Autophagy as a Possible Underlying Mechanism of Nanomaterial Toxicity


Abstract
:1. Introduction
2. Pulmonary Toxicity of Nanomaterials
2.1. Lung Remodeling Manifestations
2.2. Underlying Biological Mechanisms
2.2.1. Oxidative Stress
2.2.2. Inflammation
2.2.3. Genotoxicity
2.2.4. Interaction with the Protein Corona
2.3. Physico-Chemical Determinants
3. Autophagy
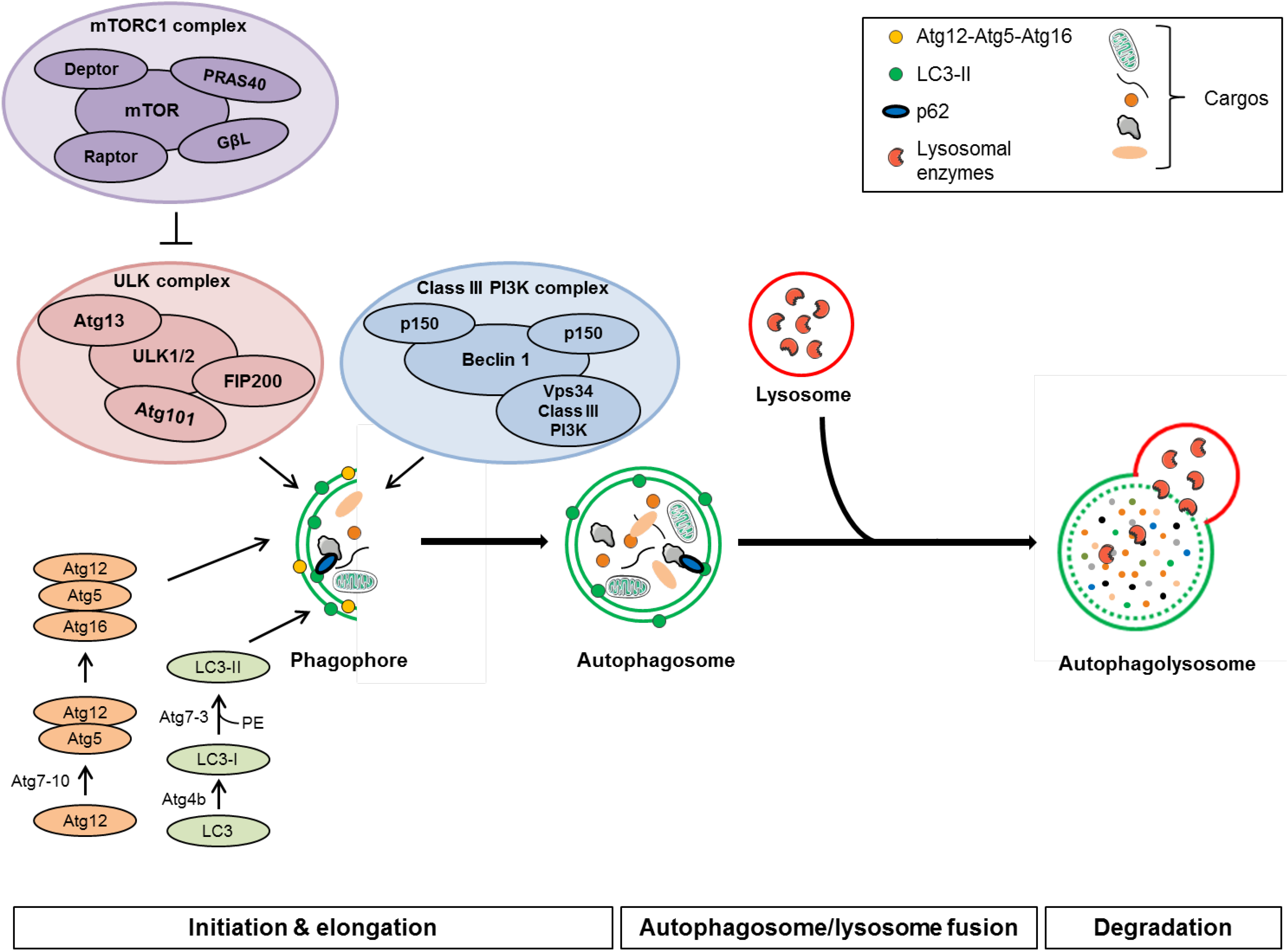
3.1. Autophagy Machinery
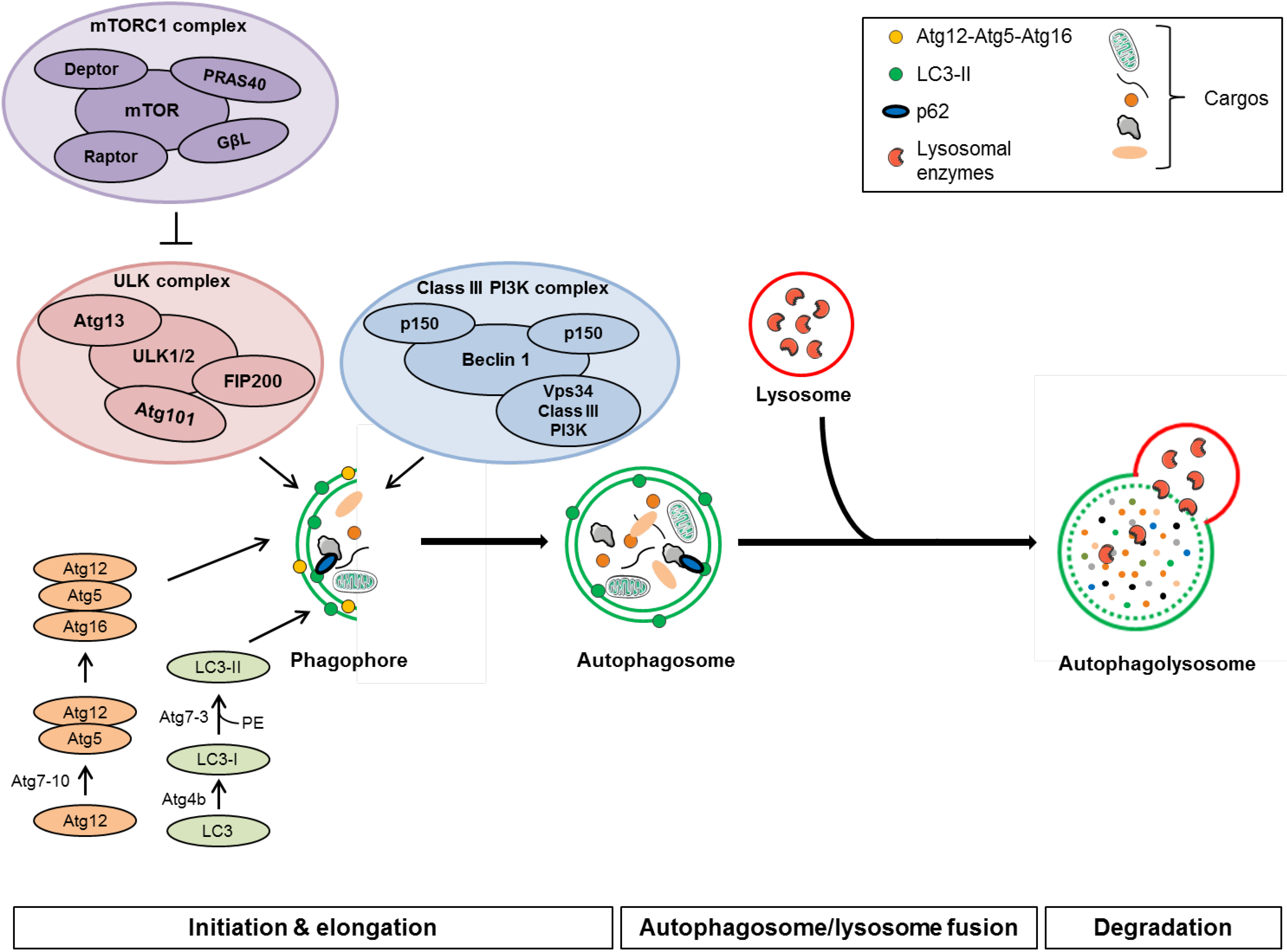
3.1.1. Initiation
3.1.2. Autophagosome Formation and Elongation
3.1.3. Autophagosome/Lysosome Fusion and Degradation
3.1.4. Evaluation of Autophagic Activity
3.2. Autophagy in Physiological and Pathological Conditions
3.2.1. Autophagy and Lung Diseases
3.2.2. Autophagy and Cancer
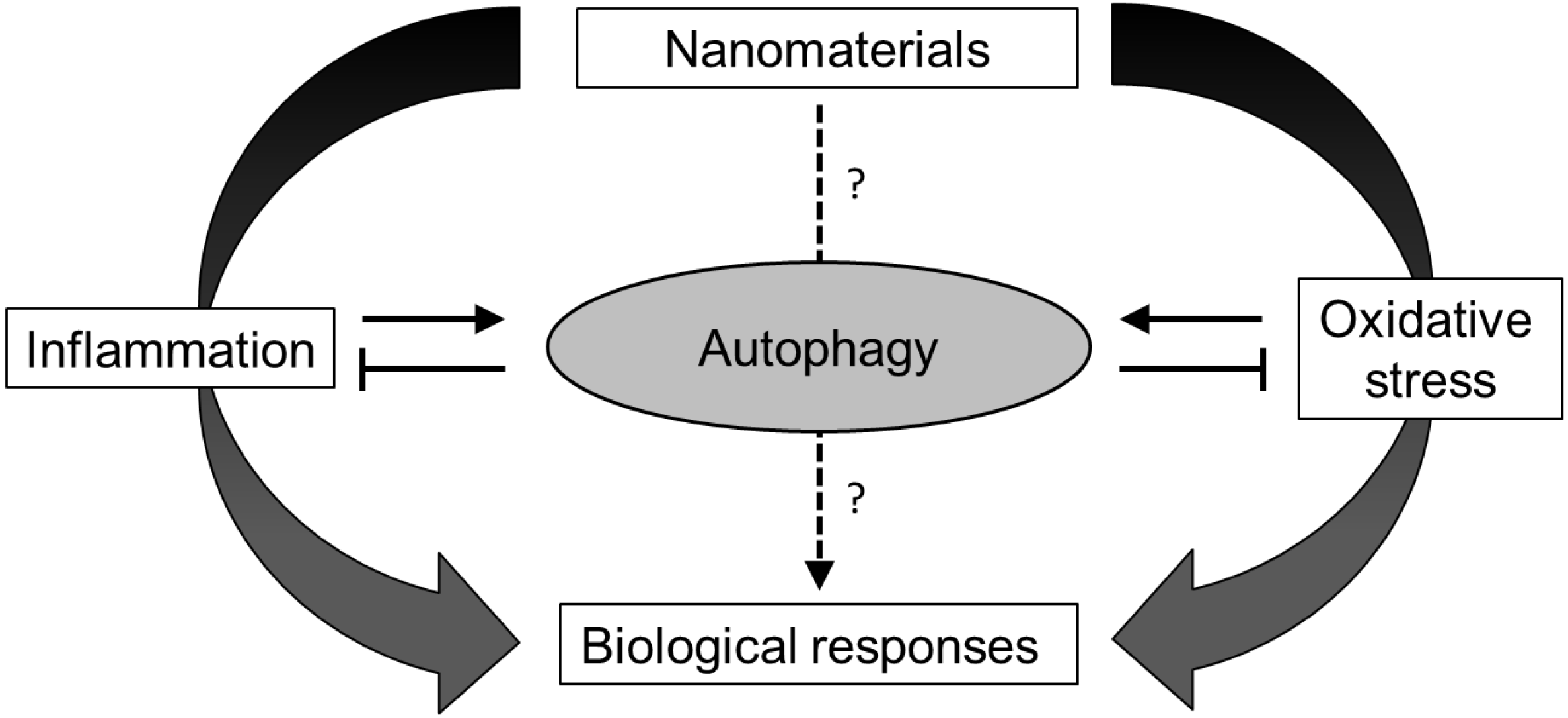
3.2.3. Autophagy, Inflammation and Oxidative Stress
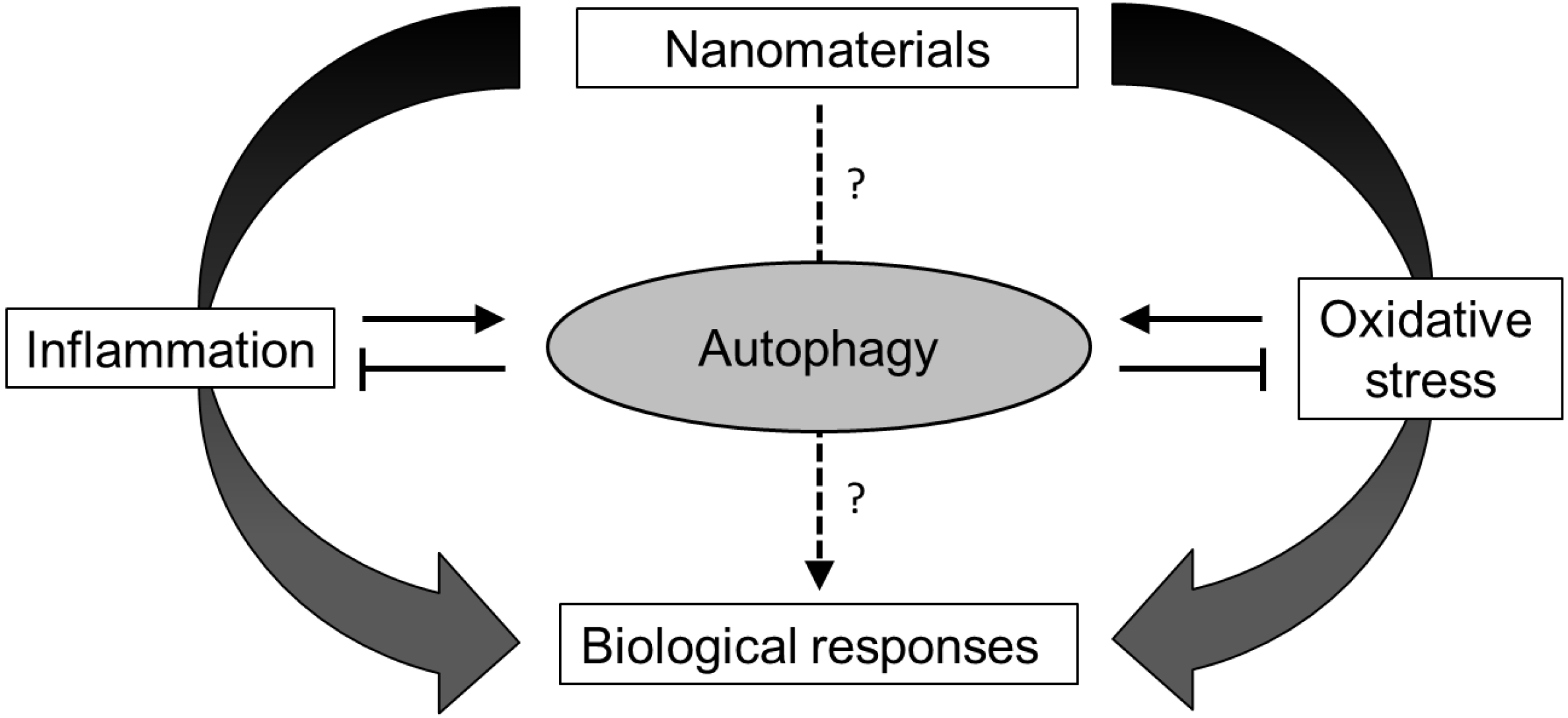
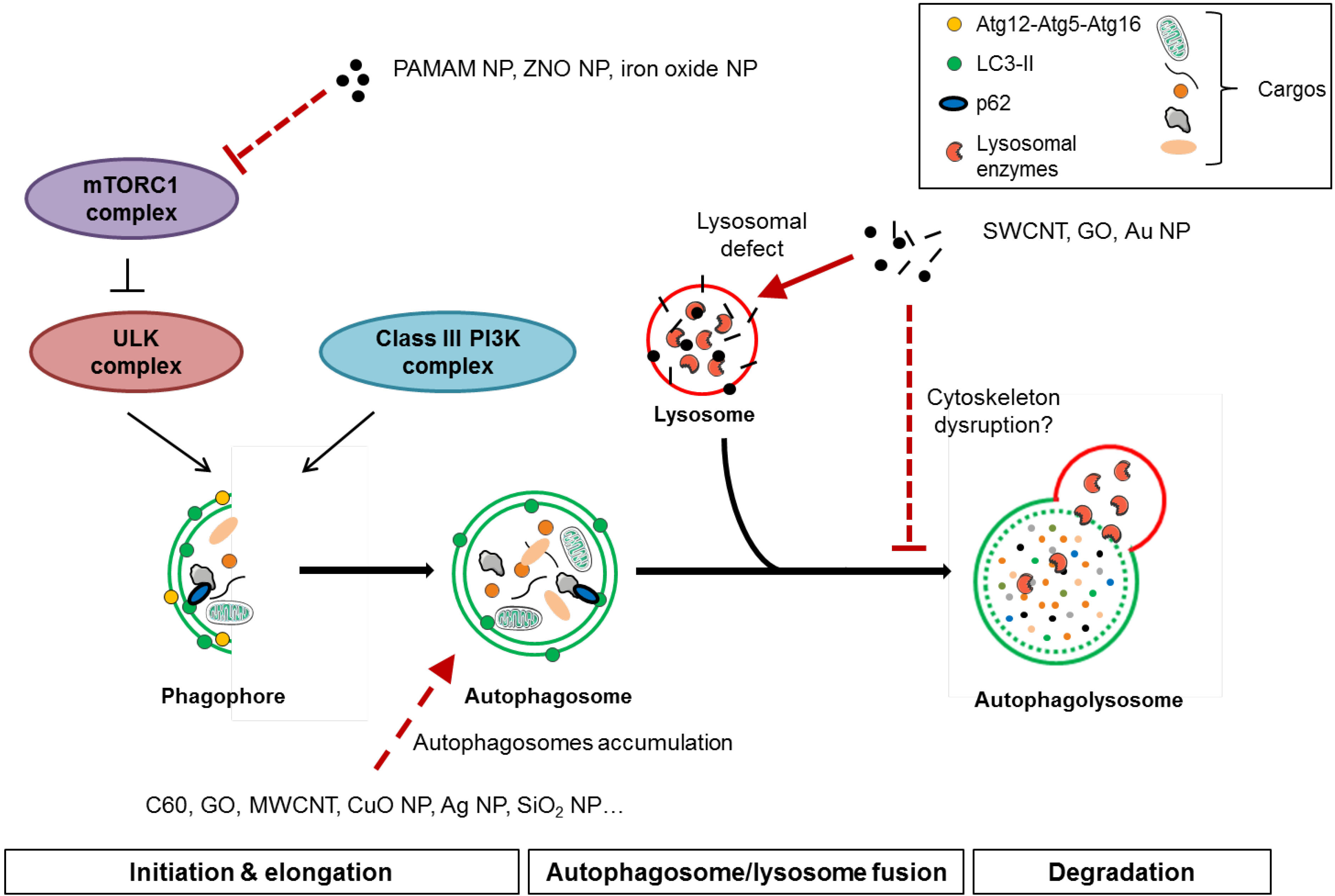
4. Nanomaterial-Induced Autophagy Perturbation
4.1. Evidences of Autophagy Perturbation by Nanomaterials

{kind=link}
{kind=link}
{kind=link}
| Nanomaterial | Model(s) | Autophagy markers | Experimental techniques | Results | Reference |
|---|---|---|---|---|---|
| Gold NP | MRC5 human lung fibroblast cell line | Beclin1, Atg5, Atg7, Atg12, LC3 | TEM, immunoblot | Increase of autophagosomes formation | [187] |
| Iron oxide NP | RAW 264.7 murine peritoneal macrophage cell line | Beclin1, Atg5, LC3, p62 | TEM, immunoblot, p62 immunostaining | Increase of autophagosomes formation | [178] |
| Silica NP | A549 lung epithelial cell line | LC3 | TEM, MDC staining, immunoblot | Increase of autophagosomes formation | [188] |
| Silver NP | NIH 3T3 mouse embryonic fibroblasts | Beclin1, LC3, p62 | TEM, acridin orange staining, immunoblot | Increase of autophagosomes formation | [177] |
| Zinc oxide NP | Mouse peritoneal macrophages | Atg5, Atg10, Atg12, LC3 | TEM, qRT-PCR, LC3 immunostaining, immunoblot | Increase of autophagosomes formation | [181] |
| Hydroxyl C60 fullerene NP | HUVEC human umbilical vein endothelial cell line | LC3 | TEM, immunoblot | Increase of autophagosomes formation | [189] |
