
Flexible NiRu Systems for CO2 Methanation: From Efficient Catalysts to Advanced Dual-Function Materials


 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Catalysts Synthesis
2.2. Material Characterisation
2.2.1. CO2 Temperature-Programmed Desorption
2.2.2. CO2 Capture Experiment
2.3. Continuous Flow Experiments
2.3.1. Reactor Setup
2.3.2. Continuous CO2 Methanation Experiment
2.3.3. Long-Term Stability Test
2.3.4. CO2 Capture and Reduction Cycle
2.4. Time-Resolved Operando DRIFTS MS Experiment
3. Results and Discussion
3.1. Promoter Effects in the Continuous Methanation of CO2 over NiRu Catalysts

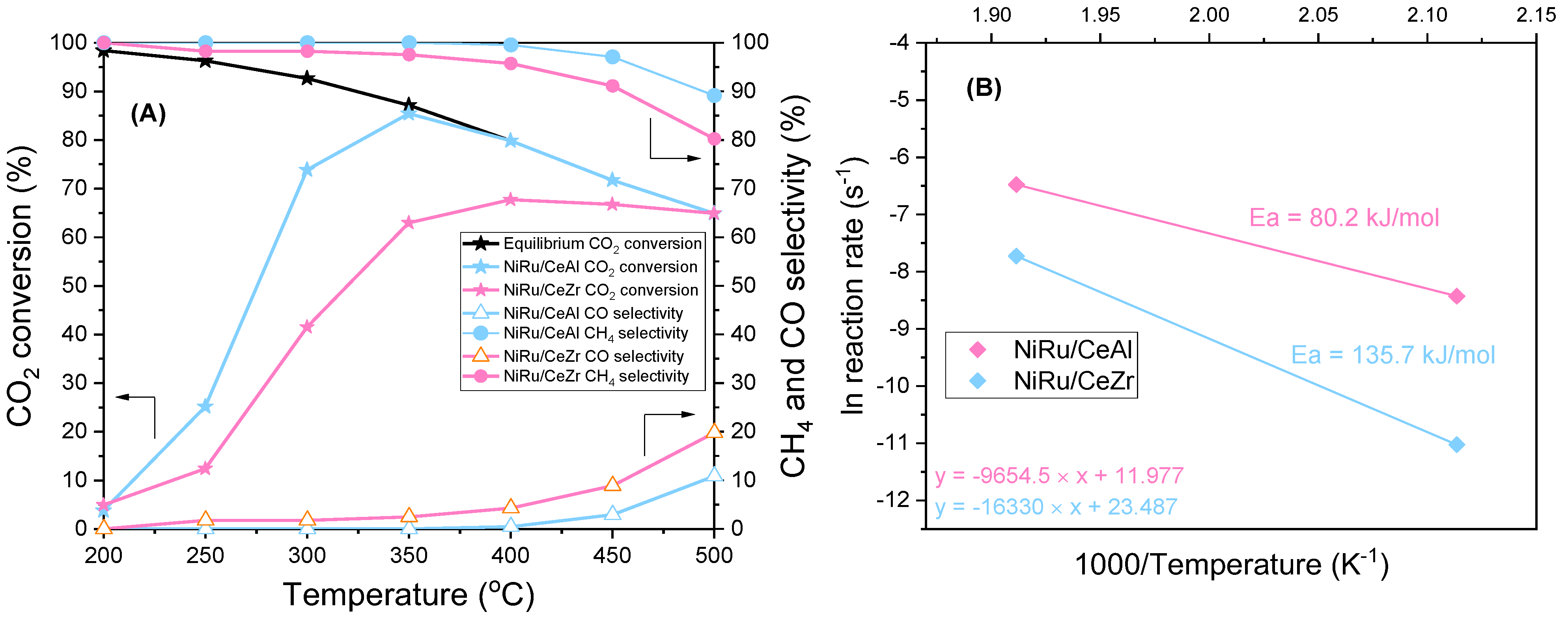{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | CO2 Conversion (%) | CH4 Selectivity (%) | Ref. |
|---|---|---|---|
| 15%Ni 1%Ru/CeO2-Al2O3 | 85 | 100 | This work |
| 15%Ni 1%Ru/Ce0.5Zr0.5O2 | 63 | 98 | This work |
| 4% Ru/Al2O3 | 80 | 99 | [55] |
| 12% Ni/Al2O3 | 55 | 98 | [55] |
| 15% CeO2 15%Ni/Al2O3 | 69 | 97 | [57] |
| 15%Ni 2%CeO2/Al2O3 | 85 | 100 | [33] |
| 5% Ni/Ce0.5Zr0.5O2 | 80 | 99 | [37] |
| 2% Ru/30% CeO2/Al2O3 | 82 | 100 | [58] |
| 15%Ni/Ce0.5Zr0.5O2 | 25 | 86 | [25] |
| 1%Ru/15%Ni/Ce0.5Zr0.5O2 | 53 | 93 | [25] |
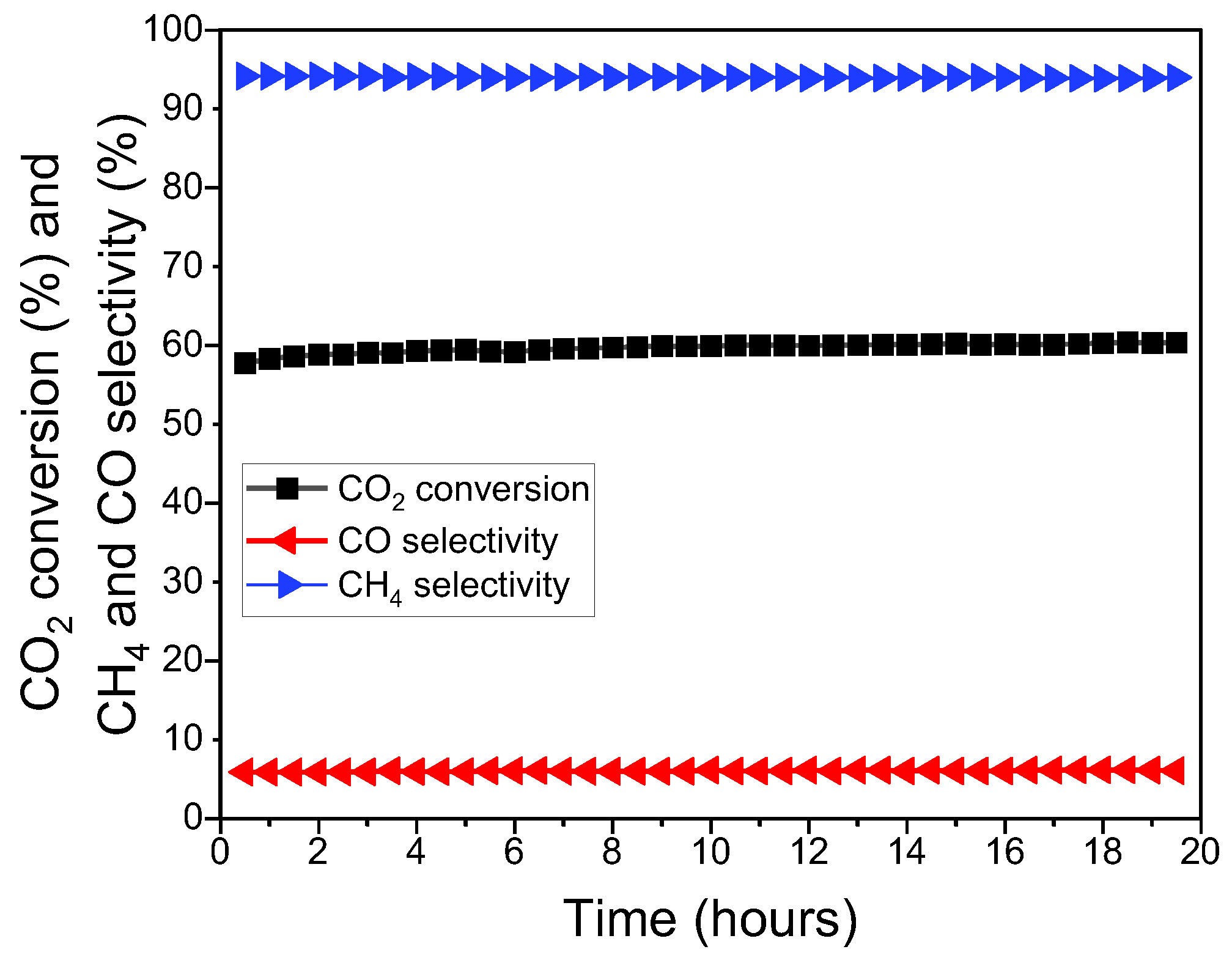
3.2. Long-Term Stability Test
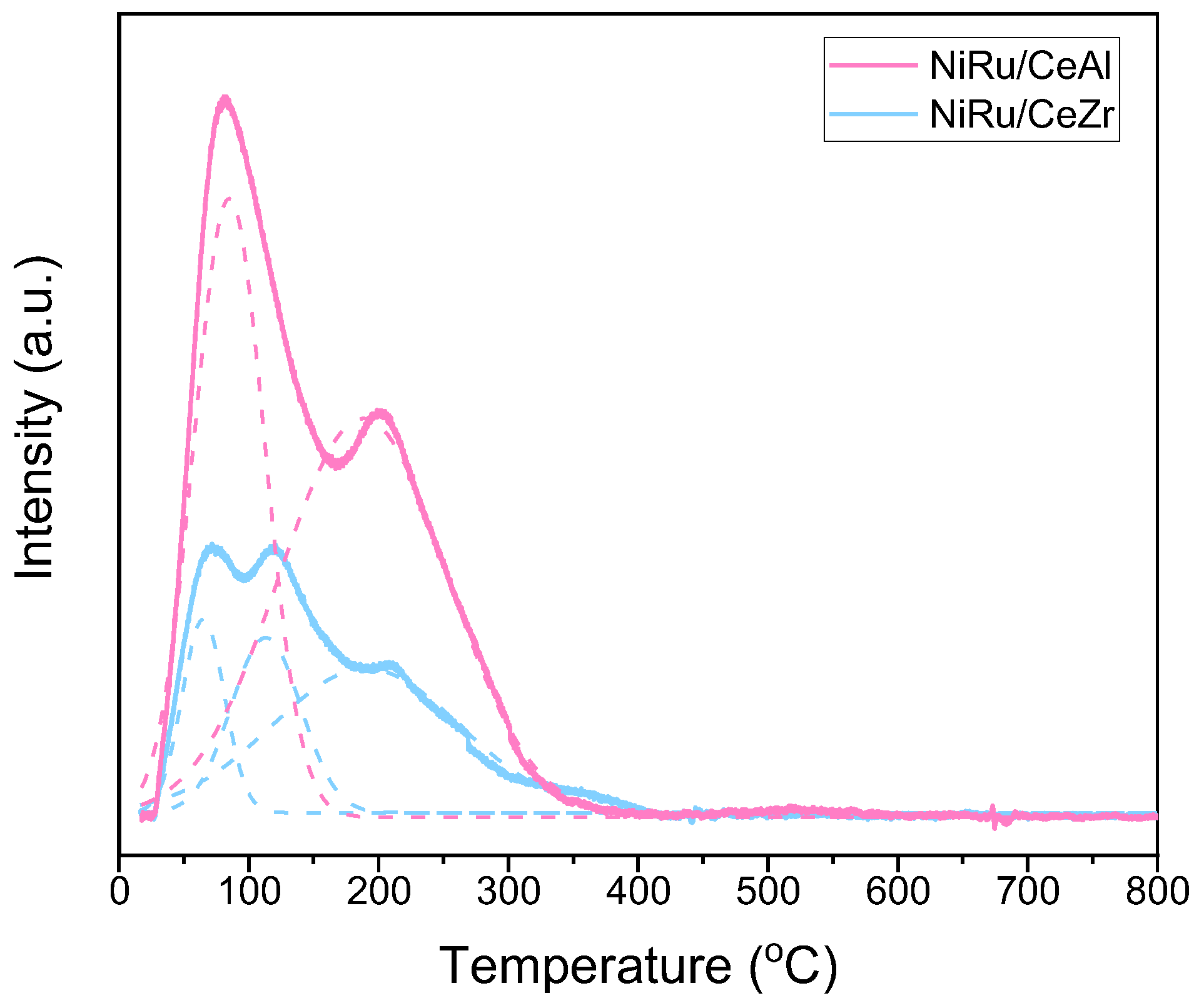
3.3. Promoter Effects on NiRu Catalysts for CO2 Methanation: The Effect of Surface Basicity
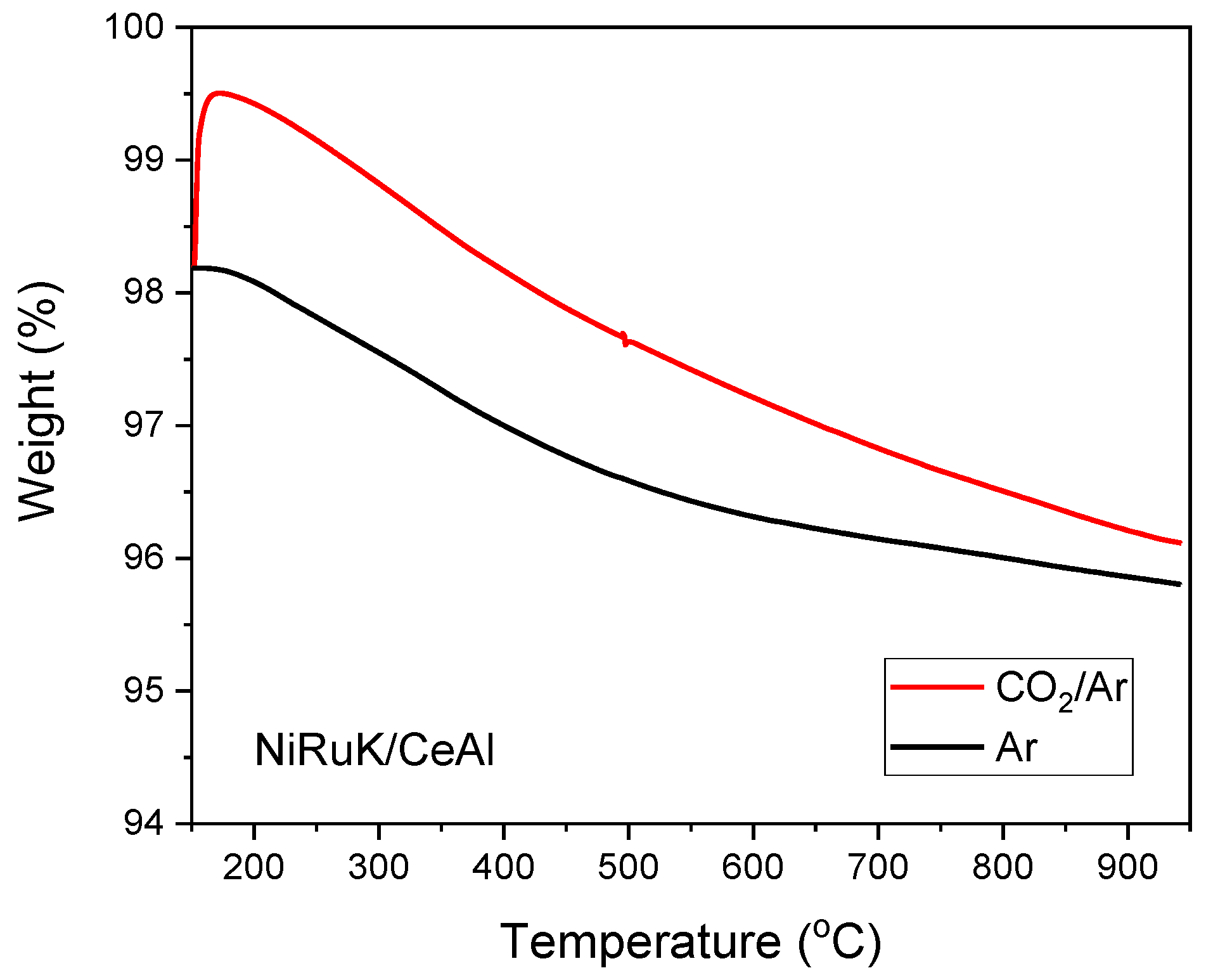
3.4. Adding CO2 Capture Functionality to Synthesise a Methanation DFM
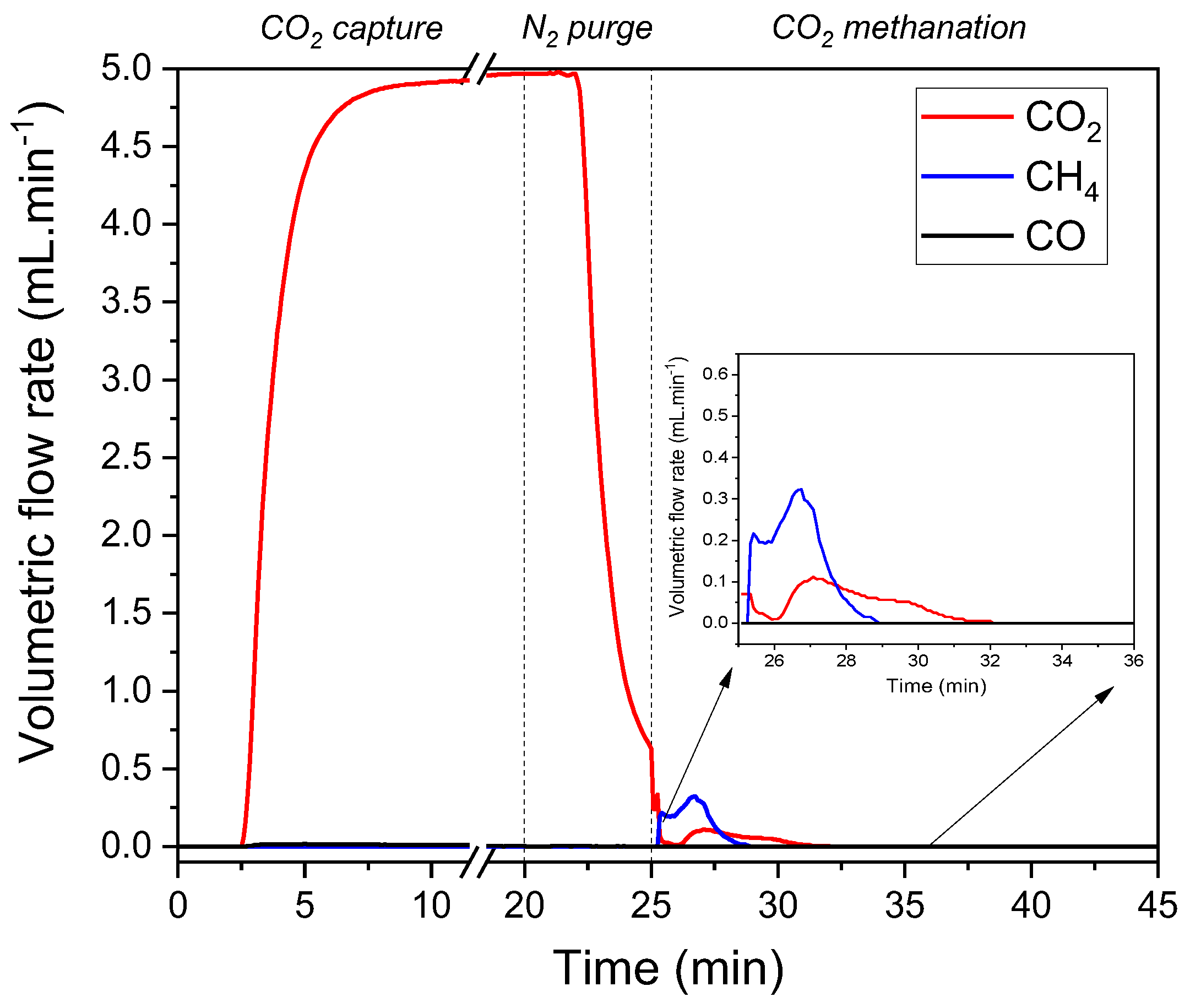
3.5. CO2 Capture and Reduction Study over NiRuK/CeAl DFM
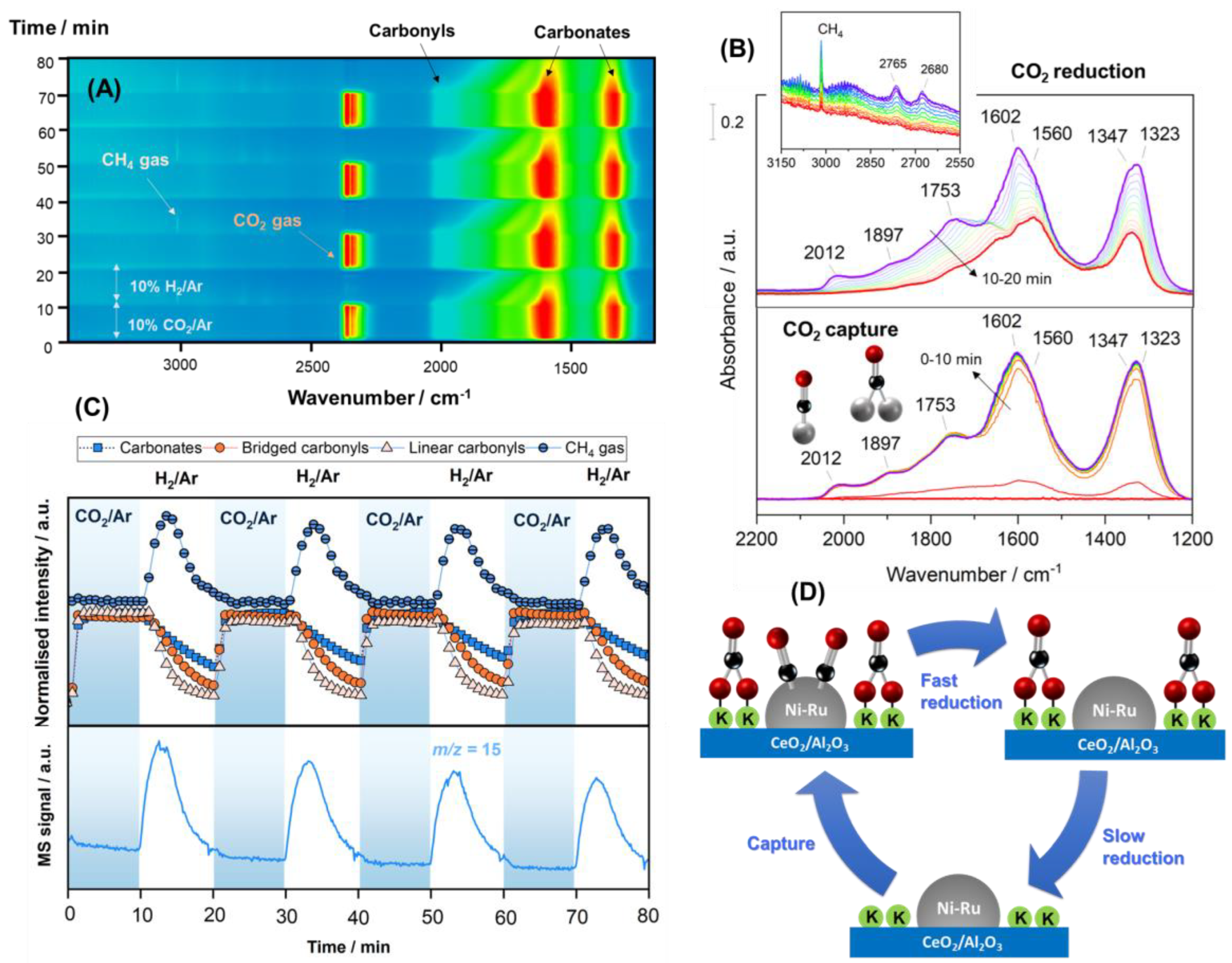
3.6. Time-Resolved Operando DRIFTS-MS Experimental Analysis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Stein, T. Carbon Dioxide Now More than 50 Higher than Pre-Industrial Levels. Available online: https://www.noaa.gov/news-release/carbon-dioxide-now-more-than-50-higher-than-pre-industrial-levels (accessed on 25 October 2022).
- Pires, J.C.M.; Martins, F.G.; Alvim-Ferraz, M.C.M.; Simões, M. Recent Developments on Carbon Capture and Storage: An Overview. Chem. Eng. Res. Des. 2011, 89, 1446–1460. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. CO2-Based Energy Vectors for the Storage of Solar Energy. Greenh. Gases: Sci. Technol. 2011, 1, 21–35. [Google Scholar] [CrossRef]
- Götz, M.; Lefebvre, J.; Mörs, F.; McDaniel Koch, A.; Graf, F.; Bajohr, S.; Reimert, R.; Kolb, T. Renewable Power-to-Gas: A Technological and Economic Review. Renew. Energy 2016, 85, 1371–1390. [Google Scholar] [CrossRef]
- Hashimoto, K.; Yamasaki, M.; Fujimura, K.; Matsui, T.; Izumiya, K.; Komori, M.; El-Moneim, A.A.; Akiyama, E.; Habazaki, H.; Kumagai, N.; et al. Global CO2 Recycling—Novel Materials and Prospect for Prevention of Global Warming and Abundant Energy Supply. Mater. Sci. Eng. A 1999, 267, 200–206. [Google Scholar] [CrossRef]
