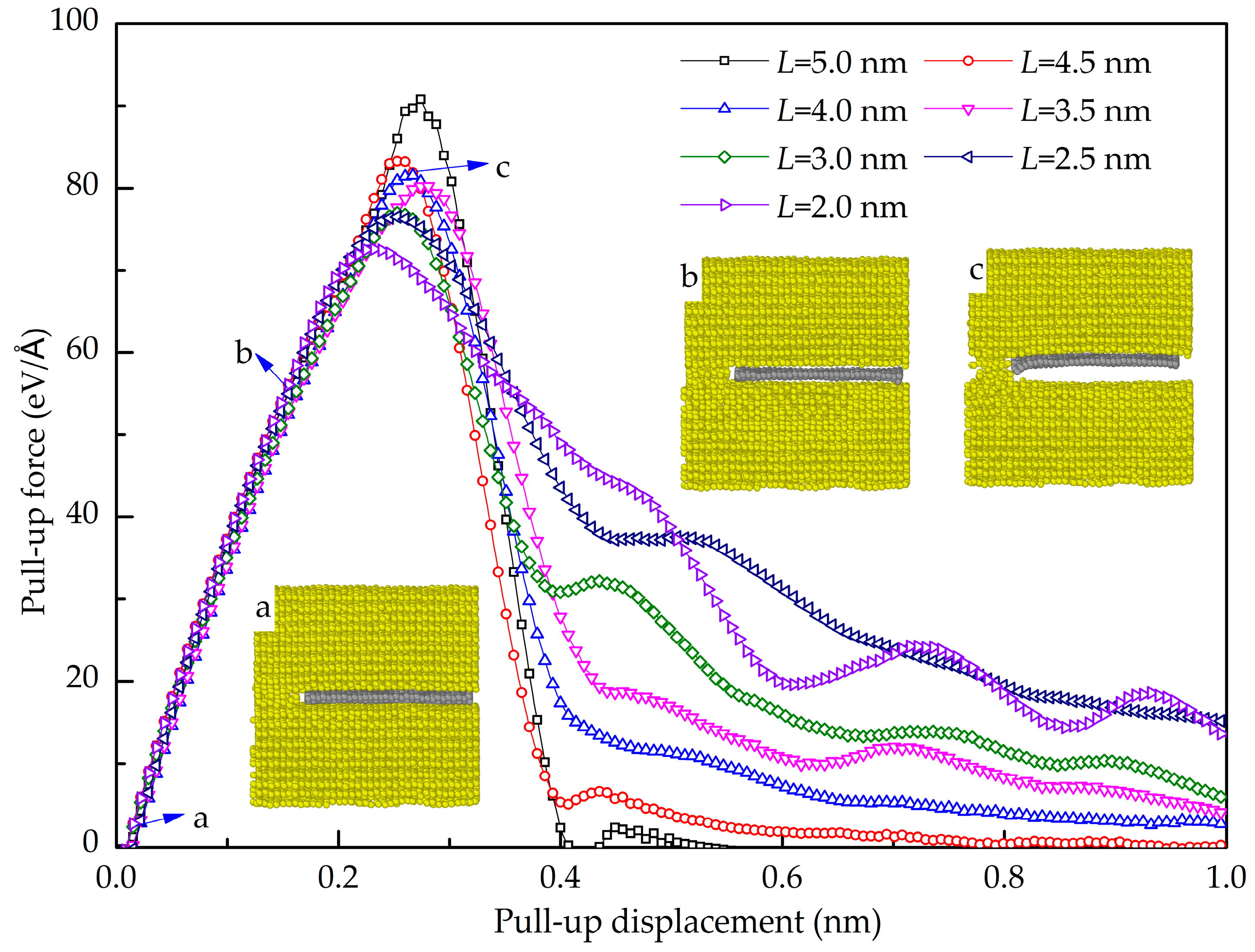
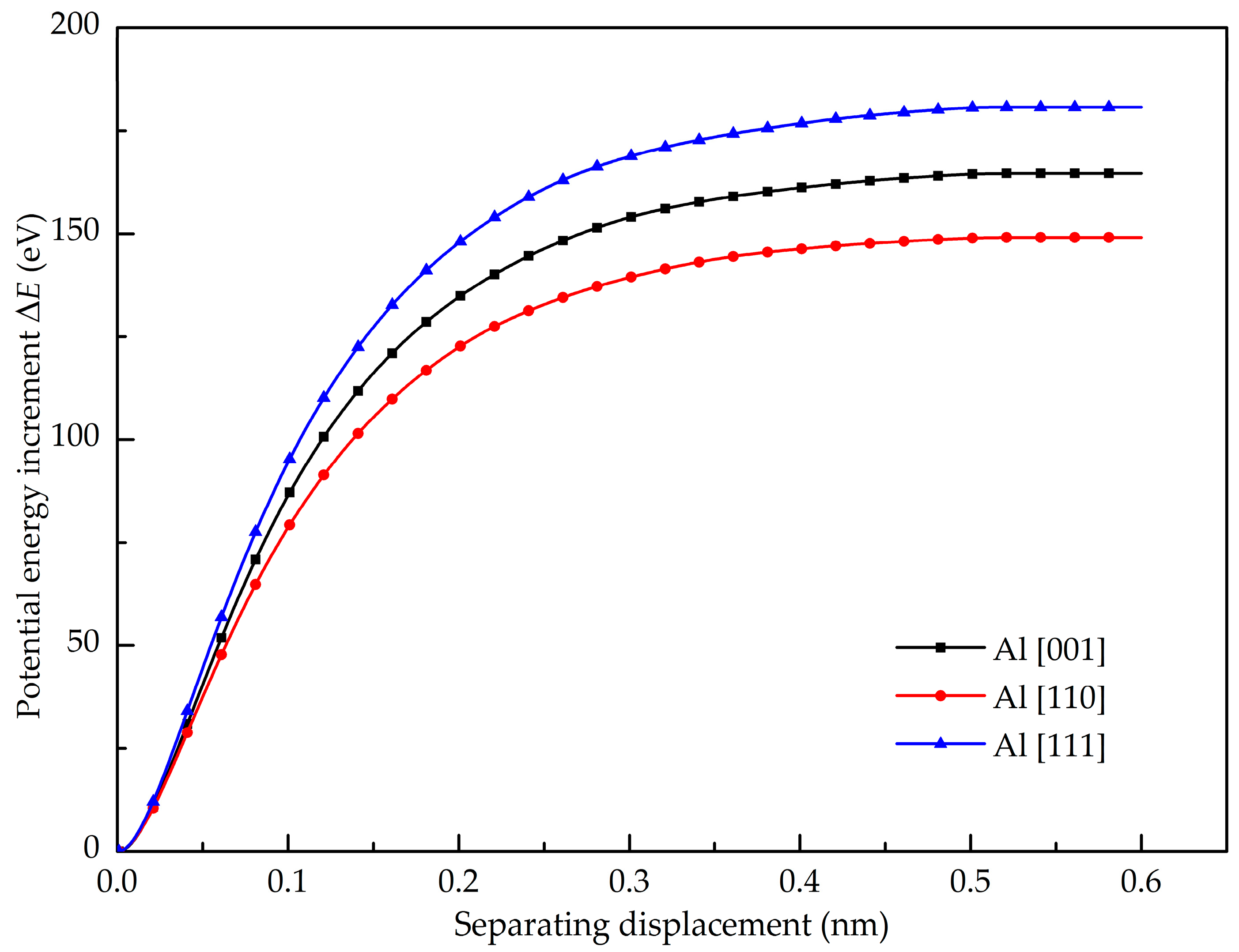
- The maximum pull-up force of the GANC is lower than that of pure Al, indicating that the presence of the Gr/Al interface reduces the tensile property of the Al. The GANC with the Al (111) exhibits the highest normal interfacial strength due to the dense arrangement of the Al atoms. In addition, the normal interfacial force decreases with the reduction of the embedded length of the graphene because all the atoms on the interface contribute to the pull-up force.