| Polymeric NP | NR8383 rat alveolar macrophage cell line | Atg16L1, LC3 | TEM, microarray, immunoblot, qRT-PCR | Increase of autophagosomes formation | [190] |
| Graphene oxide nanosheets | RAW 264.7 murine peritoneal macrophage cell line | Beclin1, LC3 | TEM, immunoblot, immunostaining | Increase of autophagosomes formation | [179] |
| Silver nanowires | THP-1 monocytic cell line, iBMM cell line | LC3 | TEM, stable GFP-LC3 transfection, immunoblot | Increase of autophagosomes formation | [180] |
| Nanomaterial | Model(s) | Autophagy markers | Experimental techniques | Results | Reference |
|---|---|---|---|---|---|
| Copper oxide NP | A549 lung epithelial cell line | Atg5, LC3 | TEM, immunoblot, GFP-LC3 transfection, Atg5 siRNA | Increase of autophagosome formation with an increase of autophagy flux | [186] |
| Iron oxide NP | A549 lung epithelial cell line | Akt signaling, Atg5, Atg12, LC3 | Immunoblot | Accumulation of autophagosomes due to a decrease in autophagy flux | [182] |
| PAMAMdendrimer | A549 lung epithelial cell line, Balb/c mice | Atg6, LC3 | TEM, immunoblot, GFP-LC3 transfection | Accumulation of autophagosomes due to a decrease in autophagy flux | [176] |
| MWCNT | A549 lung epithelial cell line | LC3 | Immunoblot, qRT-PCR, GFP-LC3 transfection | Accumulation of autophagosomes due to a decrease in autophagy flux | [191] |
| SWCNT and graphene oxides | Mouse peritoneal macrophages | LC3, p62 | GFP-LC3 transfection, immunoblot, lysotracker | Accumulation of autophagosomes due to a decrease in autophagy flux and lysosomal impairment | [185] |
| Carboxylated MWCNT | HUVEC human umbilical vein endothelial cell line | LC3, p62 | TEM, immunoblot, RFP-LC3 and GFP-LC3 transfection | Accumulation of autophagosomes due to a decrease in autophagy flux | [184] |
4.2. Mechanisms of Autophagy Perturbation by Nanomaterials

5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- The Project on Emerging Nanotechnologies. Available online: http://www.nanotechproject.org (accessed on 23 May 2014).
