
Structure and Migration Mechanisms of Small Vacancy Clusters in Cu: A Combined EAM and DFT Study

 ,
,  , , ,
, , , 
Abstract
1. Introduction
2. Methods of Calculations
2.1. Computational Details
2.1.1. Embedded Atom Method
2.1.2. DFT Calculations
2.2. Vacancy Cluster Identification
2.3. Calculation of Diffusion Barriers of Vacancy Clusters
3. Results of Calculations
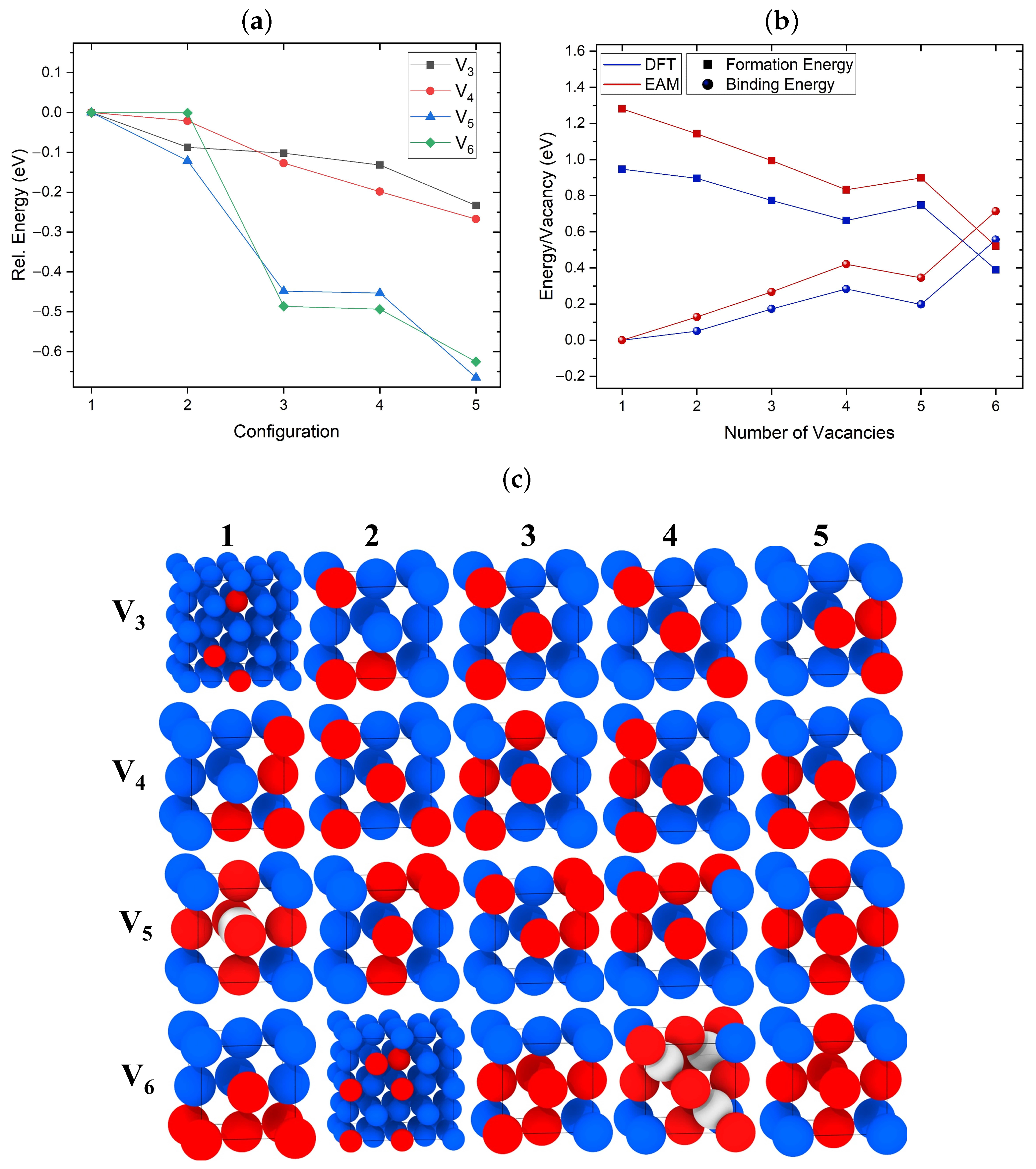
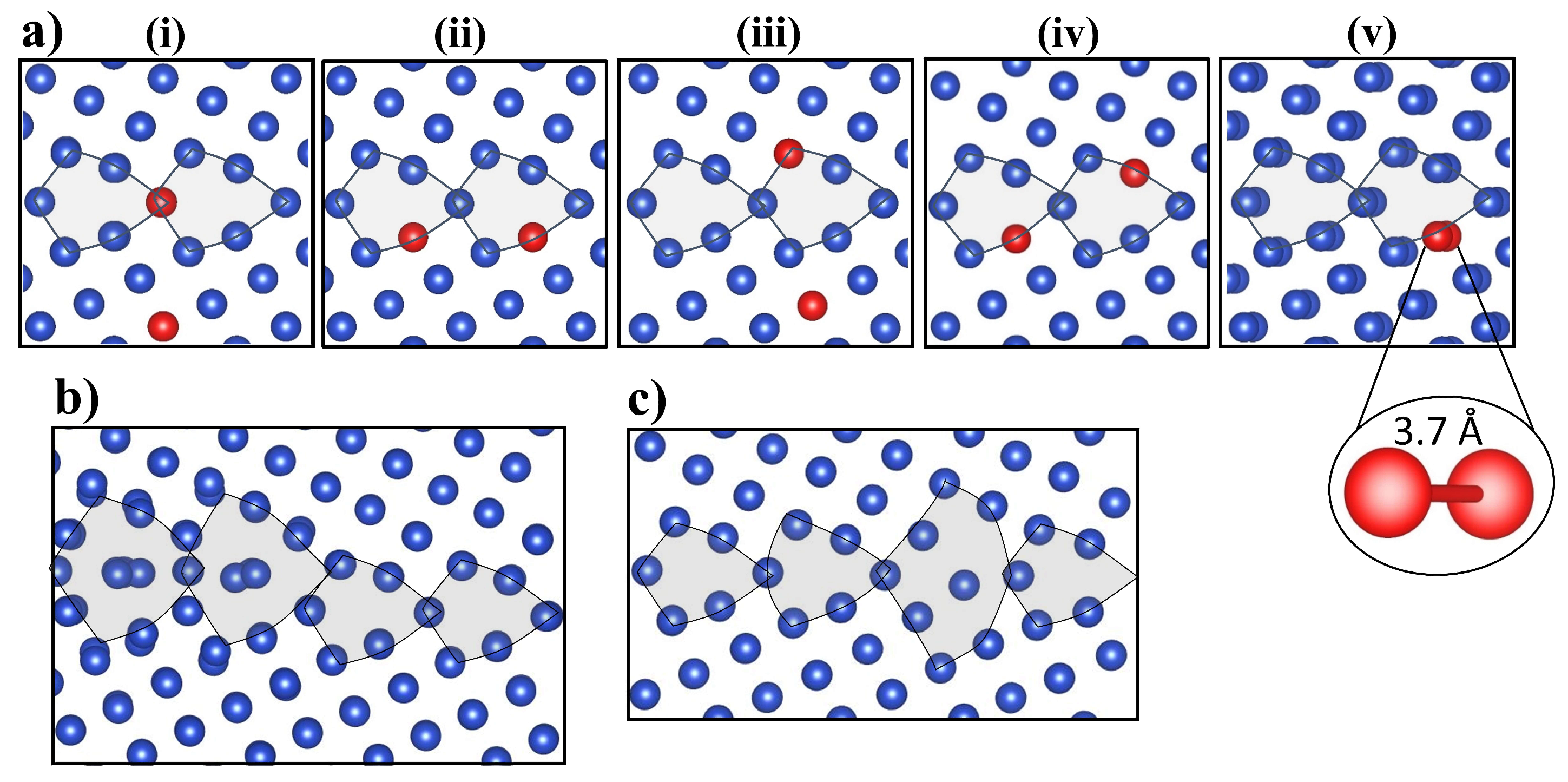
3.1. Structure of Vacancy Cluster
3.2. Formation and Binding Energies of Vacancies
3.3. Cluster Migration Paths in the Bulk Cu
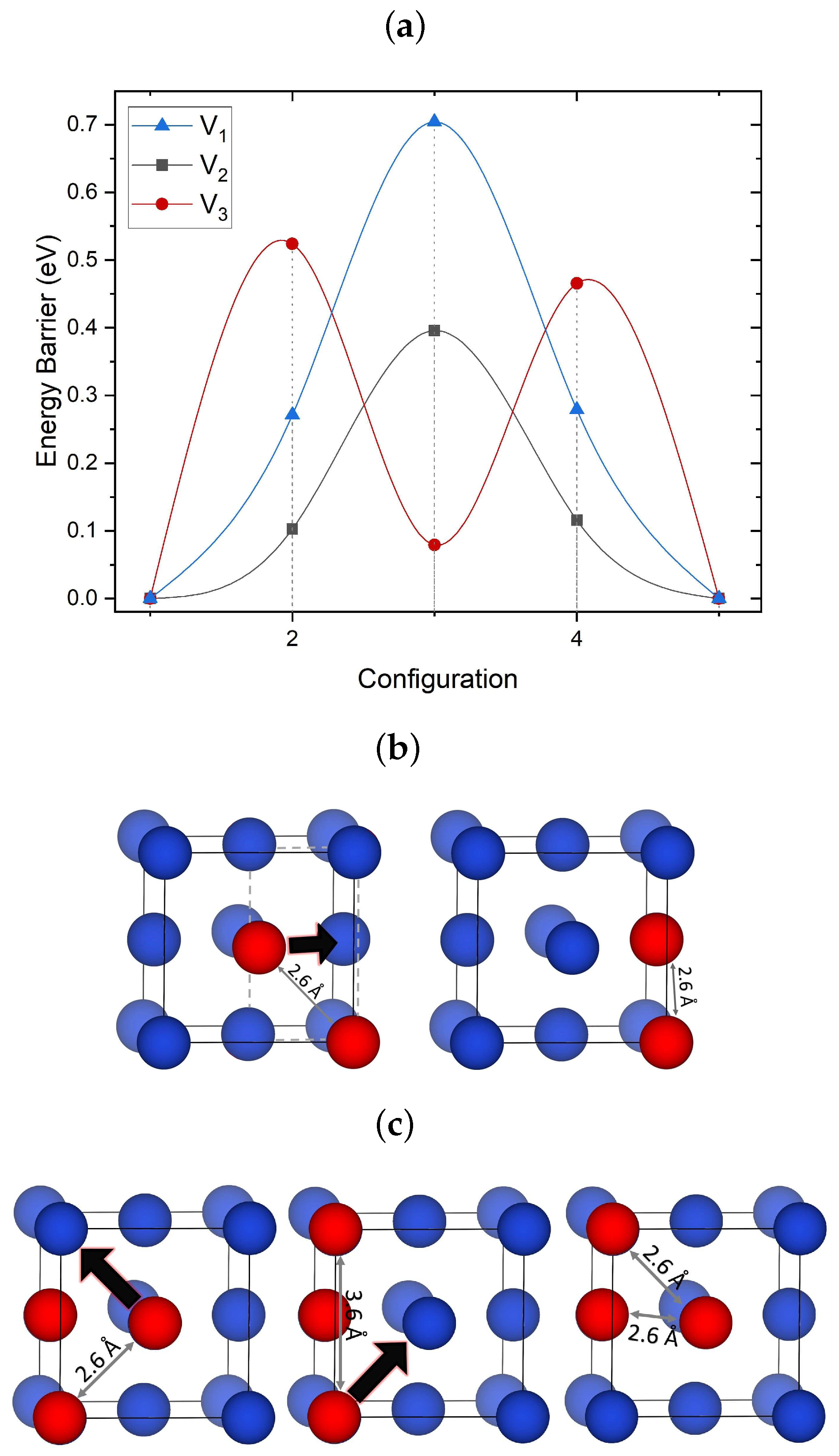
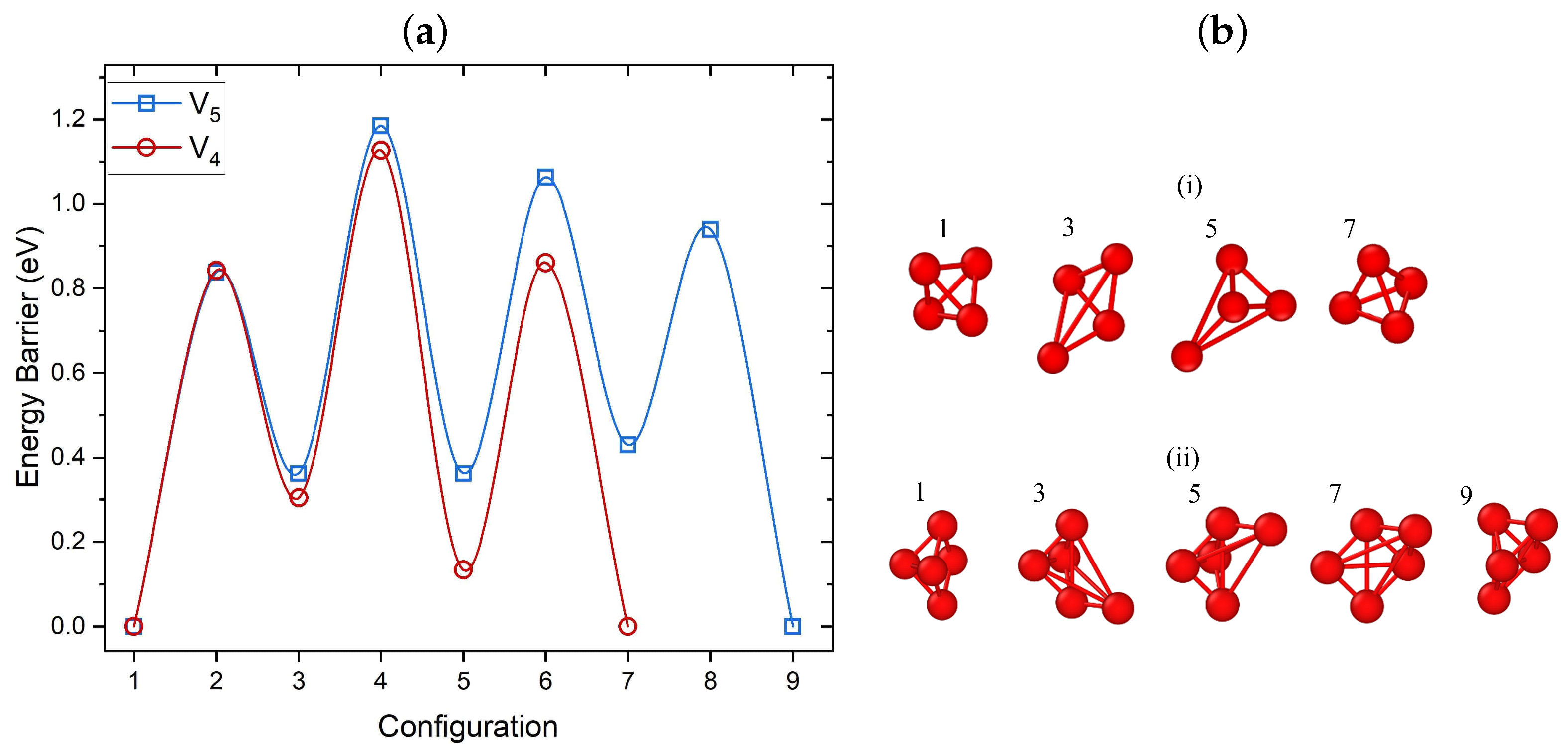
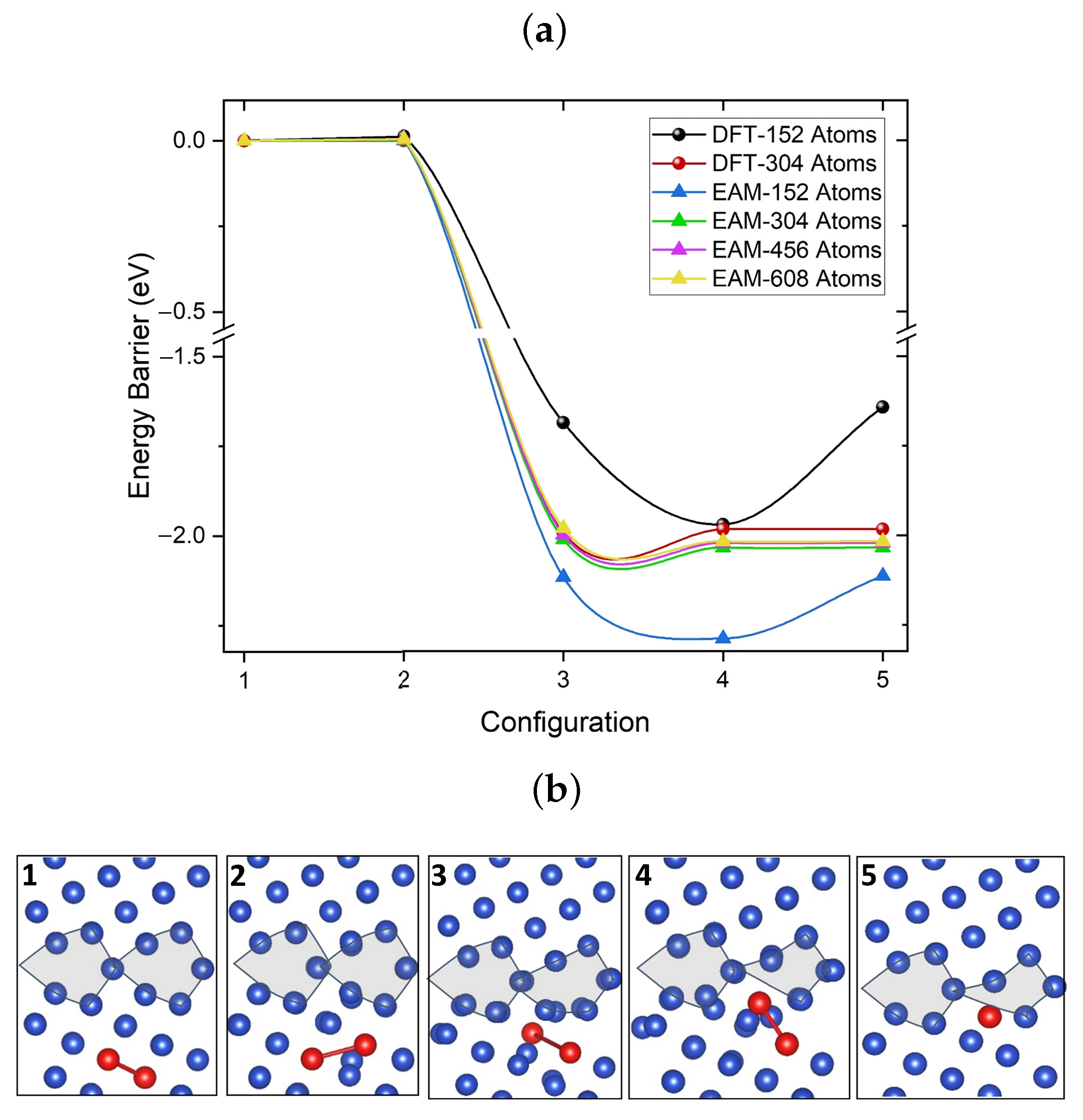
3.3.1. Migration of V–V Clusters
3.3.2. Elementary Migration Paths of V–V Clusters
3.3.3. Crowdion Motion of Vacancy Clusters
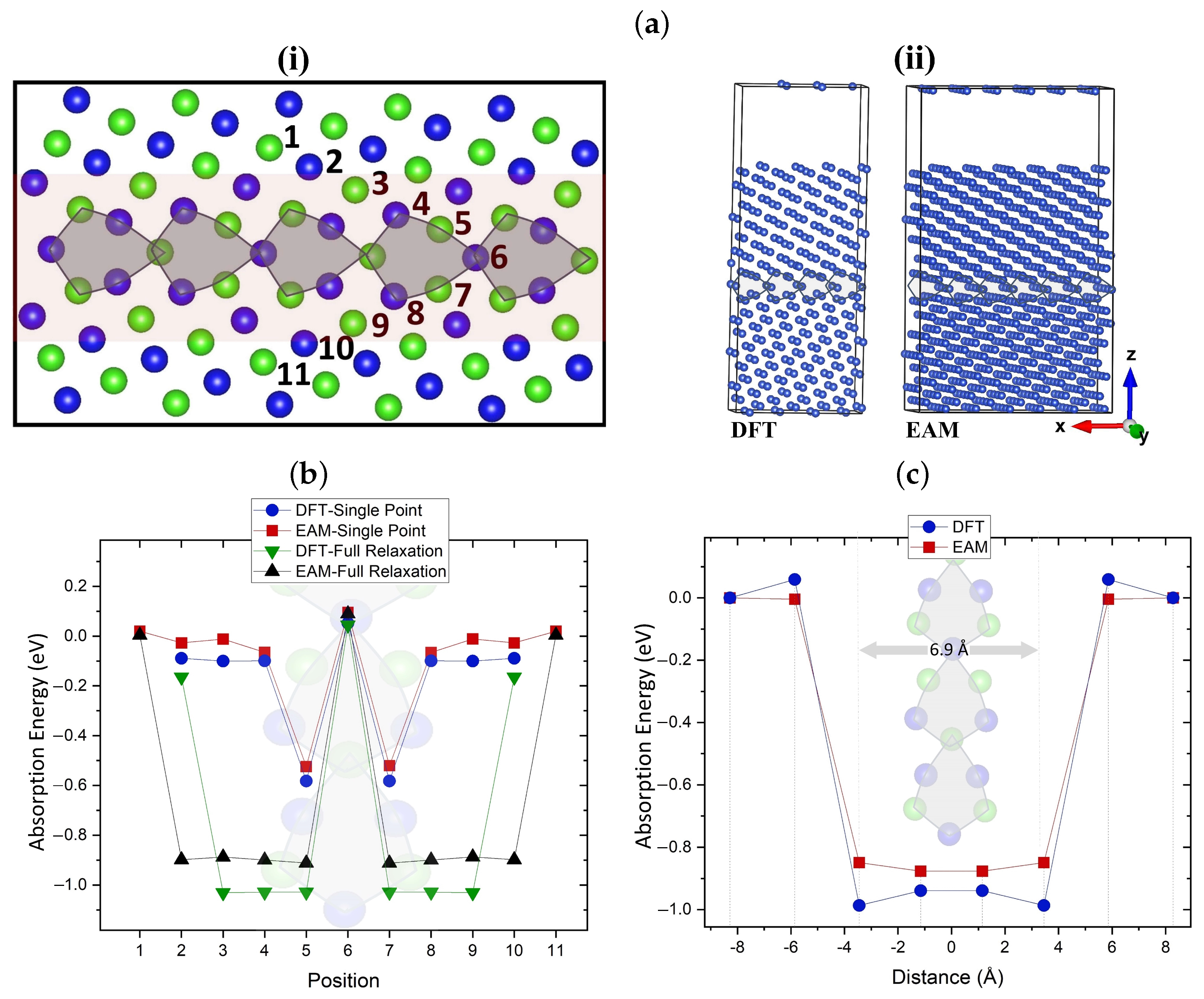
4. Vacancy Interaction with Grain Boundaries
4.1. Interaction of Mono-Vacancy with Grain Boundary
4.2. Interaction of di-Vacancy with Grain Boundary
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DFT | density functional theory |
| EAM | embedded atom method |
| LAMMPS | Atomic/Molecular Massively Parallel Simulator |
| ARTn | activation relaxation technique nouveau |
| KLMC | knowledge led master code |
| PBE | Perdew–Burke–Ernzerhof |
| GPW | Gaussian and plane wave |
| GTH | Goedecker–Teter–Hutter |
| NEB | nudged elastic band |
| CI-NEB | climbing image nudged elastic band |
| GB | grain boundary |
| SFT | stacking fault tetrahedral |
| fcc | face-centered cubic |
| FF | forcefield |
| NNN | next nearest neighbor |
| 1NN | first nearest neighbor |
| SIA | self-interstitial atom |
| CG | conjugate gradient |
| GGA | generalized gradient approximation |
| RS | real-space |
| BFGS | Broyden–Fletcher–Goldfarb–Shanno |
| DIIS | direct inversion in the iterative subspace |
| formation energy | |
| binding energy | |
| mono-vacancy formation energy | |
| formation energy of a cluster with n vacancies | |
| vacancy cluster with n vacancies |
References
- Merchant, S.M.; Kang, S.H.; Sanganeria, M.; van Schravendijk, B.; Mountsier, T. Copper interconnects for semiconductor devices. JOM 2001, 53, 43–48. [Google Scholar] [CrossRef]
- Bhalothia, D.; Lin, C.Y.; Yan, C.; Yang, Y.T.; Chen, T.Y. H2 reduction annealing induced phase transition and improvements on redox durability of Pt cluster-decorated Cu@ Pd electrocatalysts in oxygen reduction reaction. ACS Omega 2019, 4, 971–982. [Google Scholar] [CrossRef]