- Pleßmann, G.; Erdmann, M.; Hlusiak, M.; Breyer, C. Global Energy Storage Demand for a 100% Renewable Electricity Supply. Energy Procedia 2014, 46, 22–31. [Google Scholar] [CrossRef]
- Graf, F.; Götz, M.; Bajohr, S. Injection of Biogas, SNG and Hydrogen into the Gas Grid. GWF-Gas Erdgas Int. Issue 2011, 2, 30–40. [Google Scholar]
- Duyar, M.S.; Ramachandran, A.; Wang, C.; Farrauto, R.J. Kinetics of CO2 Methanation over Ru/γ-2O3 and Implications for Renewable Energy Storage Applications. J. CO2 Util. 2015, 12, 27–33. [Google Scholar] [CrossRef]
- Miguel, C.V.; Soria, M.A.; Mendes, A.; Madeira, L.M. A Sorptive Reactor for CO2 Capture and Conversion to Renewable Methane. J. Chem. Eng. 2017, 322, 590–602. [Google Scholar] [CrossRef]
- Quadrelli, E.A.; Centi, G.; Duplan, J.L.; Perathoner, S. Carbon Dioxide Recycling: Emerging Large-Scale Technologies with Industrial Potential. ChemSusChem 2011, 4, 1194–1215. [Google Scholar] [CrossRef]
- Liu, M.; Yi, Y.; Wang, L.; Guo, H.; Bogaerts, A. Hydrogenation of Carbon Dioxide to Value-Added Chemicals by Heterogeneous Catalysis and Plasma Catalysis. Catalysts 2019, 9, 275. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent Advances in Catalytic Hydrogenation of Carbon Dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef] [PubMed]
- Frontera, P.; Macario, A.; Ferraro, M.; Antonucci, P.L. Supported Catalysts for CO2 Methanation: A Review. Catalysts 2017, 7, 59. [Google Scholar] [CrossRef]
- United Nations Framework Convention on Climate Change. Adoption of the Paris Agreement; United Nations Framework Convention on Climate Change: Paris, France, 2015. [Google Scholar]
- United Nations Framework Convention on Climate Change. Glasgow Climate Pact; United Nations Framework Convention on Climate Change: Glasgow, UK, 2021. [Google Scholar]
- Vannice, M.A. Catalytic Activation of Carbon Monoxide on Metal Surfaces. Catal. Sci. Technol. 1982, 3, 139–198. [Google Scholar] [CrossRef]
- Rönsch, S.; Schneider, J.; Matthischke, S.; Schlüter, M.; Götz, M.; Lefebvre, J.; Prabhakaran, P.; Bajohr, S. Review on Methanation—From Fundamentals to Current Projects. Fuel 2016, 166, 276–296. [Google Scholar] [CrossRef]
- Gahleitner, G. Hydrogen from Renewable Electricity: An International Review of Power-to-Gas Pilot Plants for Stationary Applications. Int. J. Hydrogen Energy 2013, 38, 2039–2061. [Google Scholar] [CrossRef]
- Wang, W.; Gong, J. Methanation of Carbon Dioxide: An Overview. Front. Chem. Sci. Eng. 2011, 5, 2–10. [Google Scholar] [CrossRef]
- Pham, C.Q.; Bahari, M.B.; Kumar, P.S.; Ahmed, S.F.; Xiao, L.; Kumar, S.; Qazaq, A.S.; Siang, T.J.; Tran, H.T.; Islam, A.; et al. Carbon Dioxide Methanation on Heterogeneous Catalysts: A Review. Environ. Chem. Lett. 2022, 20, 3613–3630. [Google Scholar] [CrossRef]
- Tan, C.H.; Nomanbhay, S.; Shamsuddin, A.H.; Park, Y.K.; Hernández-Cocoletzi, H.; Show, P.L. Current Developments in Catalytic Methanation of Carbon Dioxide—A Review. Front. Energy Res. 2022, 9, 795423. [Google Scholar] [CrossRef]