- Kreyling, W.G.; Semmler-Behnke, M.; Seitz, J.; Scymczak, W.; Wenk, A.; Mayer, P.; Takenaka, S.; Oberdorster, G. Size dependence of the translocation of inhaled iridium and carbon nanoparticle aggregates from the lung of rats to the blood and secondary target organs. Inhal. Toxicol. 2009, 21, 55–60. [Google Scholar]
- Nemmar, A.; Vanbilloen, H.; Hoylaerts, M.F.; Hoet, P.H.; Verbruggen, A.; Nemery, B. Passage of intratracheally instilled ultrafine particles from the lung into the systemic circulation in hamster. Am. J. Respir. Crit. Care Med. 2001, 164, 1665–1668. [Google Scholar] [CrossRef]
- Oberdorster, G.; Oberdorster, E.; Oberdorster, J. Nanotoxicology: An emerging discipline evolving from studies of ultrafine particles. Environ. Health Perspect. 2005, 113, 823–839. [Google Scholar] [CrossRef]
- Lam, C.W.; James, J.T.; McCluskey, R.; Hunter, R.L. Pulmonary toxicity of single-wall carbon nanotubes in mice 7 and 90 days after intratracheal instillation. Toxicol. Sci. Off. J. Soc. Toxicol. 2004, 77, 126–134. [Google Scholar]
- Ho, C.C.; Chang, H.; Tsai, H.T.; Tsai, M.H.; Yang, C.S.; Ling, Y.C.; Lin, P. Quantum dot 705, a cadmium-based nanoparticle, induces persistent inflammation and granuloma formation in the mouse lung. Nanotoxicology 2013, 7, 105–115. [Google Scholar] [CrossRef]
- Mercer, R.R.; Hubbs, A.F.; Scabilloni, J.F.; Wang, L.; Battelli, L.A.; Friend, S.; Castranova, V.; Porter, D.W. Pulmonary fibrotic response to aspiration of multi-walled carbon nanotubes. Part. Fibre Toxicol. 2011, 8. [Google Scholar] [CrossRef]
- Murphy, F.A.; Poland, C.A.; Duffin, R.; Al-Jamal, K.T.; Ali-Boucetta, H.; Nunes, A.; Byrne, F.; Prina-Mello, A.; Volkov, Y.; Li, S.; et al. Length-dependent retention of carbon nanotubes in the pleural space of mice initiates sustained inflammation and progressive fibrosis on the parietal pleura. Am. J. Pathol. 2011, 178, 2587–2600. [Google Scholar] [CrossRef]
- Park, E.J.; Yoon, J.; Choi, K.; Yi, J.; Park, K. Induction of chronic inflammation in mice treated with titanium dioxide nanoparticles by intratracheal instillation. Toxicology 2009, 260, 37–46. [Google Scholar] [CrossRef]
- Tabet, L.; Bussy, C.; Setyan, A.; Simon-Deckers, A.; Rossi, M.J.; Boczkowski, J.; Lanone, S. Coating carbon nanotubes with a polystyrene-based polymer protects against pulmonary toxicity. Part. Fibre Toxicol. 2011, 8, 1–13. [Google Scholar] [CrossRef]
- Tada, Y.; Yano, N.; Takahashi, H.; Yuzawa, K.; Ando, H.; Kubo, Y.; Nagasawa, A.; Ogata, A.; Nakae, D. Acute phase pulmonary responses to a single intratracheal spray instillation of magnetite (Fe3O4) nanoparticles in fischer 344 rats. J. Toxicol. Pathol. 2012, 25, 233–239. [Google Scholar] [CrossRef]
- Coccini, T.; Barni, S.; Vaccarone, R.; Mustarelli, P.; Manzo, L.; Roda, E. Pulmonary toxicity of instilled cadmium-doped silica nanoparticles during acute and subacute stages in rats. Histol. Histopathol. 2013, 28, 195–209. [Google Scholar]
- Tada, Y.; Yano, N.; Takahashi, H.; Yuzawa, K.; Ando, H.; Kubo, Y.; Nagasawa, A.; Inomata, A.; Ogata, A.; Nakae, D. Long-term pulmonary responses to quadweekly intermittent intratracheal spray instillations of magnetite (Fe3O4) nanoparticles for 52 weeks in fischer 344 rats. J. Toxicol. Pathol. 2013, 26, 393–403. [Google Scholar] [CrossRef]
- Crouzier, D.; Follot, S.; Gentilhomme, E.; Flahaut, E.; Arnaud, R.; Dabouis, V.; Castellarin, C.; Debouzy, J.C. Carbon nanotubes induce inflammation but decrease the production of reactive oxygen species in lung. Toxicology 2010, 272, 39–45. [Google Scholar] [CrossRef]
- Lam, C.W.; James, J.T.; McCluskey, R.; Arepalli, S.; Hunter, R.L. A review of carbon nanotube toxicity and assessment of potential occupational and environmental health risks. Crit. Rev. Toxicol. 2006, 36, 189–217. [Google Scholar] [CrossRef]
- Shvedova, A.A.; Kisin, E.R.; Mercer, R.; Murray, A.R.; Johnson, V.J.; Potapovich, A.I.; Tyurina, Y.Y.; Gorelik, O.; Arepalli, S.; Schwegler-Berry, D.; et al. Unusual inflammatory and fibrogenic pulmonary responses to single-walled carbon nanotubes in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, L698–L708. [Google Scholar]
- Muller, J.; Huaux, F.; Moreau, N.; Misson, P.; Heilier, J.F.; Delos, M.; Arras, M.; Fonseca, A.; Nagy, J.B.; Lison, D. Respiratory toxicity of multi-wall carbon nanotubes. Toxicol. Appl. Pharmacol. 2005, 207, 221–231. [Google Scholar] [CrossRef]
- Chen, H.W.; Su, S.F.; Chien, C.T.; Lin, W.H.; Yu, S.L.; Chou, C.C.; Chen, J.J.; Yang, P.C. Titanium dioxide nanoparticles induce emphysema-like lung injury in mice. FASEB J. 2006, 20, 2393–2395. [Google Scholar] [CrossRef]
- Zhu, M.T.; Feng, W.Y.; Wang, Y.; Wang, B.; Wang, M.; Ouyang, H.; Zhao, Y.L.; Chai, Z.F. Particokinetics and extrapulmonary translocation of intratracheally instilled ferric oxide nanoparticles in rats and the potential health risk assessment. Toxicol. Sci. Off. J. Soc. Toxicol. 2009, 107, 342–351. [Google Scholar]
- Mossman, B.T.; Shukla, A.; Heintz, N.H.; Verschraegen, C.F.; Thomas, A.; Hassan, R. New insights into understanding the mechanisms, pathogenesis, and management of malignant mesotheliomas. Am. J. Pathol. 2013, 182, 1065–1077. [Google Scholar] [CrossRef]
- Donaldson, K.; Murphy, F.A.; Duffin, R.; Poland, C.A. Asbestos, carbon nanotubes and the pleural mesothelium: A review of the hypothesis regarding the role of long fibre retention in the parietal pleura, inflammation and mesothelioma. Part. Fibre Toxicol. 2010, 7. [Google Scholar] [CrossRef]
- Castranova, V.; Schulte, P.A.; Zumwalde, R.D. Occupational nanosafety considerations for carbon nanotubes and carbon nanofibers. Acc. Chem. Res. 2013, 46, 642–649. [Google Scholar] [CrossRef]
- Donaldson, K.; Poland, C.A.; Murphy, F.A.; Macfarlane, M.; Chernova, T.; Schinwald, A. Pulmonary toxicity of carbon nanotubes and asbestos—Similarities and differences. Adv. Drug Deliv. Rev. 2013, 65, 2078–2086. [Google Scholar] [CrossRef]
- Xu, J.; Futakuchi, M.; Shimizu, H.; Alexander, D.B.; Yanagihara, K.; Fukamachi, K.; Suzui, M.; Kanno, J.; Hirose, A.; Ogata, A.; et al. Multi-walled carbon nanotubes translocate into the pleural cavity and induce visceral mesothelial proliferation in rats. Cancer Sci. 2012, 103, 2045–2050. [Google Scholar] [CrossRef]
- Glista-Baker, E.E.; Taylor, A.J.; Sayers, B.C.; Thompson, E.A.; Bonner, J.C. Nickel nanoparticles cause exaggerated lung and airway remodeling in mice lacking the T-box transcription factor, TBX21 (T-bet). Part. Fibre Toxicol. 2014, 11, 7–22. [Google Scholar] [CrossRef]
- Ryman-Rasmussen, J.P.; Tewksbury, E.W.; Moss, O.R.; Cesta, M.F.; Wong, B.A.; Bonner, J.C. Inhaled multiwalled carbon nanotubes potentiate airway fibrosis in murine allergic asthma. Am. J. Respir. Cell Mol. Biol. 2009, 40, 349–358. [Google Scholar] [CrossRef]
- Inoue, K.; Koike, E.; Yanagisawa, R.; Hirano, S.; Nishikawa, M.; Takano, H. Effects of multi-walled carbon nanotubes on a murine allergic airway inflammation model. Toxicol. Appl. Pharmacol. 2009, 237, 306–316. [Google Scholar] [CrossRef]
- Cesta, M.F.; Ryman-Rasmussen, J.P.; Wallace, D.G.; Masinde, T.; Hurlburt, G.; Taylor, A.J.; Bonner, J.C. Bacterial lipopolysaccharide enhances PDGF signaling and pulmonary fibrosis in rats exposed to carbon nanotubes. Am. J. Respir. Cell Mol. Biol. 2010, 43, 142–151. [Google Scholar] [CrossRef]
- Shvedova, A.A.; Fabisiak, J.P.; Kisin, E.R.; Murray, A.R.; Roberts, J.R.; Tyurina, Y.Y.; Antonini, J.M.; Feng, W.H.; Kommineni, C.; Reynolds, J.; et al. Sequential exposure to carbon nanotubes and bacteria enhances pulmonary inflammation and infectivity. Am. J. Respir. Cell Mol. Biol. 2008, 38, 579–590. [Google Scholar] [CrossRef]
- Jonasson, S.; Gustafsson, A.; Koch, B.; Bucht, A. Inhalation exposure of nano-scaled titanium dioxide (TiO2) particles alters the inflammatory responses in asthmatic mice. Inhal. Toxicol. 2013, 25, 179–191. [Google Scholar] [CrossRef]