- Bhalothia, D.; Lin, C.Y.; Yan, C.; Yang, Y.T.; Chen, T.Y. Effects of Pt metal loading on the atomic restructure and oxygen reduction reaction performance of Pt-cluster decorated Cu@ Pd electrocatalysts. Sustain. Energy Fuels 2019, 3, 1668–1681. [Google Scholar] [CrossRef]
- Sharif, A. All-copper interconnects for high-temperature applications. In Harsh Environment Electronics: Interconnect Materials and Performance Assessment; Wiley: Hoboken, NJ, USA, 2019. [Google Scholar]
- Gerstein, G.; Besserer, H.B.; Nürnberger, F.; Barrales-Mora, L.A.; Shvindlerman, L.S.; Estrin, Y.; Maier, H.J. Formation and growth of voids in dual-phase steel at microscale and nanoscale levels. J. Mater. Sci. 2017, 52, 4234–4243. [Google Scholar] [CrossRef]
- Pang, W.W.; Zhang, P.; Zhang, G.C.; Xu, A.G.; Zhao, X.G. Dislocation creation and void nucleation in FCC ductile metals under tensile loading: A general microscopic picture. Sci. Rep. 2014, 4, 6981. [Google Scholar] [CrossRef]
- Feng, H.; Cheng, M.; Wang, Y.; Chang, S.; Wang, Y.; Wan, C. Mechanism for Cu void defect on various electroplated film conditions. Thin Solid Films 2006, 498, 56–59. [Google Scholar] [CrossRef]
- Katz, J.L.; Wiedersich, H. Nucleation of voids in materials supersaturated with vacancies and interstitials. J. Chem. Phys. 1971, 55, 1414–1425. [Google Scholar] [CrossRef]
- Fischer, F.; Svoboda, J. Void growth due to vacancy supersaturation—A non-equilibrium thermodynamics study. Scr. Mater. 2008, 58, 93–95. [Google Scholar] [CrossRef]
- Kovács, Z.; Chinh, N.Q. Up-hill diffusion of solute atoms towards slipped grain boundaries: A possible reason of decomposition due to severe plastic deformation. Scr. Mater. 2020, 188, 285–289. [Google Scholar] [CrossRef]
- Gondcharton, P.; Imbert, B.; Benaissa, L.; Verdier, M. Voiding phenomena in copper-copper bonded structures: Role of creep. ECS J. Solid State Sci. Technol. 2015, 4, P77. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, T.; Chen, J.; Li, X.; Song, H.; Huang, G. Investigation on strain hardening and failure in notched tension specimens of cold rolled Ti6Al4V titanium alloy. Materials 2022, 15, 3429. [Google Scholar] [CrossRef]
- Nemirovich-Danchenko, L.Y.; Lipnitskiĭ, A.G.; Kul’kova, S.E. Vacancies and their complexes in FCC metals. Phys. Solid State 2007, 49, 1079–1085. [Google Scholar] [CrossRef]