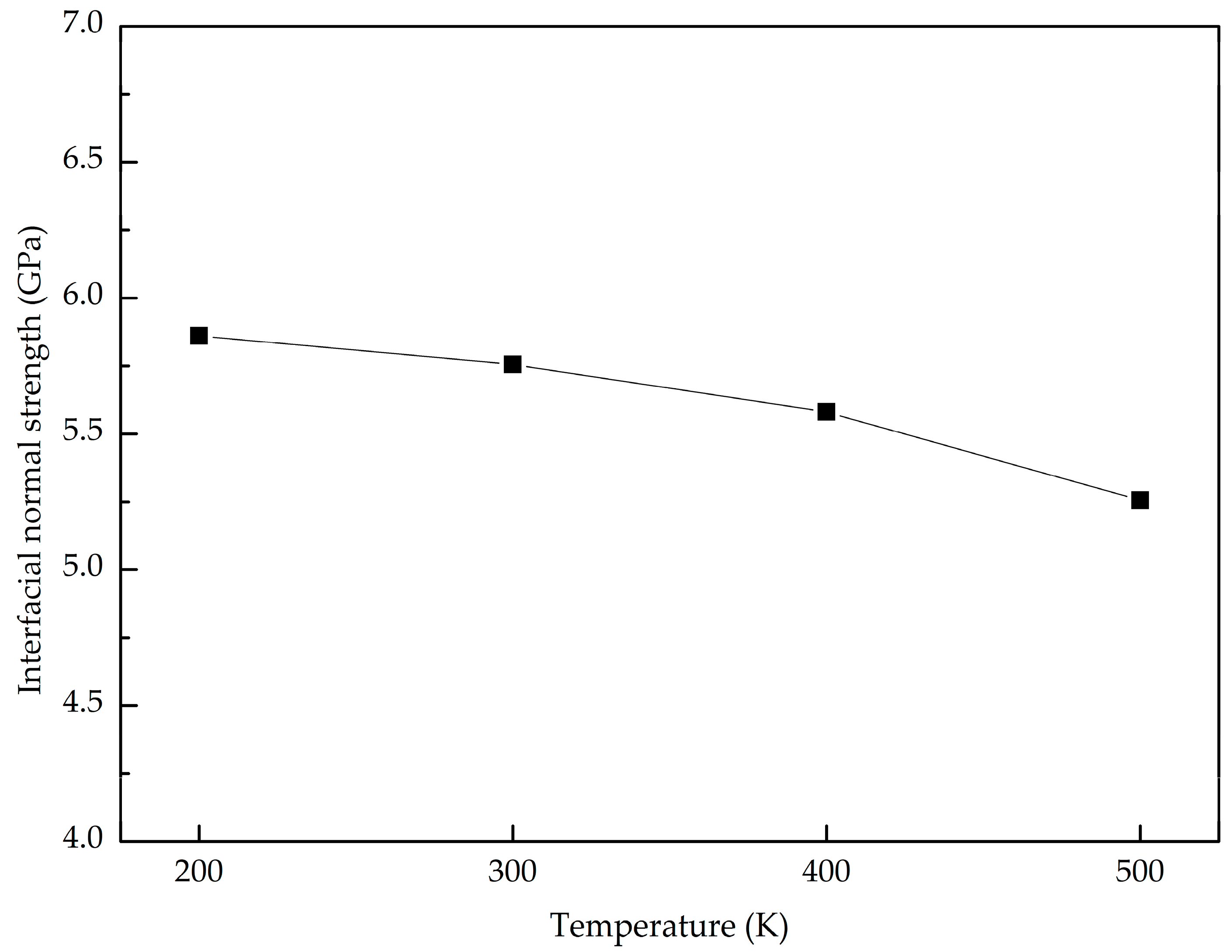
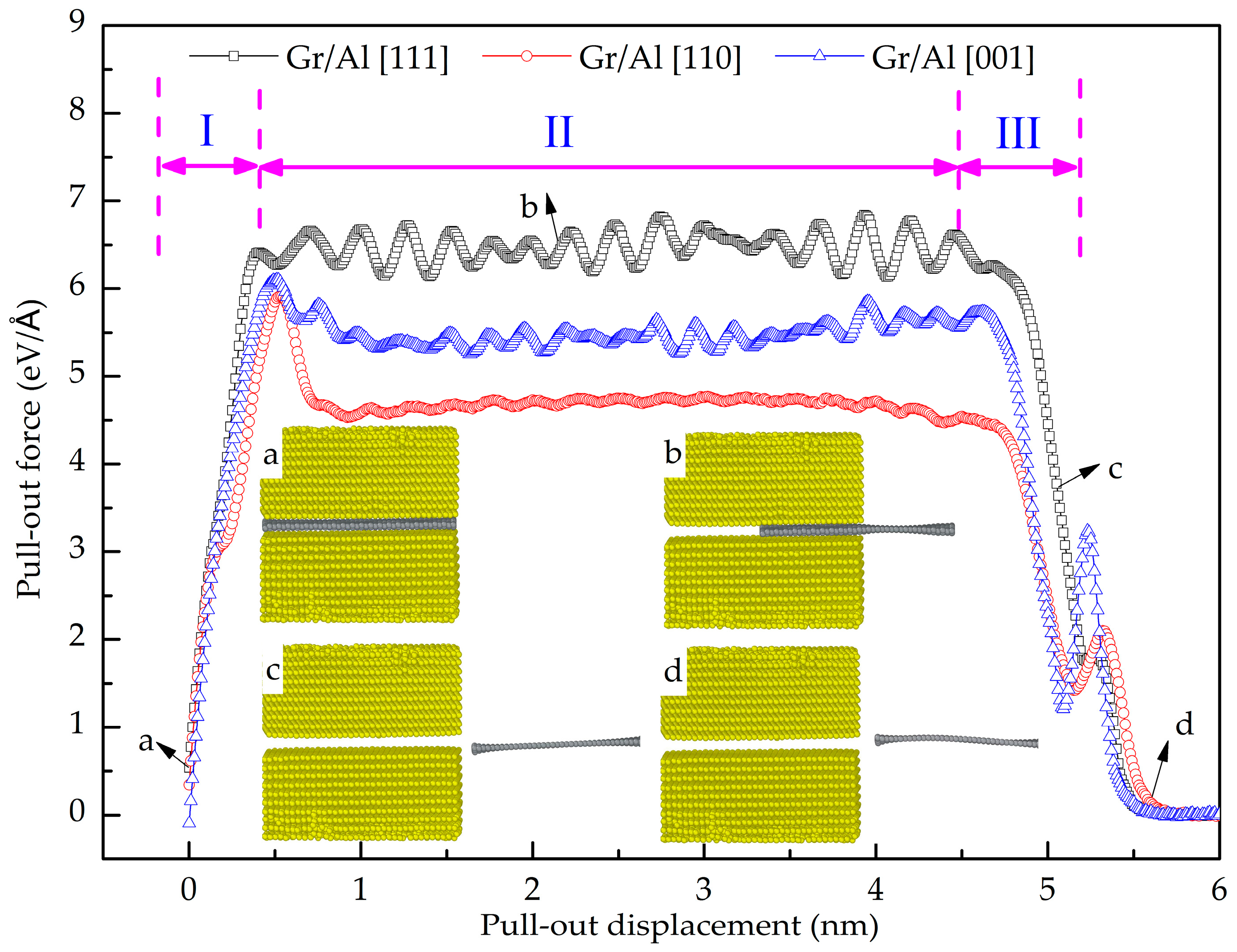
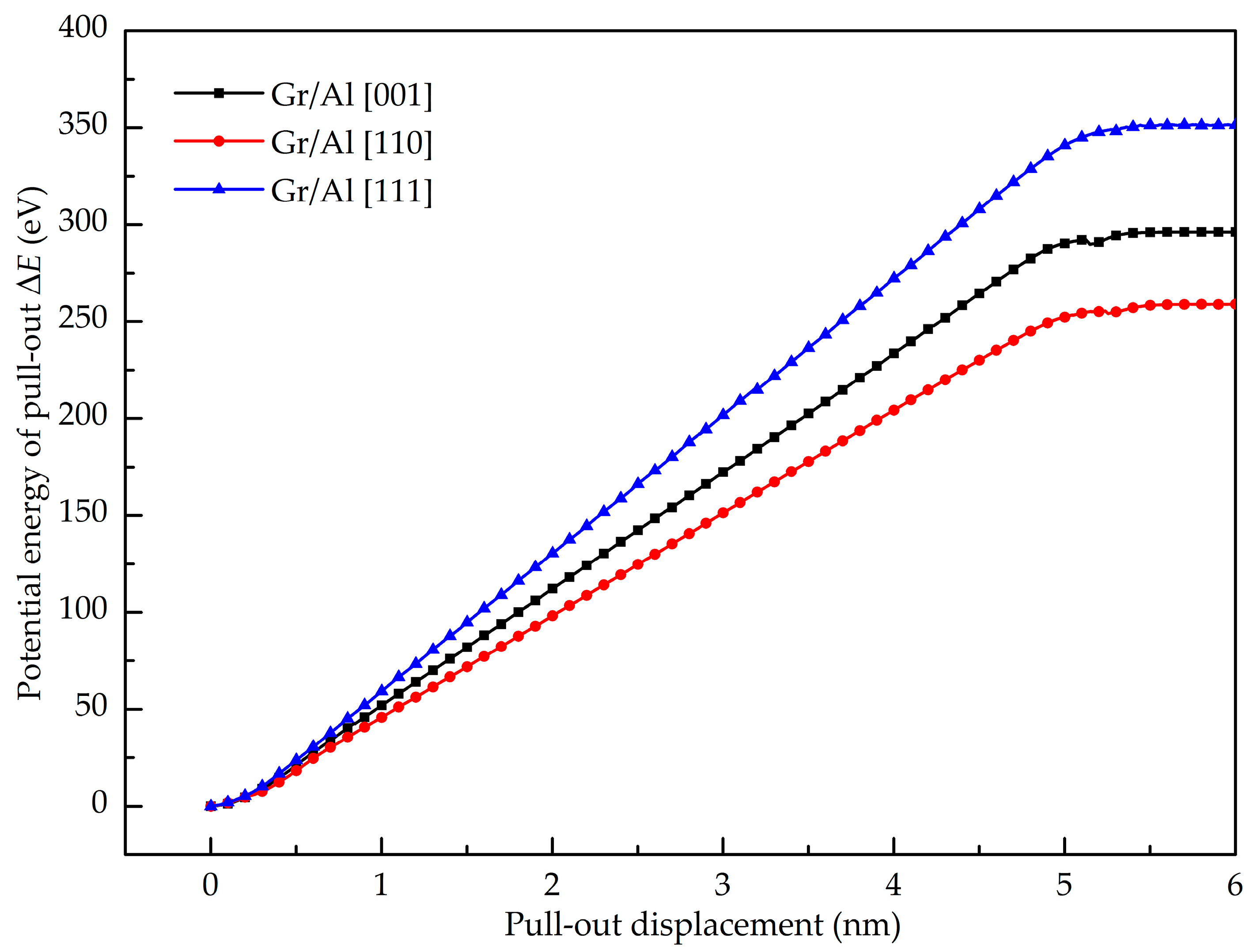
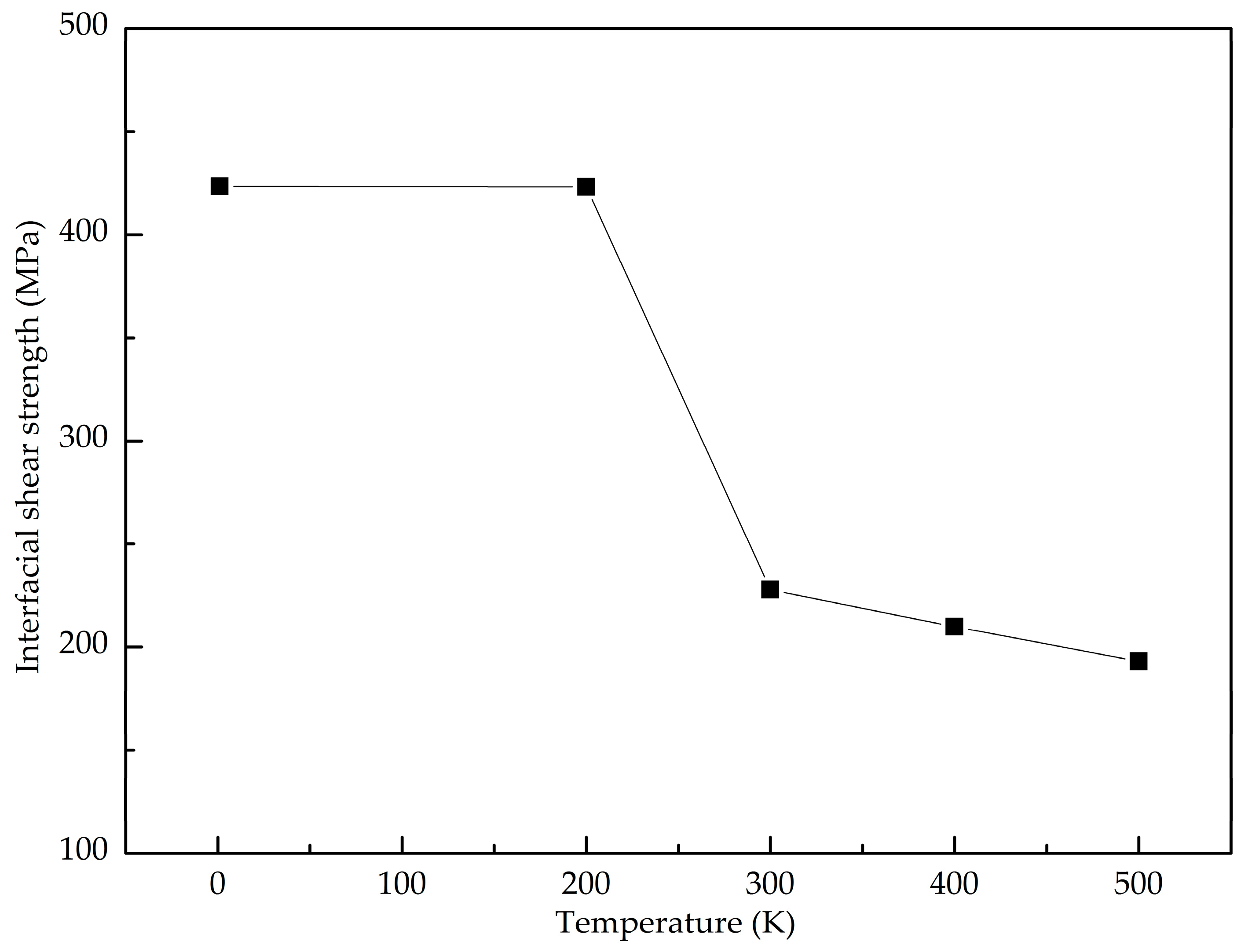
- The GANC with the Al (111) exhibits the highest normal interfacial strength because the number of atoms at the creak tip is the most. The embedded length of the graphene does not affect the pull-out force, because only a column of atoms at the crack tip acts during the pull-out process, whereas the shear interfacial strength increases with the decrease of the embedded length of graphene. Furthermore, the interface of the GANC has a better resistance at a lower temperature.
- The relation of normal stress and separating displacement of the interface was studied and a cohesive law for the finite element simulation of the interface was determined.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moghadam, A.D.; Schultz, B.F.; Ferguson, J.B.; Omrani, E.; Rohatgi, P.K.; Gupta, N. Functional metal matrix composites: Self-lubricating, self-healing, and nanocomposites-an outlook. JOM 2014, 66, 872–881. [Google Scholar] [CrossRef]
- Singh, M.; Rana, R.S.; Purohit, R.; Sahu, K. Development and analysis of al-matrix nano composites fabricated by ultrasonic assisted squeeze casting process. Mater. Today Proc. 2015, 2, 3697–3703. [Google Scholar] [CrossRef]
- Khalil, I.; Rahmati, S.; Julkapli, N.M.; Yehye, W.A. Graphene metal nanocomposites—Recent progress in electrochemical biosensing applications. J. Ind. Eng. Chem. 2018, 59, 425–439. [Google Scholar] [CrossRef]
- Kaczmar, J.W.; Pietrzak, K.; Osiński, W. The production and application of metal matrix composite materials. J. Mater. Process. Technol. 2000, 106, 58–67. [Google Scholar] [CrossRef]