- Jang, S.; Park, J.W.; Cha, H.R.; Jung, S.Y.; Lee, J.E.; Jung, S.S.; Kim, J.O.; Kim, S.Y.; Lee, C.S.; Park, H.S. Silver nanoparticles modify VEGF signaling pathway and mucus hypersecretion in allergic airway inflammation. Int. J. Nanomed. 2012, 7, 1329–1343. [Google Scholar]
- Scarino, A.; Noel, A.; Renzi, P.M.; Cloutier, Y.; Vincent, R.; Truchon, G.; Tardif, R.; Charbonneau, M. Impact of emerging pollutants on pulmonary inflammation in asthmatic rats: Ethanol vapors and agglomerated TiO2 nanoparticles. Inhal. Toxicol. 2012, 24, 528–538. [Google Scholar] [CrossRef]
- Shvedova, A.A.; Pietroiusti, A.; Fadeel, B.; Kagan, V.E. Mechanisms of carbon nanotube-induced toxicity: Focus on oxidative stress. Toxicol. Appl. Pharmacol. 2012, 261, 121–133. [Google Scholar] [CrossRef]
- Ayres, J.G.; Borm, P.; Cassee, F.R.; Castranova, V.; Donaldson, K.; Ghio, A.; Harrison, R.M.; Hider, R.; Kelly, F.; Kooter, I.M.; et al. Evaluating the toxicity of airborne particulate matter and nanoparticles by measuring oxidative stress potential—A workshop report and consensus statement. Inhal. Toxicol. 2008, 20, 75–99. [Google Scholar] [CrossRef]
- Li, J.G.; Li, W.X.; Xu, J.Y.; Cai, X.Q.; Liu, R.L.; Li, Y.J.; Zhao, Q.F.; Li, Q.N. Comparative study of pathological lesions induced by multiwalled carbon nanotubes in lungs of mice by intratracheal instillation and inhalation. Environ. Toxicol. 2007, 22, 415–421. [Google Scholar] [CrossRef]
- Noel, A.; Maghni, K.; Cloutier, Y.; Dion, C.; Wilkinson, K.J.; Halle, S.; Tardif, R.; Truchon, G. Effects of inhaled nano-TiO2 aerosols showing two distinct agglomeration states on rat lungs. Toxicol. Lett. 2012, 214, 109–119. [Google Scholar] [CrossRef]
- Park, E.J.; Kim, H.; Kim, Y.; Yi, J.; Choi, K.; Park, K. Inflammatory responses may be induced by a single intratracheal instillation of iron nanoparticles in mice. Toxicology 2010, 275, 65–71. [Google Scholar] [CrossRef]
- Sarkar, A.; Ghosh, M.; Sil, P.C. Nanotoxicity: Oxidative stress mediated toxicity of metal and metal oxide nanoparticles. J. Nanosci. Nanotechnol. 2014, 14, 730–743. [Google Scholar] [CrossRef]
- De Angelis, I.; Barone, F.; Zijno, A.; Bizzarri, L.; Russo, M.T.; Pozzi, R.; Franchini, F.; Giudetti, G.; Uboldi, C.; Ponti, J.; et al. Comparative study of ZnO and TiO2 nanoparticles: Physicochemical characterisation and toxicological effects on human colon carcinoma cells. Nanotoxicology 2013, 7, 1361–1372. [Google Scholar] [CrossRef]
- Foldbjerg, R.; Dang, D.A.; Autrup, H. Cytotoxicity and genotoxicity of silver nanoparticles in the human lung cancer cell line, a549. Arch. Toxicol. 2011, 85, 743–750. [Google Scholar] [CrossRef]
- Shvedova, A.A.; Kisin, E.R.; Murray, A.R.; Gorelik, O.; Arepalli, S.; Castranova, V.; Young, S.H.; Gao, F.; Tyurina, Y.Y.; Oury, T.D.; et al. Vitamin E deficiency enhances pulmonary inflammatory response and oxidative stress induced by single-walled carbon nanotubes in C57BL/6 mice. Toxicol. Appl. Pharmacol. 2007, 221, 339–348. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; de Haan, L.H.; Evers, N.M.; Jiang, X.; Marcelis, A.T.; Zuilhof, H.; Rietjens, I.M.; Alink, G.M. Role of surface charge and oxidative stress in cytotoxicity of organic monolayer-coated silicon nanoparticles towards macrophage NR8383 cells. Part. Fibre Toxicol. 2010, 7. [Google Scholar] [CrossRef]
- Li, J.; Li, L.; Chen, H.; Chang, Q.; Liu, X.; Wu, Y.; Wei, C.; Li, R.; Kwan, J.K.; Yeung, K.L.; et al. Application of vitamin E to antagonize SWCNTs-induced exacerbation of allergic asthma. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef]
- Shvedova, A.A.; Kisin, E.R.; Murray, A.R.; Kommineni, C.; Castranova, V.; Fadeel, B.; Kagan, V.E. Increased accumulation of neutrophils and decreased fibrosis in the lung of nadph oxidase-deficient C57BL/6 mice exposed to carbon nanotubes. Toxicol. Appl. Pharmacol. 2008, 231, 235–240. [Google Scholar] [CrossRef]
- Aalapati, S.; Ganapathy, S.; Manapuram, S.; Anumolu, G.; Prakya, B.M. Toxicity and bio-accumulation of inhaled cerium oxide nanoparticles in cd1 mice. Nanotoxicology 2014, 8, 786–798. [Google Scholar]
- Baisch, B.L.; Corson, N.M.; Wade-Mercer, P.; Gelein, R.; Kennell, A.J.; Oberdorster, G.; Elder, A. Equivalent titanium dioxide nanoparticle deposition by intratracheal instillation and whole body inhalation: The effect of dose rate on acute respiratory tract inflammation. Part. Fibre Toxicol. 2014, 11. [Google Scholar] [CrossRef]
- Brown, D.M.; Kanase, N.; Gaiser, B.; Johnston, H.; Stone, V. Inflammation and gene expression in the rat lung after instillation of silica nanoparticles: Effect of size, dispersion medium and particle surface charge. Toxicol. Lett. 2014, 224, 147–156. [Google Scholar] [CrossRef]
- Braakhuis, H.M.; Park, M.V.; Gosens, I.; De Jong, W.H.; Cassee, F.R. Physicochemical characteristics of nanomaterials that affect pulmonary inflammation. Part. Fibre Toxicol. 2014, 11. [Google Scholar] [CrossRef]
- Di, P.Y.; Tkach, A.V.; Yanamala, N.; Stanley, S.; Gao, S.; Shurin, M.R.; Kisin, E.R.; Kagan, V.E.; Shvedova, A.A. Dual acute proinflammatory and antifibrotic pulmonary effects of short palate, lung, and nasal epithelium clone-1 after exposure to carbon nanotubes. Am. J. Respir. Cell Mol. Biol. 2013, 49, 759–767. [Google Scholar] [CrossRef]
- Blum, J.L.; Rosenblum, L.K.; Grunig, G.; Beasley, M.B.; Xiong, J.Q.; Zelikoff, J.T. Short-term inhalation of cadmium oxide nanoparticles alters pulmonary dynamics associated with lung injury, inflammation, and repair in a mouse model. Inhal. Toxicol. 2014, 26, 48–58. [Google Scholar] [CrossRef]
- Lee, J.K.; Sayers, B.C.; Chun, K.S.; Lao, H.C.; Shipley-Phillips, J.K.; Bonner, J.C.; Langenbach, R. Multi-walled carbon nanotubes induce COX-2 and iNOS expression via MAP kinase-dependent and -independent mechanisms in mouse RAW264.7 macrophages. Part. Fibre Toxicol. 2012, 9. [Google Scholar] [CrossRef]
- Armand, L.; Dagouassat, M.; Belade, E.; Simon-Deckers, A.; Le Gouvello, S.; Tharabat, C.; Duprez, C.; Andujar, P.; Pairon, J.C.; Boczkowski, J.; et al. Titanium dioxide nanoparticles induce matrix metalloprotease 1 in human pulmonary fibroblasts partly via an interleukin-1beta-dependent mechanism. Am. J. Respir. Cell Mol. Biol. 2013, 48, 354–363. [Google Scholar] [CrossRef]
- Abbott Chalew, T.E.; Schwab, K.J. Toxicity of commercially available engineered nanoparticles to Caco-2 and SW480 human intestinal epithelial cells. Cell Biol. Toxicol. 2013, 29, 101–116. [Google Scholar] [CrossRef]
- Murphy, F.A.; Schinwald, A.; Poland, C.A.; Donaldson, K. The mechanism of pleural inflammation by long carbon nanotubes: Interaction of long fibres with macrophages stimulates them to amplify pro-inflammatory responses in mesothelial cells. Part. Fibre Toxicol. 2012, 9. [Google Scholar] [CrossRef]
- Donaldson, K.; Poland, C.A.; Schins, R.P. Possible genotoxic mechanisms of nanoparticles: Criteria for improved test strategies. Nanotoxicology 2010, 4, 414–420. [Google Scholar] [CrossRef]
- Hubbs, A.F.; Mercer, R.R.; Benkovic, S.A.; Harkema, J.; Sriram, K.; Schwegler-Berry, D.; Goravanahally, M.P.; Nurkiewicz, T.R.; Castranova, V.; Sargent, L.M. Nanotoxicology—A pathologist’s perspective. Toxicol. Pathol. 2011, 39, 301–324. [Google Scholar] [CrossRef]
- Kumar, A.; Dhawan, A. Genotoxic and carcinogenic potential of engineered nanoparticles: An update. Arch. Toxicol. 2013, 87, 1883–1900. [Google Scholar] [CrossRef]
- Muller, J.; Decordier, I.; Hoet, P.H.; Lombaert, N.; Thomassen, L.; Huaux, F.; Lison, D.; Kirsch-Volders, M. Clastogenic and aneugenic effects of multi-wall carbon nanotubes in epithelial cells. Carcinogenesis 2008, 29, 427–433. [Google Scholar] [CrossRef]
- Trouiller, B.; Reliene, R.; Westbrook, A.; Solaimani, P.; Schiestl, R.H. Titanium dioxide nanoparticles induce DNA damage and genetic instability in vivo in mice. Cancer Res. 2009, 69, 8784–8789. [Google Scholar] [CrossRef]