- Srivastava, K.; Rao, S.I.; El-Awady, J.A. Unveiling the role of super-jogs and dislocation induced atomic-shuffling on controlling plasticity in magnesium. Acta Mater. 2018, 161, 182–193. [Google Scholar] [CrossRef]
- Dupraz, M.; Sun, Z.; Brandl, C.; Van Swygenhoven, H. Dislocation interactions at reduced strain rates in atomistic simulations of nanocrystalline Al. Acta Mater. 2018, 144, 68–79. [Google Scholar] [CrossRef]
- Wang, H.; Rodney, D.; Xu, D.; Yang, R.; Veyssière, P. Defect kinetics on experimental timescales using atomistic simulations. Philos. Mag. 2013, 93, 186–202. [Google Scholar] [CrossRef]
- Yang, Q.; Hu, Z.; Makkonen, I.; Desgardin, P.; Egger, W.; Barthe, M.F.; Olsson, P. A combined experimental and theoretical study of small and large vacancy clusters in tungsten. J. Nucl. Mater. 2022, 571, 154019. [Google Scholar] [CrossRef]
- Ohkubo, H.; Tang, Z.; Nagai, Y.; Hasegawa, M.; Tawara, T.; Kiritani, M. Positron annihilation study of vacancy-type defects in high-speed deformed Ni, Cu and Fe. Mater. Sci. Eng. A 2003, 350, 95–101. [Google Scholar] [CrossRef]
- Ganchenkova, M.; Yagodzinskyy, Y.; Borodin, V.; Hänninen, H. Effects of hydrogen and impurities on void nucleation in copper: Simulation point of view. Philos. Mag. 2014, 94, 3522–3548. [Google Scholar] [CrossRef]
- Polfus, J.M.; Løvvik, O.M.; Bredesen, R.; Peters, T. Hydrogen induced vacancy clustering and void formation mechanisms at grain boundaries in palladium. Acta Mater. 2020, 195, 708–719. [Google Scholar] [CrossRef]
- Mahmoud, S.; Trochet, M.; Restrepo, O.A.; Mousseau, N. Study of point defects diffusion in nickel using kinetic activation-relaxation technique. Acta Mater. 2018, 144, 679–690. [Google Scholar] [CrossRef]
- McLellan, R.; Angel, Y. The thermodynamics of vacancy formation in fcc metals. Acta Metall. Mater. 1995, 43, 3721–3725. [Google Scholar] [CrossRef]
- Bartdorff, D.; Neumann, G.; Reimers, P. Self-diffusion of 64Cu in copper single crystals Monovacancy and divacancy contributions. Philos. Mag. A 1978, 38, 157–165. [Google Scholar] [CrossRef]
- Ho, G.; Ong, M.T.; Caspersen, K.J.; Carter, E.A. Energetics and kinetics of vacancy diffusion and aggregation in shocked aluminium via orbital-free density functional theory. Phys. Chem. Chem. Phys. 2007, 9, 4951–4966. [Google Scholar] [CrossRef]
- Wang, H.; Rodney, D.; Xu, D.; Yang, R.; Veyssiere, P. Pentavacancy as the key nucleus for vacancy clustering in aluminum. Phys. Rev. B 2011, 84, 220103. [Google Scholar] [CrossRef]