- Ashok, J.; Pati, S.; Hongmanorom, P.; Tianxi, Z.; Junmei, C.; Kawi, S. A Review of Recent Catalyst Advances in CO2 Methanation Processes. Catal. Today 2020, 356, 471–489. [Google Scholar] [CrossRef]
- Heraeus Precious Metal Charts. Available online: https://www.heraeus.com/en/hpm/pm_prices/charts/charts.html?currency=eur&charttype=line&dateFrom=09.02.2021&dateTo=09.08.2021&pt=1&rh=1&ir=1&ru=1&pd=1 (accessed on 7 November 2022).
- Crisafulli, C.; Scirè, S.; Minicò, S.; Solarino, L. Ni-Ru Bimetallic Catalysts for the CO2 Reforming of Methane. Appl. Catal. A Gen. 2002, 225, 1–9. [Google Scholar] [CrossRef]
- le Saché, E.; Pastor-Pérez, L.; Haycock, B.J.; Villora-Picó, J.J.; Sepúlveda-Escribano, A.; Reina, T.R. Switchable Catalysts for Chemical CO2 Recycling: A Step Forward in the Methanation and Reverse Water-Gas Shift Reactions. ACS Sustain. Chem. Eng. 2020, 8, 4614–4622. [Google Scholar] [CrossRef]
- Merkouri, L.-P.; le Sache, E.; Pastor-Perez, L.; Duyar, M.S.; Reina, T.R. Versatile Ni-Ru Catalysts for Gas Phase CO2 Conversion: Bringing Closer Dry Reforming, Reverse Water Gas Shift and Methanation to Enable End-Products Flexibility. Fuel 2022, 315, 123097. [Google Scholar] [CrossRef]
- Valdés-Martínez, O.U.; Suárez-Toriello, V.A.; de los Reyes, J.A.; Pawelec, B.; Fierro, J.L.G. Support Effect and Metals Interactions for NiRu/Al2O3, TiO2 and ZrO2 Catalysts in the Hydrodeoxygenation of Phenol. Catal. Today 2017, 296, 219–227. [Google Scholar] [CrossRef]
- Lange, F.; Armbruster, U.; Martin, A. Heterogeneously-Catalyzed Hydrogenation of Carbon Dioxide to Methane Using RuNi Bimetallic Catalysts. Energy Technol. 2015, 3, 55–62. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, S.; Zhao, G.; Yang, H.; Yuan, M.; An, X.; Zhou, H.; Qiao, Y.; Tian, Y. CO2 Methanation over Ordered Mesoporous NiRu-Doped CaO-Al2O3 Nanocomposites with Enhanced Catalytic Performance. Int. J. Hydrogen Energy 2018, 43, 239–250. [Google Scholar] [CrossRef]
- Álvarez, M.; Bobadilla, L.F.; Garcilaso, V.; Centeno, M.A.; Odriozola, J.A. CO2 Reforming of Methane over Ni-Ru Supported Catalysts: On the Nature of Active Sites by Operando DRIFTS Study. J. CO2 Util. 2018, 24, 509–515. [Google Scholar] [CrossRef]
- Trovarelli, A.; Boaro, M.; Rocchini, E.; de Leitenburg, C.; Dolcetti, G. Some Recent Developments in the Characterization of Ceria-Based Catalysts. J. Alloys Compd. 2001, 323, 584–591. [Google Scholar] [CrossRef]
- Chang, K.; Zhang, H.; Cheng, M.J.; Lu, Q. Application of Ceria in CO2 Conversion Catalysis. ACS Catal. 2020, 10, 613–631. [Google Scholar] [CrossRef]
- Liu, H.; Zou, X.; Wang, X.; Lu, X.; Ding, W. Effect of CeO2 Addition on Ni/Al2O3 Catalysts for Methanation of Carbon Dioxide with Hydrogen. J. Nat. Gas Chem. 2012, 21, 703–707. [Google Scholar] [CrossRef]
- Wang, S.; Lu, G.Q. Role of CeO2 in Ni/CeO2-Al2O3 Catalysts for Carbon Dioxide Reforming of Methane. Appl. Catal. B 1998, 19, 267–277. [Google Scholar] [CrossRef]