- Hackenberg, S.; Scherzed, A.; Kessler, M.; Hummel, S.; Technau, A.; Froelich, K.; Ginzkey, C.; Koehler, C.; Hagen, R.; Kleinsasser, N. Silver nanoparticles: Evaluation of DNA damage, toxicity and functional impairment in human mesenchymal stem cells. Toxicol. Lett. 2011, 201, 27–33. [Google Scholar] [CrossRef]
- Di Virgilio, A.L.; Reigosa, M.; Arnal, P.M.; Fernández Lorenzo de Mele, M. Comparative study of the cytotoxic and genotoxic effects of titanium oxide and aluminium oxide nanoparticles in chinese hamster ovary (CHO-K1) cells. J. Hazard Mater. 2010, 177, 711–718. [Google Scholar] [CrossRef]
- An, H.; Liu, Q.; Ji, Q.; Jin, B. DNA binding and aggregation by carbon nanoparticles. Biochem. Biophys. Res. Commun. 2010, 393, 571–576. [Google Scholar] [CrossRef]
- Jin, P.; Chen, Y.; Zhang, S.B.; Chen, Z. Interactions between Al12X (X = Al, C, N and P) nanoparticles and DNA nucleobases/base pairs: Implications for nanotoxicity. J. Mol. Model. 2012, 18, 559–568. [Google Scholar] [CrossRef]
- Kain, J.; Karlsson, H.L.; Möller, L. DNA damage induced by micro- and nanoparticles-interaction with FPG influences the detection of DNA oxidation in the comet assay. Mutagenesis 2012, 27, 491–500. [Google Scholar] [CrossRef]
- Baweja, L.; Gurbani, D.; Shanker, R.; Pandey, A.K.; Subramanian, V.; Dhawan, A. C60-fullerene binds with the ATP binding domain of human DNA topoiosmerase II alpha. J. Biomed. Nanotechnol. 2011, 7, 177–178. [Google Scholar] [CrossRef]
- Gupta, S.K.; Baweja, L.; Gurbani, D.; Pandey, A.K.; Dhawan, A. Interaction of C60 fullerene with the proteins involved in DNA mismatch repair pathway. J. Biomed. Nanotechnol. 2011, 7, 179–180. [Google Scholar] [CrossRef]
- Jugan, M.L.; Barillet, S.; Simon-Deckers, A.; Herlin-Boime, N.; Sauvaigo, S.; Douki, T.; Carriere, M. Titanium dioxide nanoparticles exhibit genotoxicity and impair DNA repair activity in A549 cells. Nanotoxicology 2012, 6, 501–513. [Google Scholar] [CrossRef]
- Hackenberg, S.; Friehs, G.; Kessler, M.; Froelich, K.; Ginzkey, C.; Koehler, C.; Scherzed, A.; Burghartz, M.; Kleinsasser, N. Nanosized titanium dioxide particles do not induce DNA damage in human peripheral blood lymphocytes. Environ. Mol. Mutagen. 2011, 52, 264–268. [Google Scholar] [CrossRef]
- Wirnitzer, U.; Herbold, B.; Voetz, M.; Ragot, J. Studies on the in vitro genotoxicity of baytubes, agglomerates of engineered multi-walled carbon-nanotubes (mwcnt). Toxicol. Lett. 2009, 186, 160–165. [Google Scholar] [CrossRef]
- Guo, Y.Y.; Zhang, J.; Zheng, Y.F.; Yang, J.; Zhu, X.Q. Cytotoxic and genotoxic effects of multi-wall carbon nanotubes on human umbilical vein endothelial cells in vitro. Mutat. Res. 2011, 721, 184–191. [Google Scholar]
- Sharma, V.; Anderson, D.; Dhawan, A. Zinc oxide nanoparticles induce oxidative DNA damage and ROS-triggered mitochondria mediated apoptosis in human liver cells (HepG2). Apoptosis 2012, 17, 852–870. [Google Scholar]
- Lynch, I.; Cedervall, T.; Lundqvist, M.; Cabaleiro-Lago, C.; Linse, S.; Dawson, K.A. The nanoparticle-protein complex as a biological entity; a complex fluids and surface science challenge for the 21st century. Adv. Colloid Interface Sci. 2007, 134–135, 167–174. [Google Scholar] [CrossRef]
- Shannahan, J.H.; Lai, X.; Ke, P.C.; Podila, R.; Brown, J.M.; Witzmann, F.A. Silver nanoparticle protein corona composition in cell culture media. PLoS One 2013, 8, e74001. [Google Scholar]
- Podila, R.; Vedantam, P.; Ke, P.C.; Brown, J.M.; Rao, A.M. Evidences for charge transfer-induced conformational changes in carbon nanostructure-protein corona. J. Phys. Chem. C Nanomater. Interfaces 2012, 116, 22098–22103. [Google Scholar] [CrossRef]
- Cedervall, T.; Lynch, I.; Lindman, S.; Berggard, T.; Thulin, E.; Nilsson, H.; Dawson, K.A.; Linse, S. Understanding the nanoparticle-protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc. Natl. Acad. Sci. USA 2007, 104, 2050–2055. [Google Scholar] [CrossRef]
- Lundqvist, M.; Stigler, J.; Elia, G.; Lynch, I.; Cedervall, T.; Dawson, K.A. Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proc. Natl. Acad. Sci. USA 2008, 105, 14265–14270. [Google Scholar] [CrossRef]
- Nel, A.E.; Madler, L.; Velegol, D.; Xia, T.; Hoek, E.M.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano-bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef]
- Johnston, H.; Brown, D.; Kermanizadeh, A.; Gubbins, E.; Stone, V. Investigating the relationship between nanomaterial hazard and physicochemical properties: Informing the exploitation of nanomaterials within therapeutic and diagnostic applications. J. Control Release 2012, 164, 307–313. [Google Scholar] [CrossRef]
- Ge, C.; Du, J.; Zhao, L.; Wang, L.; Liu, Y.; Li, D.; Yang, Y.; Zhou, R.; Zhao, Y.; Chai, Z.; et al. Binding of blood proteins to carbon nanotubes reduces cytotoxicity. Proc. Natl. Acad. Sci. USA 2011, 108, 16968–16973. [Google Scholar]
- Salvador-Morales, C.; Townsend, P.; Flahaut, E.; Venien-Bryan, C.; Vlandas, A.; Green, M.L.H.; Sim, R.B. Binding of pulmonary surfactant proteins to carbon nanotubes: Potential for damage to lung immune defense mechanisms. Carbon 2007, 45, 607–617. [Google Scholar] [CrossRef]
- Du, J.; Ge, C.; Liu, Y.; Bai, R.; Li, D.; Yang, Y.; Liao, L.; Chen, C. The interaction of serum proteins with carbon nanotubes depend on the physicochemical properties of nanotubes. J. Nanosci. Nanotechnol. 2011, 11, 10102–10110. [Google Scholar] [CrossRef]
- Banerjee, V.; Das, K.P. Structure and functional properties of a multimeric protein alphaa-crystallin adsorbed on silver nanoparticle surface. Langmuir 2014, 30, 4775–4783. [Google Scholar] [CrossRef]
- Zhang, B.; Xing, Y.; Li, Z.; Zhou, H.; Mu, Q.; Yan, B. Functionalized carbon nanotubes specifically bind to alpha-chymotrypsin's catalytic site and regulate its enzymatic function. Nano Lett. 2009, 9, 2280–2284. [Google Scholar] [CrossRef]
- Treuel, L.; Brandholt, S.; Maffre, P.; Wiegele, S.; Shang, L.; Nienhaus, G.U. Impact of protein modification on the protein corona on nanoparticles and nanoparticle-cell interactions. ACS Nano 2014, 8, 503–513. [Google Scholar] [CrossRef]
- Saptarshi, S.R.; Duschl, A.; Lopata, A.L. Interaction of nanoparticles with proteins: Relation to bio-reactivity of the nanoparticle. J. Nanobiotechnol. 2013, 11, 26–37. [Google Scholar] [CrossRef]
- Donaldson, K. Resolving the nanoparticles paradox. Nanomed. Lond. 2006, 1, 229–234. [Google Scholar] [CrossRef]
- Lanone, S.; Rogerieux, F.; Geys, J.; Dupont, A.; Maillot-Marechal, E.; Boczkowski, J.; Lacroix, G.; Hoet, P. Comparative toxicity of 24 manufactured nanoparticles in human alveolar epithelial and macrophage cell lines. Part. Fibre Toxicol. 2009, 6, 14–25. [Google Scholar] [CrossRef]
- Tian, F.; Cui, D.; Schwarz, H.; Estrada, G.G.; Kobayashi, H. Cytotoxicity of single-wall carbon nanotubes on human fibroblasts. Toxicol. In Vitro 2006, 20, 1202–1212. [Google Scholar] [CrossRef]
- Petkovic, J.; Zegura, B.; Stevanovic, M.; Drnovsek, N.; Uskokovic, D.; Novak, S.; Filipic, M. DNA damage and alterations in expression of DNA damage responsive genes induced by TiO2 nanoparticles in human hepatoma HepG2 cells. Nanotoxicology 2011, 5, 341–353. [Google Scholar] [CrossRef]
- Bussy, C.; Pinault, M.; Cambedouzou, J.; Landry, M.J.; Jegou, P.; Mayne-L’hermite, M.; Launois, P.; Boczkowski, J.; Lanone, S. Critical role of surface chemical modifications induced by length shortening on multi-walled carbon nanotubes-induced toxicity. Part. Fibre Toxicol. 2012, 9. [Google Scholar] [CrossRef]
- Kagan, V.E.; Tyurina, Y.Y.; Tyurin, V.A.; Konduru, N.V.; Potapovich, A.I.; Osipov, A.N.; Kisin, E.R.; Schwegler-Berry, D.; Mercer, R.; Castranova, V.; et al. Direct and indirect effects of single walled carbon nanotubes on RAW 264.7 macrophages: Role of iron. Toxicol. Lett. 2006, 165, 88–100. [Google Scholar] [CrossRef]