- Shimomura, Y.; Nishiguchi, R. Vacancy clustering to faulted loop, stacking fault tetrahedron and void in fcc metals. Radiat. Eff. Defects Solids 1997, 141, 311–324. [Google Scholar] [CrossRef]
- Xv, H.; Zhao, J.; Ye, F.; Tong, K. Strain-induced transformation between vacancy voids and stacking fault tetrahedra in Cu. Comput. Mater. Sci. 2019, 158, 359–368. [Google Scholar] [CrossRef]
- Balluffi, R. Grain boundary diffusion mechanisms in metals. Metall. Trans. B 1982, 13, 527–553. [Google Scholar] [CrossRef]
- Yu, W.; Demkowicz, M. Non-coherent Cu grain boundaries driven by continuous vacancy loading. J. Mater. Sci. 2015, 50, 4047–4065. [Google Scholar] [CrossRef]
- Chen, K.; Xue, D. Cu-based materials as high-performance electrodes toward electrochemical energy storage. Funct. Mater. Lett. 2014, 7, 1430001. [Google Scholar] [CrossRef]
- Wan, L.; Geng, W.T.; Ishii, A.; Du, J.P.; Mei, Q.; Ishikawa, N.; Kimizuka, H.; Ogata, S. Hydrogen embrittlement controlled by reaction of dislocation with grain boundary in alpha-iron. Int. J. Plast. 2019, 112, 206–219. [Google Scholar] [CrossRef]
- Fröhlking, T.; Bernetti, M.; Calonaci, N.; Bussi, G. Toward empirical force fields that match experimental observables. J. Chem. Phys. 2020, 152, 230902. [Google Scholar] [CrossRef]
- Kashefolgheta, S.; Oliveira, M.P.; Rieder, S.R.; Horta, B.A.; Acree, W.E., Jr.; Hunenberger, P.H. Evaluating classical force fields against experimental cross-solvation free energies. J. Chem. Theory Comput. 2020, 16, 7556–7580. [Google Scholar] [CrossRef]
- Fotopoulos, V.; Grau-Crespo, R.; Shluger, A. Thermodynamic analysis of the interaction between metal vacancies and hydrogen in bulk Cu. Phys. Chem. Chem. Phys. 2023, 25, 9168–9175. [Google Scholar] [CrossRef]
- Bodlos, R.; Fotopoulos, V.; Spitaler, J.; Shluger, A.; Romaner, L. Energies and structures of Cu/Nb and Cu/W interfaces from density functional theory and semi-empirical calculations. Materialia 2022, 21, 101362. [Google Scholar] [CrossRef]
- Grau-Crespo, R.; Hamad, S.; Catlow, C.R.A.; De Leeuw, N. Symmetry-adapted configurational modelling of fractional site occupancy in solids. J. Phys. Condens. Matter 2007, 19, 256201. [Google Scholar] [CrossRef]
- Jay, A.; Gunde, M.; Salles, N.; Poberžnik, M.; Martin-Samos, L.; Richard, N.; de Gironcoli, S.; Mousseau, N.; Hémeryck, A. Activation–Relaxation Technique: An efficient way to find minima and saddle points of potential energy surfaces. Comp. Mater. Sci. 2022, 209, 111363. [Google Scholar] [CrossRef]