- Rawal, S.P. Metal-matrix composites for space applications. JOM 2001, 53, 14–17. [Google Scholar] [CrossRef]
- Kashfipour, M.A.; Mehra, N.; Zhu, J. A review on the role of interface in mechanical, thermal, and electrical properties of polymer composites. Adv. Compos. Hybrid Mater. 2018, 1, 1–25. [Google Scholar] [CrossRef]
- Awasthi, A.P.; Lagoudas, D.C.; Hammerand, D.C. Modeling of graphene-polymer interfacial mechanical behavior using molecular dynamics. Model. Simul. Mater. Sci. Eng. 2009, 17, 15002. [Google Scholar] [CrossRef]
- Chen, Y.L.; Liu, B.; Huang, Y.; Hwang, K.C. Fracture toughness of carbon nanotube-reinforced metal- and ceramic-matrix composites. J. Nanomater. 2011, 2011, 1–9. [Google Scholar] [CrossRef]
- Chowdhury, S.C.; Okabe, T. Computer simulation of carbon nanotube pull-out from polymer by the molecular dynamics method. Compos. Part A-Appl. Sci. Manuf. 2007, 38, 747–754. [Google Scholar] [CrossRef]
- Coto, B.; Antia, I.; Barriga, J.; Blanco, M.; Sarasua, J. Influence of the geometrical properties of the carbon nanotubes on the interfacial behavior of epoxy/cnt composites: A molecular modelling approach. Comput. Mater. Sci. 2013, 79, 99–104. [Google Scholar] [CrossRef]
- Peng, X.; Meguid, S.A. Molecular simulations of the influence of defects and functionalization on the shear strength of carbon nanotube-epoxy polymer interfaces. Comput. Mater. Sci. 2017, 126, 204–216. [Google Scholar] [CrossRef]
- Sindu, B.S.; Sasmal, S. Evaluation of mechanical characteristics of nano modified epoxy based polymers using molecular dynamics. Comput. Mater. Sci. 2015, 96, 146–158. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Peng, X.; Yan, C.; Liu, S.; Hu, N. Pull-out simulations on interfacial properties of carbon nanotube-reinforced polymer nanocomposites. Comput. Mater. Sci. 2011, 50, 1854–1860. [Google Scholar] [CrossRef]