- Shvedova, A.A.; Castranova, V.; Kisin, E.R.; Schwegler-Berry, D.; Murray, A.R.; Gandelsman, V.Z.; Maynard, A.; Baron, P. Exposure to carbon nanotube material: Assessment of nanotube cytotoxicity using human keratinocyte cells. J. Toxicol. Environ. Health A 2003, 66, 1909–1926. [Google Scholar] [CrossRef]
- Bussy, C.; Paineau, E.; Cambedouzou, J.; Brun, N.; Mory, C.; Fayard, B.; Salome, M.; Pinault, M.; Huard, M.; Belade, E.; et al. Intracellular fate of carbon nanotubes inside murine macrophages: Ph-dependent detachment of iron catalyst nanoparticles. Part. Fibre Toxicol. 2013, 10. [Google Scholar] [CrossRef]
- Hamzeh, M.; Sunahara, G.I. In vitro cytotoxicity and genotoxicity studies of titanium dioxide (TiO2) nanoparticles in chinese hamster lung fibroblast cells. Toxicol. In Vitro 2013, 27, 864–873. [Google Scholar] [CrossRef]
- Patnaik, A. Structure and dynamics in self-organized C60 fullerenes. J. Nanosci. Nanotechnol. 2007, 7, 1111–1150. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Ryter, S.W.; Nakahira, K.; Haspel, J.A.; Choi, A.M. Autophagy in pulmonary diseases. Annu. Rev. Physiol. 2012, 74, 377–401. [Google Scholar] [CrossRef]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef]
- Yla-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 2009, 5, 1180–1185. [Google Scholar] [CrossRef]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef]
- Geng, J.; Nair, U.; Yasumura-Yorimitsu, K.; Klionsky, D.J. Post-Golgi Sec proteins are required for autophagy in saccharomyces cerevisiae. Mol. Biol. Cell 2010, 21, 2257–2269. [Google Scholar] [CrossRef]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef]
- Burman, C.; Ktistakis, N.T. Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett. 2010, 584, 1302–1312. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Fimia, G.M.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007, 447, 1121–1125. [Google Scholar]
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.H.; Jung, J.U. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 2006, 8, 688–699. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mulé, J.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar] [CrossRef]
- Kuma, A.; Mizushima, N.; Ishihara, N.; Ohsumi, Y. Formation of the approximately 350-kDa Apg12-Apg5.Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J. Biol. Chem. 2002, 277, 18619–18625. [Google Scholar] [CrossRef]
- Mizushima, N.; Kuma, A.; Kobayashi, Y.; Yamamoto, A.; Matsubae, M.; Takao, T.; Natsume, T.; Ohsumi, Y.; Yoshimori, T. Mouse Apg16l, a novel wd-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J. Cell Sci. 2003, 116, 1679–1688. [Google Scholar] [CrossRef]
- Tanida, I.; Sou, Y.S.; Ezaki, J.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. J. Biol. Chem. 2004, 279, 36268–36276. [Google Scholar]
- Tanida, I.; Ueno, T.; Kominami, E. Lc3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2503–2518. [Google Scholar] [CrossRef]
- Hanada, T.; Noda, N.N.; Satomi, Y.; Ichimura, Y.; Fujioka, Y.; Takao, T.; Inagaki, F.; Ohsumi, Y. The Atg12-Atg5 conjugate has a novel e3-like activity for protein lipidation in autophagy. J. Biol. Chem. 2007, 282, 37298–37302. [Google Scholar]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16l complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef]
- Jahreiss, L.; Menzies, F.M.; Rubinsztein, D.C. The itinerary of autophagosomes: From peripheral formation to kiss-and-run fusion with lysosomes. Traffic 2008, 9, 574–587. [Google Scholar] [CrossRef]
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Funct. 2008, 33, 109–122. [Google Scholar] [CrossRef]
- Ravikumar, B.; Acevedo-Arozena, A.; Imarisio, S.; Berger, Z.; Vacher, C.; O’Kane, C.J.; Brown, S.D.; Rubinsztein, D.C. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat. Genet. 2005, 37, 771–776. [Google Scholar] [CrossRef]
- Webb, J.L.; Ravikumar, B.; Rubinsztein, D.C. Microtubule disruption inhibits autophagosome-lysosome fusion: Implications for studying the roles of aggresomes in polyglutamine diseases. Int. J. Biochem. Cell Biol. 2004, 36, 2541–2550. [Google Scholar] [CrossRef]
- Gutierrez, M.G.; Vazquez, C.L.; Munafo, D.B.; Zoppino, F.C.; Beron, W.; Rabinovitch, M.; Colombo, M.I. Autophagy induction favours the generation and maturation of the coxiella-replicative vacuoles. Cell. Microbiol. 2005, 7, 981–993. [Google Scholar] [CrossRef]
- Jager, S.; Bucci, C.; Tanida, I.; Ueno, T.; Kominami, E.; Saftig, P.; Eskelinen, E.L. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004, 117, 4837–4848. [Google Scholar] [CrossRef]
- Liang, C.; Lee, J.S.; Inn, K.S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 2008, 10, 776–787. [Google Scholar] [CrossRef]
- Renna, M.; Schaffner, C.; Winslow, A.R.; Menzies, F.M.; Peden, A.A.; Floto, R.A.; Rubinsztein, D.C. Autophagic substrate clearance requires activity of the syntaxin-5 snare complex. J. Cell Sci. 2011, 124, 469–482. [Google Scholar] [CrossRef]
- Tanaka, Y.; Guhde, G.; Suter, A.; Eskelinen, E.L.; Hartmann, D.; Lullmann-Rauch, R.; Janssen, P.M.; Blanz, J.; von Figura, K.; Saftig, P. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 2000, 406, 902–906. [Google Scholar] [CrossRef]
- Tanida, I.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005, 1, 84–91. [Google Scholar] [CrossRef]
- Berg, T.O.; Fengsrud, M.; Stromhaug, P.E.; Berg, T.; Seglen, P.O. Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J. Biol. Chem. 1998, 273, 21883–21892. [Google Scholar]
- Lee, J.A.; Beigneux, A.; Ahmad, S.T.; Young, S.G.; Gao, F.B. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr. Biol. 2007, 17, 1561–1567. [Google Scholar]
- Rusten, T.E.; Stenmark, H. How do ESCRT proteins control autophagy? J. Cell Sci. 2009, 122, 2179–2183. [Google Scholar]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef]
- Mizushima, N. Methods for monitoring autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2491–2502. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef]
- Bjorkoy, G.; Lamark, T.; Pankiv, S.; Overvatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef]
- Chen, Z.H.; Kim, H.P.; Sciurba, F.C.; Lee, S.J.; Feghali-Bostwick, C.; Stolz, D.B.; Dhir, R.; Landreneau, R.J.; Schuchert, M.J.; Yousem, S.A.; et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One 2008, 3, e3316. [Google Scholar] [CrossRef]
- Hwang, J.W.; Chung, S.; Sundar, I.K.; Yao, H.; Arunachalam, G.; McBurney, M.W.; Rahman, I. Cigarette smoke-induced autophagy is regulated by SIRT1-PARP-1-dependent mechanism: Implication in pathogenesis of COPD. Arch. Biochem. Biophys. 2010, 500, 203–209. [Google Scholar] [CrossRef]
- Kim, H.P.; Wang, X.; Chen, Z.H.; Lee, S.J.; Huang, M.H.; Wang, Y.; Ryter, S.W.; Choi, A.M. Autophagic proteins regulate cigarette smoke-induced apoptosis: Protective role of heme oxygenase-1. Autophagy 2008, 4, 887–895. [Google Scholar]
- Chen, Z.H.; Lam, H.C.; Jin, Y.; Kim, H.P.; Cao, J.; Lee, S.J.; Ifedigbo, E.; Parameswaran, H.; Ryter, S.W.; Choi, A.M. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc. Natl. Acad. Sci. USA 2010, 107, 18880–18885. [Google Scholar] [CrossRef]
- Monick, M.M.; Powers, L.S.; Walters, K.; Lovan, N.; Zhang, M.; Gerke, A.; Hansdottir, S.; Hunninghake, G.W. Identification of an autophagy defect in smokers’ alveolar macrophages. J. Immunol. 2010, 185, 5425–5435. [Google Scholar] [CrossRef]
- Luciani, A.; Villella, V.R.; Esposito, S.; Brunetti-Pierri, N.; Medina, D.; Settembre, C.; Gavina, M.; Pulze, L.; Giardino, I.; Pettoello-Mantovani, M.; et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat. Cell Biol. 2010, 12, 863–875. [Google Scholar] [CrossRef]
- Abdulrahman, B.A.; Khweek, A.A.; Akhter, A.; Caution, K.; Kotrange, S.; Abdelaziz, D.H.; Newland, C.; Rosales-Reyes, R.; Kopp, B.; McCoy, K.; et al. Autophagy stimulation by rapamycin suppresses lung inflammation and infection by burkholderia cenocepacia in a model of cystic fibrosis. Autophagy 2011, 7, 1359–1370. [Google Scholar] [CrossRef]