- Woodley, S.M. Knowledge led master code search for atomic and electronic structures of LaF3 nanoclusters on hybrid rigid ion–shell model–DFT landscapes. J. Phys. Chem. C 2013, 117, 24003–24014. [Google Scholar] [CrossRef]
- Larsen, A.H.; Mortensen, J.J.; Blomqvist, J.; Castelli, I.E.; Christensen, R.; Dułak, M.; Friis, J.; Groves, M.N.; Hammer, B.; Hargus, C.; et al. The atomic simulation environment—A Python library for working with atoms. J. Phys. Condens. Matter 2017, 29, 273002. [Google Scholar] [CrossRef]
- Mishin, Y.; Mehl, M.; Papaconstantopoulos, D.; Voter, A.; Kress, J. Structural stability and lattice defects in copper: Ab initio, tight-binding, and embedded-atom calculations. Phys. Rev. B 2001, 63, 224106. [Google Scholar] [CrossRef]
- Zhang, B.; Hu, W.; Shu, X. Theory of Embedded Atom Method and Its Application to Materials Science-Atomic Scale Materials Design Theory; Hunan University Publication Press: Changsha, China, 2003. [Google Scholar]
- Jin, H.S.; An, J.D.; Jong, Y.S. EAM potentials for BCC, FCC and HCP metals with farther neighbor atoms. Appl. Phys. A 2015, 120, 189–197. [Google Scholar] [CrossRef]
- Asadi, E.; Zaeem, M.A.; Moitra, A.; Tschopp, M.A. Effect of vacancy defects on generalized stacking fault energy of fcc metals. J. Phys. Condens. Matter 2014, 26, 115404. [Google Scholar] [CrossRef] [PubMed]
- Kühne, T.D.; Iannuzzi, M.; Del Ben, M.; Rybkin, V.V.; Seewald, P.; Stein, F.; Laino, T.; Khaliullin, R.Z.; Schütt, O.; Schiffmann, F.; et al. CP2K: An electronic structure and molecular dynamics software package-Quickstep: Efficient and accurate electronic structure calculations. J. Chem. Phys. 2020, 152, 194103. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671. [Google Scholar] [CrossRef] [PubMed]
- Lippert, B.G.; Hutter, J.; Parrinello, M. A hybrid Gaussian and plane wave density functional scheme. Mol. Phys. 1997, 92, 477–488. [Google Scholar] [CrossRef]
- Fletcher, R. Practical Methods of Optimization; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Broyden, C.G. A class of methods for solving nonlinear simultaneous equations. Math. Comput. 1965, 19, 577–593. [Google Scholar] [CrossRef]
- VandeVondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef]
- Goedecker, S.; Teter, M.; Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 1996, 54, 1703. [Google Scholar] [CrossRef]