- Hartmann, S.; Blaudeck, T.; Hölck, O.; Hermann, S.; Schulz, S.E.; Gessner, T.; Wunderle, B. Quantitativein-situ scanning electron microscope pull-out experiments and molecular dynamics simulations of carbon nanotubes embedded in palladium. J. Appl. Phys. 2014, 115, 144301. [Google Scholar] [CrossRef]
- Hartmann, S.; Wunderle, B.; Hölck, O. Pull-out testing of swcnts simulated by molecular dynamics. Int. J. Theor. Appl. Nanotechnol. 2012, 1. [Google Scholar] [CrossRef]
- Bartolucci, S.F.; Paras, J.; Rafiee, M.A.; Rafiee, J.; Lee, S.; Kapoor, D.; Koratkar, N. Graphene–aluminum nanocomposites. Mater. Sci. Eng. A 2011, 528, 7933–7937. [Google Scholar] [CrossRef]
- Silvestre, N.; Faria, B.; Canongia Lopes, J.N. Compressive behavior of cnt-reinforced aluminum composites using molecular dynamics. Compos. Sci. Technol. 2014, 90, 16–24. [Google Scholar] [CrossRef]
- Song, H.; Zha, X. Influence of nickel coating on the interfacial bonding characteristics of carbon nanotube–aluminum composites. Comput. Mater. Sci. 2010, 49, 899–903. [Google Scholar] [CrossRef]
- Liu, S.; Hu, N.; Yamamoto, G.; Cai, Y.; Zhang, Y.; Liu, Y.; Li, Y.; Hashida, T.; Fukunaga, H. Investigation on cnt/alumina interface properties using molecular mechanics simulations. Carbon 2011, 49, 3701–3704. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, J.; Yeom, M.S.; Shin, J.W.; Kim, H.; Cui, Y.; Kysar, J.W.; Hone, J.; Jung, Y.; Jeon, S.; et al. Strengthening effect of single-atomic-layer graphene in metal–graphene nanolayered composites. Nat. Commun. 2013, 4, 2114. [Google Scholar] [CrossRef] [PubMed]