- Stroud, T.; Smith, T.J.; Le Saché, E.; Santos, J.L.; Centeno, M.A.; Arellano-Garcia, H.; Odriozola, J.A.; Reina, T.R. Chemical CO2 Recycling via Dry and Bi Reforming of Methane Using Ni-Sn/Al2O3 and Ni-Sn/CeO2-Al2O3 Catalysts. Appl. Catal. B 2018, 224, 125–135. [Google Scholar] [CrossRef]
- Pastor-Pérez, L.; Le Saché, E.; Jones, C.; Gu, S.; Arellano-Garcia, H.; Reina, T.R. Synthetic Natural Gas Production from CO2 over Ni-x/CeO2-ZrO2 (x = Fe, Co) Catalysts: Influence of Promoters and Space Velocity. Catal. Today 2018, 317, 108–113. [Google Scholar] [CrossRef]
- Ocampo, F.; Louis, B.; Kiwi-Minsker, L.; Roger, A.C. Effect of Ce/Zr Composition and Noble Metal Promotion on Nickel Based CexZr1-XO2 Catalysts for Carbon Dioxide Methanation. Appl. Catal. A Gen. 2011, 392, 36–44. [Google Scholar] [CrossRef]
- Ocampo, F.; Louis, B.; Kiennemann, A.; Roger, A.C. CO2 Methanation over Ni-Ceria-Zirconia Catalysts: Effect of Preparation and Operating Conditions. IOP Conf. Ser. Mater. Sci. Eng. 2011, 19, 012007. [Google Scholar] [CrossRef]
- Duyar, M.S.; Treviño, M.A.A.; Farrauto, R.J. Dual Function Materials for CO2 Capture and Conversion Using Renewable H2. Appl. Catal. B 2015, 168, 370–376. [Google Scholar] [CrossRef]
- Merkouri, L.P.; Reina, T.R.; Duyar, M.S. Closing the Carbon Cycle with Dual Function Materials. Energy Fuels 2021, 35, 19859–19880. [Google Scholar] [CrossRef]
- Sun, S.; Sun, H.; Williams, P.T.; Wu, C. Recent Advances in Integrated CO2 Capture and Utilization: A Review. Sustain. Energy Fuels 2021, 5, 4546–4559. [Google Scholar] [CrossRef]
- Omodolor, I.S.; Otor, H.O.; Andonegui, J.A.; Allen, B.J.; Alba-Rubio, A.C. Dual-Function Materials for CO2 Capture and Conversion: A Review. Ind. Eng. Chem. Res. 2020, 59, 17612–17631. [Google Scholar] [CrossRef]
- Melo Bravo, P.; Debecker, D.P. Combining CO2 Capture and Catalytic Conversion to Methane. Waste Dispos. Sustain Energy 2019, 1, 53–65. [Google Scholar] [CrossRef]
- Tsiotsias, A.I.; Charisiou, N.D.; Yentekakis, I.V.; Goula, M.A. The Role of Alkali and Alkaline Earth Metals in the CO2 Methanation Reaction and the Combined Capture and Methanation of CO2. Catalysts 2020, 10, 35. [Google Scholar] [CrossRef]
- Proaño, L.; Tello, E.; Arellano-Trevino, M.A.; Wang, S.; Farrauto, R.J.; Cobo, M. In-Situ DRIFTS Study of Two-Step CO2 Capture and Catalytic Methanation over Ru,“Na2O”/Al2O3 Dual Functional Material. Appl. Surf. Sci. 2019, 479, 25–30. [Google Scholar] [CrossRef]
- Proaño, L.; Arellano-Treviño, M.A.; Farrauto, R.J.; Figueredo, M.; Jeong-Potter, C.; Cobo, M. Mechanistic Assessment of Dual Function Materials, Composed of Ru-Ni, Na2O/Al2O3 and Pt-Ni, Na2O/Al2O3, for CO2 Capture and Methanation by in-Situ DRIFTS. Appl. Surf. Sci. 2020, 533, 147469. [Google Scholar] [CrossRef]
- Sun, H.; Wang, J.; Zhao, J.; Shen, B.; Shi, J.; Huang, J.; Wu, C. Dual Functional Catalytic Materials of Ni over Ce-Modified CaO Sorbents for Integrated CO2 Capture and Conversion. Appl. Catal. B 2019, 244, 63–75. [Google Scholar] [CrossRef]