- Huang, Z.; Chen, F.; Shen, Q.; Zhang, L.; Rupert, T.J. Uncovering the influence of common nonmetallic impurities on the stability and strength of a Σ5 (310) grain boundary in Cu. Acta Mater. 2018, 148, 110–122. [Google Scholar] [CrossRef]
- Bodlos, R.; Scheiber, D.; Spitaler, J.; Romaner, L. Modification of the Cu/W interface cohesion by segregation. Metals 2023, 13, 346. [Google Scholar] [CrossRef]
- Tkatchenko, A.; Romaner, L.; Hofmann, O.T.; Zojer, E.; Ambrosch-Draxl, C.; Scheffler, M. Van der Waals interactions between organic adsorbates and at organic/inorganic interfaces. MRS Bull. 2010, 35, 435–442. [Google Scholar] [CrossRef]
- Yu, Y.X. High storage capacity and small volume change of potassium-intercalation into novel vanadium oxychalcogenide monolayers V2S2O, V2Se2O and V2Te2O: An ab initio DFT investigation. Appl. Surf. Sci. 2021, 546, 149062. [Google Scholar] [CrossRef]
- Adeleke, A.A.; Johnson, E.R. Effects of dispersion corrections on the theoretical description of bulk metals. Phys. Rev. B 2023, 107, 064101. [Google Scholar] [CrossRef]
- Chiter, F.; Nguyen, V.B.; Tarrat, N.; Benoit, M.; Tang, H.; Lacaze-Dufaure, C. Effect of van der Waals corrections on DFT-computed metallic surface properties. Mater. Res. Express 2016, 3, 046501. [Google Scholar] [CrossRef]
- Wu, X.; You, Y.W.; Kong, X.S.; Chen, J.L.; Luo, G.N.; Lu, G.H.; Liu, C.; Wang, Z. First-principles determination of grain boundary strengthening in tungsten: Dependence on grain boundary structure and metallic radius of solute. Acta Mater. 2016, 120, 315–326. [Google Scholar] [CrossRef]
- Scheiber, D. Segregation and embrittlement of gold grain boundaries. Comput. Mater. Sci. 2021, 187, 110110. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA: A three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2009, 18, 015012. [Google Scholar] [CrossRef]
- Hirel, P. Atomsk: A tool for manipulating and converting atomic data files. Comput. Phys. Commun. 2015, 197, 212–219. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Pulay, P. Convergence acceleration of iterative sequences. The case of SCF iteration. Chem. Phys. Lett. 1980, 73, 393–398. [Google Scholar] [CrossRef]
- Lazauskas, T.; Sokol, A.A.; Woodley, S.M. An efficient genetic algorithm for structure prediction at the nanoscale. Nanoscale 2017, 9, 3850–3864. [Google Scholar] [CrossRef] [PubMed]
- Barkema, G.; Mousseau, N. The activation–relaxation technique: An efficient algorithm for sampling energy landscapes. Comput. Mater. Sci. 2001, 20, 285–292. [Google Scholar] [CrossRef]