- Lee, S.J.; Smith, A.; Guo, L.; Alastalo, T.P.; Li, M.; Sawada, H.; Liu, X.; Chen, Z.H.; Ifedigbo, E.; Jin, Y.; et al. Autophagic protein LC3B confers resistance against hypoxia-induced pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2011, 183, 649–658. [Google Scholar] [CrossRef]
- Haspel, J.A.; Choi, A.M. Autophagy: A core cellular process with emerging links to pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 184, 1237–1246. [Google Scholar] [CrossRef]
- Mizumura, K.; Cloonan, S.M.; Haspel, J.A.; Choi, A.M. The emerging importance of autophagy in pulmonary diseases. Chest 2012, 142, 1289–1299. [Google Scholar] [CrossRef]
- Ryter, S.W. Bile pigments in pulmonary and vascular disease. Front. Pharmacol. 2012, 3, 39–46. [Google Scholar] [CrossRef]
- Arico, S.; Petiot, A.; Bauvy, C.; Dubbelhuis, P.F.; Meijer, A.J.; Codogno, P.; Ogier-Denis, E. The tumor suppressor pten positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 2001, 276, 35243–35246. [Google Scholar]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by Beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the Beclin 1 autophagy gene. J. Clin. Invest. 2003, 112, 1809–1820. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef]
- Karantza-Wadsworth, V.; Patel, S.; Kravchuk, O.; Chen, G.; Mathew, R.; Jin, S.; White, E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007, 21, 1621–1635. [Google Scholar] [CrossRef]
- Levine, B. Cell biology: Autophagy and cancer. Nature 2007, 446, 745–747. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16l1 enhances endotoxin-induced IL-1beta production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Ling, J.; Kang, Y.; Zhao, R.; Xia, Q.; Lee, D.F.; Chang, Z.; Li, J.; Peng, B.; Fleming, J.B.; Wang, H.; et al. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 105–120. [Google Scholar] [CrossRef]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef]
- Moscat, J.; Diaz-Meco, M.T. P62: A versatile multitasker takes on cancer. Trends Biochem. Sci. 2012, 37, 230–236. [Google Scholar] [CrossRef]
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007, 447, 661–678. [Google Scholar] [CrossRef]
- Henckaerts, L.; Cleynen, I.; Brinar, M.; John, J.M.; Van Steen, K.; Rutgeerts, P.; Vermeire, S. Genetic variation in the autophagy gene ULK1 and risk of crohn’s disease. Inflamm. Bowel Dis. 2011, 17, 1392–1397. [Google Scholar] [CrossRef]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009, 16, 1040–1052. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Zhang, H.; Kong, X.; Kang, J.; Su, J.; Li, Y.; Zhong, J.; Sun, L. Oxidative stress induces parallel autophagy and mitochondria dysfunction in human glioma U251 cells. Toxicol. Sci. Off. J. Soc. Toxicol. 2009, 110, 376–388. [Google Scholar] [CrossRef]
- Farombi, E.O. Genotoxicity of chloroquine in rat liver cells: Protective role of free radical scavengers. Cell Biol. Toxicol. 2006, 22, 159–167. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Park, B.C.; Park, S.H.; Paek, S.H.; Park, S.Y.; Kwak, M.K.; Choi, H.G.; Yong, C.S.; Yoo, B.K.; Kim, J.A. Chloroquine-induced nitric oxide increase and cell death is dependent on cellular GSH depletion in A172 human glioblastoma cells. Toxicol. Lett. 2008, 178, 52–60. [Google Scholar] [CrossRef]
- Park, J.; Choi, K.; Jeong, E.; Kwon, D.; Benveniste, E.N.; Choi, C. Reactive oxygen species mediate chloroquine-induced expression of chemokines by human astroglial cells. Glia 2004, 47, 9–20. [Google Scholar] [CrossRef]
- Yamasaki, R.; Zhang, J.; Koshiishi, I.; Sastradipura Suniarti, D.F.; Wu, Z.; Peters, C.; Schwake, M.; Uchiyama, Y.; Kira, J.; Saftig, P.; et al. Involvement of lysosomal storage-induced p38 map kinase activation in the overproduction of nitric oxide by microglia in cathepsin D-deficient mice. Mol. Cell. Neurosci. 2007, 35, 573–584. [Google Scholar] [CrossRef]
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef]
- Kurihara, Y.; Kanki, T.; Aoki, Y.; Hirota, Y.; Saigusa, T.; Uchiumi, T.; Kang, D. Mitophagy plays an essential role in reducing mitochondrial production of reactive oxygen species and mutation of mitochondrial DNA by maintaining mitochondrial quantity and quality in yeast. J. Biol. Chem. 2012, 287, 3265–3272. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Autophagy as a cell-repair mechanism: Activation of chaperone-mediated autophagy during oxidative stress. Mol. Aspects Med. 2006, 27, 444–454. [Google Scholar] [CrossRef]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of chaperone-mediated autophagy during oxidative stress. Mol. Biol. Cell 2004, 15, 4829–4840. [Google Scholar] [CrossRef]
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. P62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor NRF2 through inactivation of KEAP1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar]
- Li, C.; Liu, H.; Sun, Y.; Wang, H.; Guo, F.; Rao, S.; Deng, J.; Zhang, Y.; Miao, Y.; Guo, C.; et al. Pamam nanoparticles promote acute lung injury by inducing autophagic cell death through the Akt-TSC2-mTOR signaling pathway. J. Mol. Cell Biol. 2009, 1, 37–45. [Google Scholar] [CrossRef]
- Lee, Y.H.; Cheng, F.Y.; Chiu, H.W.; Tsai, J.C.; Fang, C.Y.; Chen, C.W.; Wang, Y.J. Cytotoxicity, oxidative stress, apoptosis and the autophagic effects of silver nanoparticles in mouse embryonic fibroblasts. Biomaterials 2014, 35, 4706–4715. [Google Scholar] [CrossRef]
- Park, E.J.; Umh, H.N.; Kim, S.W.; Cho, M.H.; Kim, J.H.; Kim, Y. ERK pathway is activated in bare-FeNPs-induced autophagy. Arch. Toxicol. 2014, 88, 323–336. [Google Scholar] [CrossRef]
- Chen, G.Y.; Yang, H.J.; Lu, C.H.; Chao, Y.C.; Hwang, S.M.; Chen, C.L.; Lo, K.W.; Sung, L.Y.; Luo, W.Y.; Tuan, H.Y.; et al. Simultaneous induction of autophagy and toll-like receptor signaling pathways by graphene oxide. Biomaterials 2012, 33, 6559–6569. [Google Scholar] [CrossRef]
- Verma, N.K.; Conroy, J.; Lyons, P.E.; Coleman, J.; O’Sullivan, M.P.; Kornfeld, H.; Kelleher, D.; Volkov, Y. Autophagy induction by silver nanowires: A new aspect in the biocompatibility assessment of nanocomposite thin films. Toxicol. Appl. Pharmacol. 2012, 264, 451–461. [Google Scholar] [CrossRef]
- Roy, R.; Singh, S.K.; Chauhan, L.K.; Das, M.; Tripathi, A.; Dwivedi, P.D. Zinc oxide nanoparticles induce apoptosis by enhancement of autophagy via PI3K/Akt/mTOR inhibition. Toxicol. Lett. 2014, 227, 29–40. [Google Scholar] [CrossRef]
- Khan, M.I.; Mohammad, A.; Patil, G.; Naqvi, S.A.; Chauhan, L.K.; Ahmad, I. Induction of ROS, mitochondrial damage and autophagy in lung epithelial cancer cells by iron oxide nanoparticles. Biomaterials 2012, 33, 1477–1488. [Google Scholar]
- Ma, X.; Wu, Y.; Jin, S.; Tian, Y.; Zhang, X.; Zhao, Y.; Yu, L.; Liang, X.J. Gold nanoparticles induce autophagosome accumulation through size-dependent nanoparticle uptake and lysosome impairment. ACS Nano 2011, 5, 8629–8639. [Google Scholar] [CrossRef]
- Orecna, M.; De Paoli, S.H.; Janouskova, O.; Tegegn, T.Z.; Filipova, M.; Bonevich, J.E.; Holada, K.; Simak, J. Toxicity of carboxylated carbon nanotubes in endothelial cells is attenuated by stimulation of the autophagic flux with the release of nanomaterial in autophagic vesicles. Nanomedicine 2014. [Google Scholar] [CrossRef]
- Wan, B.; Wang, Z.X.; Lv, Q.Y.; Dong, P.X.; Zhao, L.X.; Yang, Y.; Guo, L.H. Single-walled carbon nanotubes and graphene oxides induce autophagosome accumulation and lysosome impairment in primarily cultured murine peritoneal macrophages. Toxicol. Lett. 2013, 221, 118–127. [Google Scholar] [CrossRef]
- Sun, T.; Yan, Y.; Zhao, Y.; Guo, F.; Jiang, C. Copper oxide nanoparticles induce autophagic cell death in A549 cells. PLoS One 2012, 7, e43442. [Google Scholar]