- King, A.; Johnson, G.; Engelberg, D.; Ludwig, W.; Marrow, J. Observations of intergranular stress corrosion cracking in a grain-mapped polycrystal. Science 2008, 321, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Wei, X.; Kysar, J.W.; Hone, J. Measurement of the elastic properties and intrinsic strength of monolayer graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, Z.; Cui, Y.; Wei, B.; Xu, S.; Sheng, J.; Wang, M.; Zhu, Y.; Fei, W. Graphene-copper composite with micro-layered grains and ultrahigh strength. Sci. Rep. 2017, 7, 41896. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Murali, S.; Cai, W.; Li, X.; Suk, J.W.; Potts, J.R.; Ruoff, R.S. Graphene and graphene oxide: Synthesis, properties, and applications. Adv. Mater. 2010, 22, 3906–3924. [Google Scholar] [CrossRef] [PubMed]
- Mas-Balleste, R.; Gomez-Navarro, C.; Gomez-Herrero, J.; Zamora, F. 2d materials: To graphene and beyond. Nanoscale 2011, 3, 20–30. [Google Scholar] [CrossRef]
- Lu, Q.; Huang, R. Nonlinear mechanics of single-atomic-layer graphene sheets. Int. J. Appl. Mech. 2009, 1, 443–467. [Google Scholar] [CrossRef]
- Cao, M.; Xiong, D.; Tan, Z.; Ji, G.; Amin-Ahmadi, B.; Guo, Q.; Fan, G.; Guo, C.; Li, Z.; Zhang, D. Aligning graphene in bulk copper: Nacre-inspired nanolaminated architecture coupled with in-situ processing for enhanced mechanical properties and high electrical conductivity. Carbon 2017, 117, 65–74. [Google Scholar] [CrossRef]
- Shin, S.E.; Choi, H.J.; Shin, J.H.; Bae, D.H. Strengthening behavior of few-layered graphene/aluminum composites. Carbon 2015, 82, 143–151. [Google Scholar] [CrossRef]