- Barkema, G.; Mousseau, N. Event-based relaxation of continuous disordered systems. Phys. Rev. Lett. 1996, 77, 4358. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.H.; Zhang, C.G.; Li, Y.G.; Zeng, Z. Creeping motion of self interstitial atom clusters in tungsten. Sci. Rep. 2014, 4, 5096. [Google Scholar] [CrossRef]
- Carlberg, M.; Münger, E.; Hultman, L. Dynamics of self-interstitial structures in body-centred-cubic W studied by molecular dynamics simulation. J. Phys. Condens. Matter 2000, 12, 79. [Google Scholar] [CrossRef]
- Xu, D.; Wang, H.; Yang, R.; Veyssière, P. Point defect formation by dislocation reactions in TiAl. In Proceedings of the IOP Conference Series: Materials Science and Engineering; IOP Publishing: Bristol, UK, 2009; Volume 3, p. 012024. [Google Scholar]
- Martínez, E.; Uberuaga, B.P. Mobility and coalescence of stacking fault tetrahedra in Cu. Sci. Rep. 2015, 5, 9084. [Google Scholar] [CrossRef]
- Ackland, G.J.; Vitek, V. Many-body potentials and atomic-scale relaxations in noble-metal alloys. Phys. Rev. B 1990, 41, 10324. [Google Scholar] [CrossRef]
- Osetsky, Y.N.; Barashev, A.; Zhang, Y. On the mobility of defect clusters and their effect on microstructure evolution in fcc Ni under irradiation. Materialia 2018, 4, 139–146. [Google Scholar] [CrossRef]
- Fitzgerald, S. Crowdion–solute interactions: Analytical modelling and stochastic simulation. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2015, 352, 14–17. [Google Scholar] [CrossRef]
- Derlet, P.M.; Nguyen-Manh, D.; Dudarev, S. Multiscale modeling of crowdion and vacancy defects in body-centered-cubic transition metals. Phys. Rev. B 2007, 76, 054107. [Google Scholar] [CrossRef]
- Hehenkamp, T.; Berger, W.; Kluin, J.E.; Lüdecke, C.; Wolff, J. Equilibrium vacancy concentrations in copper investigated with the absolute technique. Phys. Rev. B 1992, 45, 1998. [Google Scholar] [CrossRef]
- Razumovskiy, V.; Divinski, S.; Romaner, L. Solute segregation in Cu: DFT vs. Experiment. Acta Mater. 2018, 147, 122–132. [Google Scholar] [CrossRef]
- Wurmshuber, M.; Burtscher, M.; Doppermann, S.; Bodlos, R.; Scheiber, D.; Romaner, L.; Kiener, D. Mechanical performance of doped W–Cu nanocomposites. Mater. Sci. Eng. A 2022, 857, 144102. [Google Scholar] [CrossRef]
- Ebner, A.S.; Jakob, S.; Clemens, H.; Pippan, R.; Maier-Kiener, V.; He, S.; Ecker, W.; Scheiber, D.; Razumovskiy, V.I. Grain boundary segregation in Ni-base alloys: A combined atom probe tomography and first principles study. Acta Mater. 2021, 221, 117354. [Google Scholar] [CrossRef]