- Li, J.J.; Hartono, D.; Ong, C.N.; Bay, B.H.; Yung, L.Y. Autophagy and oxidative stress associated with gold nanoparticles. Biomaterials 2010, 31, 5996–6003. [Google Scholar] [CrossRef]
- Nowak, J.S.; Mehn, D.; Nativo, P.; Garcia, C.P.; Gioria, S.; Ojea-Jimenez, I.; Gilliland, D.; Rossi, F. Silica nanoparticle uptake induces survival mechanism in A549 cells by the activation of autophagy but not apoptosis. Toxicol. Lett. 2013, 224, 84–92. [Google Scholar]
- Yamawaki, H.; Iwai, N. Cytotoxicity of water-soluble fullerene in vascular endothelial cells. Am. J. Physiol. Cell Physiol. 2006, 290, 1495–1502. [Google Scholar] [CrossRef]
- Eidi, H.; Joubert, O.; Nemos, C.; Grandemange, S.; Mograbi, B.; Foliguet, B.; Tournebize, J.; Maincent, P.; Le Faou, A.; Aboukhamis, I.; et al. Drug delivery by polymeric nanoparticles induces autophagy in macrophages. Int. J. Pharm. 2012, 422, 495–503. [Google Scholar] [CrossRef]
- Tsukahara, T.; Matsuda, Y.; Usui, Y.; Haniu, H. Highly purified, multi-wall carbon nanotubes induce light-chain 3B expression in human lung cells. Biochem. Biophys. Res. Commun. 2013, 440, 348–353. [Google Scholar] [CrossRef]
- Monastyrska, I.; Rieter, E.; Klionsky, D.J.; Reggiori, F. Multiple roles of the cytoskeleton in autophagy. Biol. Rev. Camb. Philos. Soc. 2009, 84, 431–448. [Google Scholar] [CrossRef]
- Aplin, A.; Jasionowski, T.; Tuttle, D.L.; Lenk, S.E.; Dunn, W.A., Jr. Cytoskeletal elements are required for the formation and maturation of autophagic vacuoles. J. Cell. Physiol. 1992, 152, 458–466. [Google Scholar] [CrossRef]
- Seglen, P.O.; Berg, T.O.; Blankson, H.; Fengsrud, M.; Holen, I.; Stromhaug, P.E. Structural aspects of autophagy. Adv. Exp. Med. Biol. 1996, 389, 103–111. [Google Scholar] [CrossRef]
- Kochl, R.; Hu, X.W.; Chan, E.Y.; Tooze, S.A. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic 2006, 7, 129–145. [Google Scholar] [CrossRef]
- Lee, J.Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010, 29, 969–980. [Google Scholar] [CrossRef]
- Choudhury, D.; Xavier, P.L.; Chaudhari, K.; John, R.; Dasgupta, A.K.; Pradeep, T.; Chakrabarti, G. Unprecedented inhibition of tubulin polymerization directed by gold nanoparticles inducing cell cycle arrest and apoptosis. Nanoscale 2013, 5, 4476–4489. [Google Scholar] [CrossRef]
- Gheshlaghi, Z.N.; Riazi, G.H.; Ahmadian, S.; Ghafari, M.; Mahinpour, R. Toxicity and interaction of titanium dioxide nanoparticles with microtubule protein. Acta Biochim. Biophys. Sinica 2008, 40, 777–782. [Google Scholar] [CrossRef]
- Ratnikova, T.A.; Govindan, P.N.; Salonen, E.; Ke, P.C. In vitro polymerization of microtubules with a fullerene derivative. ACS Nano 2011, 5, 6306–6314. [Google Scholar] [CrossRef]
- Shams, H.; Holt, B.D.; Mahboobi, S.H.; Jahed, Z.; Islam, M.F.; Dahl, K.N.; Mofrad, M.R. Actin reorganization through dynamic interactions with single-wall carbon nanotubes. ACS Nano 2014, 8, 188–197. [Google Scholar] [CrossRef]
- Okoturo-Evans, O.; Dybowska, A.; Valsami-Jones, E.; Cupitt, J.; Gierula, M.; Boobis, A.R.; Edwards, R.J. Elucidation of toxicity pathways in lung epithelial cells induced by silicon dioxide nanoparticles. PLoS One 2013, 8, e72633. [Google Scholar]
- Pernodet, N.; Fang, X.; Sun, Y.; Bakhtina, A.; Ramakrishnan, A.; Sokolov, J.; Ulman, A.; Rafailovich, M. Adverse effects of citrate/gold nanoparticles on human dermal fibroblasts. Small 2006, 2, 766–773. [Google Scholar] [CrossRef]
- Mironava, T.; Hadjiargyrou, M.; Simon, M.; Jurukovski, V.; Rafailovich, M.H. Gold nanoparticles cellular toxicity and recovery: Effect of size, concentration and exposure time. Nanotoxicology 2010, 4, 120–137. [Google Scholar] [CrossRef]
- Pisanic, T.R.; Blackwell, J.D.; Shubayev, V.I.; Finones, R.R.; Jin, S. Nanotoxicity of iron oxide nanoparticle internalization in growing neurons. Biomaterials 2007, 28, 2572–2581. [Google Scholar] [CrossRef]
- Dadras, A.; Riazi, G.H.; Afrasiabi, A.; Naghshineh, A.; Ghalandari, B.; Mokhtari, F. In vitro study on the alterations of brain tubulin structure and assembly affected by magnetite nanoparticles. JBIC Publ. Soc. Biol. Inorg. Chem. 2013, 18, 357–369. [Google Scholar] [CrossRef]
- Wu, X.; Tan, Y.; Mao, H.; Zhang, M. Toxic effects of iron oxide nanoparticles on human umbilical vein endothelial cells. Int. J. Nanomed. 2010, 5, 385–399. [Google Scholar]
- Soenen, S.J.; Nuytten, N.; De Meyer, S.F.; De Smedt, S.C.; De Cuyper, M. High intracellular iron oxide nanoparticle concentrations affect cellular cytoskeleton and focal adhesion kinase-mediated signaling. Small 2010, 6, 832–842. [Google Scholar] [CrossRef]
- Johnson-Lyles, D.N.; Peifley, K.; Lockett, S.; Neun, B.W.; Hansen, M.; Clogston, J.; Stern, S.T.; McNeil, S.E. Fullerenol cytotoxicity in kidney cells is associated with cytoskeleton disruption, autophagic vacuole accumulation, and mitochondrial dysfunction. Toxicol. Appl. Pharmacol. 2010, 248, 249–258. [Google Scholar] [CrossRef]
- Sohaebuddin, S.K.; Thevenot, P.T.; Baker, D.; Eaton, J.W.; Tang, L. Nanomaterial cytotoxicity is composition, size, and cell type dependent. Part. Fibre Toxicol. 2010, 7, 22. [Google Scholar] [CrossRef]
- Thomas, T.P.; Majoros, I.; Kotlyar, A.; Mullen, D.; Holl, M.M.; Baker, J.R., Jr. Cationic poly(amidoamine) dendrimer induces lysosomal apoptotic pathway at therapeutically relevant concentrations. Biomacromolecules 2009, 10, 3207–3214. [Google Scholar] [CrossRef]
- Baltazar, G.C.; Guha, S.; Lu, W.; Lim, J.; Boesze-Battaglia, K.; Laties, A.M.; Tyagi, P.; Kompella, U.B.; Mitchell, C.H. Acidic nanoparticles are trafficked to lysosomes and restore an acidic lysosomal pH and degradative function to compromised ARPE-19 cells. PLoS One 2012, 7, e49635. [Google Scholar] [CrossRef]
- Hussain, S.; Thomassen, L.C.; Ferecatu, I.; Borot, M.C.; Andreau, K.; Martens, J.A.; Fleury, J.; Baeza-Squiban, A.; Marano, F.; Boland, S. Carbon black and titanium dioxide nanoparticles elicit distinct apoptotic pathways in bronchial epithelial cells. Part. Fibre Toxicol. 2010, 7, 10–26. [Google Scholar] [CrossRef]
- Jin, C.Y.; Zhu, B.S.; Wang, X.F.; Lu, Q.H. Cytotoxicity of titanium dioxide nanoparticles in mouse fibroblast cells. Chem. Res. Toxicol. 2008, 21, 1871–1877. [Google Scholar] [CrossRef]
- Cho, W.S.; Duffin, R.; Howie, S.E.; Scotton, C.J.; Wallace, W.A.; Macnee, W.; Bradley, M.; Megson, I.L.; Donaldson, K. Progressive severe lung injury by zinc oxide nanoparticles; the role of Zn2+ dissolution inside lysosomes. Part. Fibre Toxicol. 2011, 8, 27–42. [Google Scholar] [CrossRef]
- Hamilton, R.F.; Wu, N.; Porter, D.; Buford, M.; Wolfarth, M.; Holian, A. Particle length-dependent titanium dioxide nanomaterials toxicity and bioactivity. Part. Fibre Toxicol. 2009, 6. [Google Scholar] [CrossRef]
- Lunov, O.; Syrovets, T.; Loos, C.; Nienhaus, G.U.; Mailander, V.; Landfester, K.; Rouis, M.; Simmet, T. Amino-functionalized polystyrene nanoparticles activate the NLRP3 inflammasome in human macrophages. ACS Nano 2011, 5, 9648–9657. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cohignac, V.; Landry, M.J.; Boczkowski, J.; Lanone, S. Autophagy as a Possible Underlying Mechanism of Nanomaterial Toxicity. Nanomaterials 2014, 4, 548-582. https://doi.org/10.3390/nano4030548
Cohignac V, Landry MJ, Boczkowski J, Lanone S. Autophagy as a Possible Underlying Mechanism of Nanomaterial Toxicity. Nanomaterials. 2014; 4(3):548-582. https://doi.org/10.3390/nano4030548
Chicago/Turabian StyleCohignac, Vanessa, Marion Julie Landry, Jorge Boczkowski, and Sophie Lanone. 2014. "Autophagy as a Possible Underlying Mechanism of Nanomaterial Toxicity" Nanomaterials 4, no. 3: 548-582. https://doi.org/10.3390/nano4030548
APA StyleCohignac, V., Landry, M. J., Boczkowski, J., & Lanone, S. (2014). Autophagy as a Possible Underlying Mechanism of Nanomaterial Toxicity. Nanomaterials, 4(3), 548-582. https://doi.org/10.3390/nano4030548