- Hu, L.; Urakawa, A. Continuous CO2 Capture and Reduction in One Process: CO2 Methanation over Unpromoted and Promoted Ni/ZrO2. J. CO2 Util. 2018, 25, 323–329. [Google Scholar] [CrossRef]
- Hyakutake, T.; van Beek, W.; Urakawa, A. Unravelling the Nature, Evolution and Spatial Gradients of Active Species and Active Sites in the Catalyst Bed of Unpromoted and K/Ba-Promoted Cu/Al2O3 during CO2 Capture-Reduction. J. Mater. Chem. A Mater. 2016, 4, 6878–6885. [Google Scholar] [CrossRef]
- Zhou, Z.; Sun, N.; Wang, B.; Han, Z.; Cao, S.; Hu, D.; Zhu, T.; Shen, Q.; Wei, W. 2D-Layered Ni–MgO–Al2O3 Nanosheets for Integrated Capture and Methanation of CO2. ChemSusChem 2020, 13, 360–368. [Google Scholar] [CrossRef]
- Merkouri, L.-P.; Reina, R.; Duyar, M.S. Feasibility of Switchable Dual Function Materials as a Flexible Technology for CO2 Capture and Utilisation and Evidence of Passive Direct Air Capture. Nanoscale 2022, 14, 12620–12637. [Google Scholar] [CrossRef]
- Janke, C.; Duyar, M.S.; Hoskins, M.; Farrauto, R. Catalytic and Adsorption Studies for the Hydrogenation of CO2 to Methane. Appl. Catal. B 2014, 152, 184–191. [Google Scholar] [CrossRef]
- Kwak, J.H.; Kovarik, L.; Szanyi, J. CO2 Reduction on Supported Ru/Al2O3 Catalysts: Cluster Size Dependence of Product Selectivity. ACS Catal. 2013, 3, 2449–2455. [Google Scholar] [CrossRef]
- van Herwijnen, T.; van Doesburg, H.; de Jong, W.A. Kinetics of the Methanation of CO and CO2 on a Nickel Catalyst. J. Catal. 1973, 28, 391–402. [Google Scholar] [CrossRef]
- Quindimil, A.; De-La-Torre, U.; Pereda-Ayo, B.; Davó-Quiñonero, A.; Bailón-García, E.; Lozano-Castelló, D.; González-Marcos, J.A.; Bueno-López, A.; González-Velasco, J.R. Effect of Metal Loading on the CO2 Methanation: A Comparison between Alumina Supported Ni and Ru Catalysts. Catal. Today 2020, 356, 419–432. [Google Scholar] [CrossRef]
- Pan, Q.; Peng, J.; Sun, T.; Wang, S.; Wang, S. Insight into the Reaction Route of CO2 Methanation: Promotion Effect of Medium Basic Sites. Catal. Commun. 2014, 45, 74–78. [Google Scholar] [CrossRef]
- Kim, M.J.; Youn, J.R.; Kim, H.J.; Seo, M.W.; Lee, D.; Go, K.S.; Lee, K.B.; Jeon, S.G. Effect of Surface Properties Controlled by Ce Addition on CO2 Methanation over Ni/Ce/Al2O3 Catalyst. Int. J. Hydrogen Energy 2020, 45, 24595–24603. [Google Scholar] [CrossRef]
- Tada, S.; Ochieng, O.J.; Kikuchi, R.; Haneda, T.; Kameyama, H. Promotion of CO2 Methanation Activity and CH4 Selectivity at Low Temperatures over Ru/CeO2/Al2O 3 Catalysts. Int. J. Hydrogen Energy 2014, 39, 10090–10100. [Google Scholar] [CrossRef]
- Bermejo-López, A.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R. Ni Loading Effects on Dual Function Materials for Capture and In-Situ Conversion of CO2 to CH4 Using CaO or Na2CO3. J. CO2 Util. 2019, 34, 576–587. [Google Scholar] [CrossRef]
- Di Cosimo, J.I.; Apesteguía, C.R.; Ginés, M.J.L.; Iglesia, E. Structural Requirements and Reaction Pathways in Condensation Reactions of Alcohols on MgyAlOx Catalysts. J Catal. 2000, 190, 261–275. [Google Scholar] [CrossRef]