- Duan, K.; Li, L.; Hu, Y.; Wang, X. Interface mechanical properties of graphene reinforced copper nanocomposites. Mater. Res. Express 2017, 4, 115020. [Google Scholar] [CrossRef]
- Chen, S.J.; Li, C.Y.; Wang, Q.; Duan, W.H. Reinforcing mechanism of graphene at atomic level: Friction, crack surface adhesion and 2d geometry. Carbon 2017, 114, 557–565. [Google Scholar] [CrossRef]
- Shi, X.; Yin, Q.; Wei, Y. A theoretical analysis of the surface dependent binding, peeling and folding of graphene on single crystal copper. Carbon 2012, 50, 3055–3063. [Google Scholar] [CrossRef]
- Chu, K.; Wang, F.; Wang, X.; Huang, D. Anisotropic mechanical properties of graphene/copper composites with aligned graphene. Mater. Sci. Eng. A 2018, 713, 269–277. [Google Scholar] [CrossRef]
- Jiang, W.; Wu, Y.; Qin, Q.; Li, D.; Liu, X.; Fu, M. A molecular dynamics based cohesive zone model for predicting interfacial properties between graphene coating and aluminum. Comput. Mater. Sci. 2018, 151, 117–123. [Google Scholar] [CrossRef]
- Daw, M.S.; Baskes, M.I. Embedded-atom method—Derivation and application to impurities, surfaces, and other defects in metals. Phys. Rev. B 1984, 29, 6443–6453. [Google Scholar] [CrossRef]
- Brenner, D.W.; Shenderova, O.A.; Harrison, J.A.; Stuart, S.J.; Ni, B.; Sinnott, S.B. A second-generation reactive empirical bond order (rebo) potential energy expression for hydrocarbons. J. Phys. Condens. Mattter 2002, 14, 783–802. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with ovito-the open visualization tool. Model. Simul. Mater. Sci. Eng. 2010, 18, 2154–2162. [Google Scholar] [CrossRef]
- Duan, K.; Zhu, F.; Tang, K.; He, L.; Chen, Y.; Liu, S. Effects of chirality and number of graphene layers on the mechanical properties of graphene-embedded copper nanocomposites. Comput. Mater. Sci. 2016, 117, 294–299. [Google Scholar] [CrossRef]