- Huang, Z.; Chen, F.; Shen, Q.; Zhang, L.; Rupert, T.J. Combined effects of nonmetallic impurities and planned metallic dopants on grain boundary energy and strength. Acta Mater. 2019, 166, 113–125. [Google Scholar] [CrossRef]
- Chen, N.; Niu, L.L.; Zhang, Y.; Shu, X.; Zhou, H.B.; Jin, S.; Ran, G.; Lu, G.H.; Gao, F. Energetics of vacancy segregation to [100] symmetric tilt grain boundaries in bcc tungsten. Sci. Rep. 2016, 6, 36955. [Google Scholar] [CrossRef]
- Suzuki, A.; Mishin, Y. Interaction of point defects with grain boundaries in fcc Metals. Interface Sci. 2003, 11, 425–437. [Google Scholar] [CrossRef]
- Zhang, L.; Shibuta, Y.; Lu, C.; Huang, X. Interaction between nano-voids and migrating grain boundary by molecular dynamics simulation. Acta Mater. 2019, 173, 206–224. [Google Scholar] [CrossRef]
- Han, J.; Thomas, S.L.; Srolovitz, D.J. Grain-boundary kinetics: A unified approach. Prog. Mater. Sci. 2018, 98, 386–476. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | Barrier (eV) | Method |
|---|---|---|
| V | 0.65 | DFT/NEB |
| V | 0.40 | DFT/NEB |
| V | 0.52 | DFT/NEB |
| V | 0.84 | DFT/EAM/ARTn |
| V | 0.84 | DFT/EAM/ARTn |
| V | 0.96 | EAM/ARTn |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fotopoulos, V.; Mora-Fonz, D.; Kleinbichler, M.; Bodlos, R.; Kozeschnik, E.; Romaner, L.; Shluger, A.L. Structure and Migration Mechanisms of Small Vacancy Clusters in Cu: A Combined EAM and DFT Study. Nanomaterials 2023, 13, 1464. https://doi.org/10.3390/nano13091464
Fotopoulos V, Mora-Fonz D, Kleinbichler M, Bodlos R, Kozeschnik E, Romaner L, Shluger AL. Structure and Migration Mechanisms of Small Vacancy Clusters in Cu: A Combined EAM and DFT Study. Nanomaterials. 2023; 13(9):1464. https://doi.org/10.3390/nano13091464
Chicago/Turabian StyleFotopoulos, Vasileios, David Mora-Fonz, Manuel Kleinbichler, Rishi Bodlos, Ernst Kozeschnik, Lorenz Romaner, and Alexander L. Shluger. 2023. "Structure and Migration Mechanisms of Small Vacancy Clusters in Cu: A Combined EAM and DFT Study" Nanomaterials 13, no. 9: 1464. https://doi.org/10.3390/nano13091464
APA StyleFotopoulos, V., Mora-Fonz, D., Kleinbichler, M., Bodlos, R., Kozeschnik, E., Romaner, L., & Shluger, A. L. (2023). Structure and Migration Mechanisms of Small Vacancy Clusters in Cu: A Combined EAM and DFT Study. Nanomaterials, 13(9), 1464. https://doi.org/10.3390/nano13091464