- Wierzbicki, D.; Baran, R.; Dębek, R.; Motak, M.; Grzybek, T.; Gálvez, M.E.; Da Costa, P. The Influence of Nickel Content on the Performance of Hydrotalcite-Derived Catalysts in CO2 Methanation Reaction. Int. J. Hydrogen Energy 2017, 42, 23548–23555. [Google Scholar] [CrossRef]
- Bobadilla, L.F.; Riesco-García, J.M.; Penelás-Pérez, G.; Urakawa, A. Enabling Continuous Capture and Catalytic Conversion of Flue Gas CO2 to Syngas in One Process. J. CO2 Util. 2016, 14, 106–111. [Google Scholar] [CrossRef]
- Samanta, A.; Zhao, A.; Shimizu, G.K.H.; Sarkar, P.; Gupta, R. Post-Combustion CO2 Capture Using Solid Sorbents: A Review. Ind. Eng. Chem. Res. 2012, 51, 1438–1463. [Google Scholar] [CrossRef]
- Lux, S.; Baldauf-Sommerbauer, G.; Siebenhofer, M. Hydrogenation of Inorganic Metal Carbonates: A Review on Its Potential for Carbon Dioxide Utilization and Emission Reduction. ChemSusChem 2018, 11, 3357–3375. [Google Scholar] [CrossRef]
- Borhani, T.N.G.; Azarpour, A.; Akbari, V.; Wan Alwi, S.R.; Manan, Z.A. CO2 Capture with Potassium Carbonate Solutions: A State-of-the-Art Review. Int. J. Greenh. Gas Control 2015, 41, 142–162. [Google Scholar] [CrossRef]
- Lee, S.C.; Choi, B.Y.; Lee, T.J.; Ryu, C.K.; Ahn, Y.S.; Kim, J.C. CO2 Absorption and Regeneration of Alkali Metal-Based Solid Sorbents. Catal. Today 2006, 111, 385–390. [Google Scholar] [CrossRef]
- Duyar, M.S.; Wang, S.; Arellano-Treviño, M.A.; Farrauto, R.J. CO2 Utilization with a Novel Dual Function Material (DFM) for Capture and Catalytic Conversion to Synthetic Natural Gas: An Update. J. CO2 Util. 2016, 15, 65–71. [Google Scholar] [CrossRef]
- Al-Mamoori, A.; Lawson, S.; Rownaghi, A.A.; Rezaei, F. Improving Adsorptive Performance of CaO for High-Temperature CO 2 Capture through Fe and Ga Doping. Energy Fuels 2019, 33, 1404–1413. [Google Scholar] [CrossRef]
- Chen, C.; Cheng, W.; Lin, S. Study of Reverse Water Gas Shift Reaction by TPD, TPR and CO2 Hydrogenation over Potassium-Promoted Cu/SiO2. Catalyst 2003, 238, 55–67. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Baldwin, J.W.; Peng, X.; Mpourmpakis, G.; Willauer, H.D. Potassium-Promoted Molybdenum Carbide as a Highly Active and Selective Catalyst for CO2 Conversion to CO. ChemSusChem 2017, 10, 2408–2415. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kuhn, J.N. CO2 Conversion by Reverse Water Gas Shift Catalysis: Comparison of Catalysts, Mechanisms and Their Consequences for CO2 Conversion to Liquid Fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Yang, X.; Su, X.; Chen, X.; Duan, H.; Liang, B.; Liu, Q.; Liu, X.; Ren, Y.; Huang, Y.; Zhang, T. Environmental Promotion Effects of Potassium on the Activity and Selectivity of Pt/Zeolite Catalysts for Reverse Water Gas Shift Reaction. Appl. Catal. B 2017, 216, 95–105. [Google Scholar] [CrossRef]