- Lv, C.; Xue, Q.; Xia, D.; Ma, M.; Xie, J.; Chen, H. Effect of chemisorption on the interfacial bonding characteristics of graphene−polymer composites. J. Phys. Chem. C 2010, 114, 6588–6594. [Google Scholar] [CrossRef]
- Jiang, L.; Tan, H.; Wu, J.; Huang, Y.; Hwang, K. Continuum modeling of interfaces in polymer matrix composites reinforced by carbon nanotubes. Nano 2007, 2, 139–148. [Google Scholar] [CrossRef]
- Tan, H.; Jiang, L.Y.; Huang, Y.; Liu, B.; Hwang, K.C. The effect of van der waals-based interface cohesive law on carbon nanotube-reinforced composite materials. Compos. Sci. Technol. 2007, 67, 2941–2946. [Google Scholar] [CrossRef]

















© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, J.-Q.; Yang, Q.-S.; He, X.-Q.; Fu, K.-K. Micro-Mechanism of Interfacial Separation and Slippage of Graphene/Aluminum Nanolaminated Composites. Nanomaterials 2018, 8, 1046. https://doi.org/10.3390/nano8121046
Zhu J-Q, Yang Q-S, He X-Q, Fu K-K. Micro-Mechanism of Interfacial Separation and Slippage of Graphene/Aluminum Nanolaminated Composites. Nanomaterials. 2018; 8(12):1046. https://doi.org/10.3390/nano8121046
Chicago/Turabian StyleZhu, Jia-Qi, Qing-Sheng Yang, Xiao-Qiao He, and Kun-Kun Fu. 2018. "Micro-Mechanism of Interfacial Separation and Slippage of Graphene/Aluminum Nanolaminated Composites" Nanomaterials 8, no. 12: 1046. https://doi.org/10.3390/nano8121046
APA StyleZhu, J.-Q., Yang, Q.-S., He, X.-Q., & Fu, K.-K. (2018). Micro-Mechanism of Interfacial Separation and Slippage of Graphene/Aluminum Nanolaminated Composites. Nanomaterials, 8(12), 1046. https://doi.org/10.3390/nano8121046