- Liang, B.; Duan, H.; Su, X.; Chen, X.; Huang, Y.; Chen, X.; Delgado, J.J.; Zhang, T. Promoting Role of Potassium in the Reverse Water Gas Shift Reaction on Pt/Mullite Catalyst. Catal. Today 2017, 281, 319–326. [Google Scholar] [CrossRef]
- Arellano-Treviño, M.A.; He, Z.; Libby, M.C.; Farrauto, R.J. Catalysts and Adsorbents for CO2 Capture and Conversion with Dual Function Materials: Limitations of Ni-Containing DFMs for Flue Gas Applications. J. CO2 Util. 2019, 31, 143–151. [Google Scholar] [CrossRef]
- Arellano-Treviño, M.A.; Kanani, N.; Jeong-Potter, C.W.; Farrauto, R.J. Bimetallic Catalysts for CO2 Capture and Hydrogenation at Simulated Flue Gas Conditions. J. Chem. Eng. 2019, 375, 121953. [Google Scholar] [CrossRef]
- Cimino, S.; Cepollaro, E.M.; Lisi, L. Sulfur Tolerance and Self-Regeneration Mechanism of Na-Ru/Al2O3 Dual Function Material during the Cyclic CO2 Capture and Catalytic Methanation. Appl. Catal. B 2022, 317, 121705. [Google Scholar] [CrossRef]
- Porta, A.; Visconti, C.G.; Castoldi, L.; Matarrese, R.; Jeong-Potter, C.; Farrauto, R.; Lietti, L. Ru-Ba Synergistic Effect in Dual Functioning Materials for Cyclic CO2 Capture and Methanation. Appl. Catal. B 2021, 283, 119654. [Google Scholar] [CrossRef]
- Bermejo-López, A.; Pereda-Ayo, B.; Onrubia-Calvo, J.A.; González-Marcos, J.A.; González-Velasco, J.R. How the Presence of O2 and NOx Influences the Alternate Cycles of CO2 Adsorption and Hydrogenation to CH4 on Ru-Na-Ca/Al2O3 Dual Function Material. J. CO2 Util. 2023, 67, 102343. [Google Scholar] [CrossRef]
- Azancot, L.; Bobadilla, L.F.; Centeno, M.A.; Odriozola, J.A. IR Spectroscopic Insights into the Coking-Resistance Effect of Potassium on Nickel-Based Catalyst during Dry Reforming of Methane. Appl. Catal. B 2021, 285, 119822. [Google Scholar] [CrossRef]
- Zhang, S.T.; Yan, H.; Wei, M.; Evans, D.G.; Duan, X. Hydrogenation Mechanism of Carbon Dioxide and Carbon Monoxide on Ru(0001) Surface: A Density Functional Theory Study. RSC Adv. 2014, 4, 30241–30249. [Google Scholar] [CrossRef]
- Zhu, M.; Tian, P.; Cao, X.; Chen, J.; Pu, T.; Shi, B.; Xu, J.; Moon, J.; Wu, Z.; Han, Y.F. Vacancy Engineering of the Nickel-Based Catalysts for Enhanced CO2 Methanation. Appl. Catal. B 2021, 282, 119561. [Google Scholar] [CrossRef]
- Lavalley, J.C. Infrared Spectrometric Studies of the Surface Basicity of Metal Oxides and Zeolites Using Adsorbed Probe Molecules. Catal. Today 1996, 27, 377–401. [Google Scholar] [CrossRef]
- Föttinger, K.; Schlögl, R.; Rupprechter, G. The Mechanism of Carbonate Formation on Pd-Al2O3 Catalysts. Chem. Comm. 2008, 320–322. [Google Scholar] [CrossRef]
- Navarro-Jaén, S.; Szego, A.; Bobadilla, L.F.; Laguna, Ó.H.; Romero-Sarria, F.; Centeno, M.A.; Odriozola, J.A. Operando Spectroscopic Evidence of the Induced Effect of Residual Species in the Reaction Intermediates during CO2 Hydrogenation over Ruthenium Nanoparticles. ChemCatChem 2019, 11, 2063–2068. [Google Scholar] [CrossRef]
- Vogt, C.; Wijten, J.; Madeira, C.L.; Kerkenaar, O.; Xu, K.; Holzinger, R.; Monai, M.; Weckhuysen, B.M. Alkali Promotion in the Formation of CH4 from CO2 and Renewably Produced H2 over Supported Ni Catalysts. ChemCatChem 2020, 12, 2792–2800. [Google Scholar] [CrossRef]
- Vogt, C.; Kranenborg, J.; Monai, M.; Weckhuysen, B.M. Structure Sensitivity in Steam and Dry Methane Reforming over Nickel: Activity and Carbon Formation. ACS Catal. 2020, 10, 1428–1438. [Google Scholar] [CrossRef]
- Solymosi, F.; Knozinger, H. Infrared Spectroscopic Study of the Adsorption and Reactions of CO2 on K-Modified Rh/SiO2. J. Catal. 1990, 122, 166–177. [Google Scholar] [CrossRef]
- Solis-Garcia, A.; Zepeda, T.A.; Fierro-Gonzalez, J.C. Spectroscopic Evidence of Surface Species during CO2 Methanation Catalyzed by Supported Metals: A Review. Catal. Today 2022, 394, 2–12. [Google Scholar] [CrossRef]
- Falbo, L.; Visconti, C.G.; Lietti, L.; Szanyi, J. The Effect of CO on CO2 Methanation over Ru/2O3 Catalysts: A Combined Steady-State Reactivity and Transient DRIFT Spectroscopy Study. Appl. Catal. B 2019, 256, 117791. [Google Scholar] [CrossRef]
- Vogt, C.; Groeneveld, E.; Kamsma, G.; Nachtegaal, M.; Lu, L.; Kiely, C.J.; Berben, P.H.; Meirer, F.; Weckhuysen, B.M. Unravelling Structure Sensitivity in CO2 Hydrogenation over Nickel. Nat. Catal. 2018, 1, 127–134. [Google Scholar] [CrossRef]






| Material | Metal Loading (%) | Adsorbent Loading (%) | BET (m2/g) | Pore Volume (cm3/g) | Ni Particle Size (nm) 1 | Crystalline Phases 2 | H2-TPR Main Reduction Peaks (°C) |
|---|---|---|---|---|---|---|---|
| 15%Ni 1%Ru/CeO2-Al2O3 | 15-1 | - | 141 | 0.29 | 12 | Ni0, Ru0, Al2O3 | 130, 380 |
| 15%Ni 1%Ru/Ce0.5Zr0.5O2 | 15-1 | - | 60 | 0.18 | 34 | Ni0, NiO, Ce0.5Zr0.5O2 | 150, 320 |
| 15%Ni 1%Ru 10%K2O/CeO2-Al2O3 | 15-1 | 10 | 170 | 0.40 | 10 | Ni0, CeO2, Al2O3 | 190, 350, 460 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merkouri, L.-P.; Martín-Espejo, J.L.; Bobadilla, L.F.; Odriozola, J.A.; Duyar, M.S.; Reina, T.R. Flexible NiRu Systems for CO2 Methanation: From Efficient Catalysts to Advanced Dual-Function Materials. Nanomaterials 2023, 13, 506. https://doi.org/10.3390/nano13030506
Merkouri L-P, Martín-Espejo JL, Bobadilla LF, Odriozola JA, Duyar MS, Reina TR. Flexible NiRu Systems for CO2 Methanation: From Efficient Catalysts to Advanced Dual-Function Materials. Nanomaterials. 2023; 13(3):506. https://doi.org/10.3390/nano13030506
Chicago/Turabian StyleMerkouri, Loukia-Pantzechroula, Juan Luis Martín-Espejo, Luis Francisco Bobadilla, José Antonio Odriozola, Melis Seher Duyar, and Tomas Ramirez Reina. 2023. "Flexible NiRu Systems for CO2 Methanation: From Efficient Catalysts to Advanced Dual-Function Materials" Nanomaterials 13, no. 3: 506. https://doi.org/10.3390/nano13030506
APA StyleMerkouri, L.-P., Martín-Espejo, J. L., Bobadilla, L. F., Odriozola, J. A., Duyar, M. S., & Reina, T. R. (2023). Flexible NiRu Systems for CO2 Methanation: From Efficient Catalysts to Advanced Dual-Function Materials. Nanomaterials, 13(3), 506. https://doi.org/10.3390/nano13030506