1. Introduction
Despite recent improvements in breast cancer mortality, many patients relapse after initial response to conventional therapy and chemotherapy. Several alternative hypotheses have been proposed to explain this treatment failure and recurrence. Of particular interest is the cancer stem cell theory, which suggests that a small subpopulation of cells within tumors possess the unique ability to self-renew and generate the diverse cells that comprise the tumor [
1]. Often referred to as cancer stem cells (CSCs), these cells are found to be resistant to therapy and, hence, may reinitiate tumor growth after treatment [
1,
2,
3]. As such, isolation of this tumorigenic population from tumor tissues will be crucial for therapy development. Breast CSCs were first identified and isolated by Al-Hajj
et al. [
4]. When human breast tumors were propagated in non-obese diabetic severe combined immunodeficiency (NOD/SCID) mice, it was observed that as few as 100 of the CD44
+CD24
−/low cells were able to give rise to new tumors. Thus, breast CSCs are postulated to have a CD44
+CD24
−/low phenotype, strongly expressing the adhesion CD44 molecule, while very weakly expressing the adhesion CD24 molecule. Subsequent studies were conducted to validate Al-Hajj’s findings [
5,
6,
7].
The CSC fractions in solid tumors have been observed to be highly impure, and thus, the reported frequencies for the same tumor types have varied enormously between different research groups [
1]. Thus, more definitive markers are required to better characterize the CSCs. In the breast CSC field, it was observed from Abraham’s work that the proportion of CD44
+CD24
−/low cells ranged from 0 to 40% in normal tissues [
5]. Human breast cancer cell lines also differ quantitatively in the proportion of CD44
+CD24
− cells [
7]. The percentage of CD44
+CD24
− cells within breast cancer cell lines was found to be uncorrelated with tumorigenicity [
8]. In addition, studies have found that the epithelial specific antigen (ESA) cell surface molecule is overexpressed by the majority of human epithelial carcinomas, including breast carcinomas [
8,
9,
10]. The tumorigenic activity of the CD44
+CD24
−/low population was further enhanced when the CD44
+CD24
−/lowESA
+ cell population was used [
4]. Therefore, CD44
+CD24
−/lowESA
+ is a potential breast CSC marker for further investigation of this group of breast cancer cells with tumorigenic properties. The methods used to isolate CSCs have generally revolved around the use of labels, such as selective expression of surface markers [
1,
4,
11]. However, while this method enables the identification and isolation of a specific sub-group of CSC, the marker may not be able to serve as a universal marker for CSC.
A second method of CSC isolation is the Hoechst dye exclusion. These cells are referred to as side population cells and have been found to exhibit CSC characteristics of tumorigenicity and chemotherapeutic drug resistance [
12,
13]. A third method based on the observation that CSCs lack 26S proteasome function has emerged [
3,
14]. Cells with reduced 26S proteasome activity express CSC markers and appear to be more tumorigenic than the control cells. They have also been demonstrated to characterize a sub-population of the CD44
+CD24
−/low breast CSCs [
15].
Label-free methods exploiting the differences in physical properties, such as cell size, density, cell adhesion and dielectric properties, are also being explored. Advantages conferred by label-free methods include less laborious and time-consuming procedures, as the preparation for staining before and after cell separation will not be needed. Nonetheless, one potential drawback of these methods is that these physical differences might be insufficient for accurate cell separation. Substrates act as intelligent surfaces capable of providing biochemical and topographical signals to guide cell adhesion, spreading, morphology, proliferation, and eventually, cell differentiation [
16]. Numerous studies have demonstrated that cell adhesion behavior is significantly affected by surface nanotopography [
17,
18]. Cell adhesion can be increased or decreased by changing the material or geometry used to construct the surface structure [
19]. Kwon and co-workers [
19] used a nanotopographic substrate in a microfluidic approach to separate human breast cancer cells using cell adhesion as a physical marker. The breast cancer cell line (MCF7) and normal human breast epithelial cells (MCF10A) were observed to display different adhesion properties when cultured on substrates with different topographies. However, the cell adhesion analysis of the cell line mono-culture may differ from the heterogenicity of breast cancer tissues, and therefore, the cell adhesion properties of a heterogeneous population would need to be further investigated.
Thus, we hypothesize that different cell populations in the heterogeneous breast cancer tissue will respond differently to nanotopographical cues, potentially providing a means of enriching breast CSCs for isolation. The information can be used to develop a fast and easy method to isolate cancer stem cells, as well as different cell populations from cancer tissue, via a label-free method employing the differences in cell adhesion. Here, we investigated the adhesion characteristics of human mammary epithelial cells (HMEC), invasive breast cancer cell line MCF7 and invasive ductal carcinoma (IDC) primary breast tumor cells on nanopatterned poly-L-lactic acid (PLLA) films, as well as studying the effectiveness of these nanopatterns in preferential adhesion and isolation of breast CSCs.
3. Discussion
Cancer stem cells (CSC) are a population of proliferating cells with the capability of self-renewal and re-establishment of tumor heterogeneity [
22]. Since the establishment of the cancer stem cell theory, increasing evidence indicates that cancer stem cells play an important role in tumorigenesis and potential relapse of cancer. However, in general, current cancer therapies, such as chemotherapy, target rapidly proliferating cells instead of CSCs. As such, many have suggested that in order to reduce cancer relapse, eradication of CSCs is crucial.
As cells are surrounded by submicron features in their microenvironment, it has been shown that nanotopography influences cell adhesion, proliferation and differentiation [
23]. We hypothesize that different cell populations in breast cancer tissue exhibit different adhesion characteristics on substrates with nanotopography and investigated the adhesion characteristics of three cell populations, HMEC, breast cancer cell line MCF7 and primary IDC cells, on biomaterial substrates with nanotopographical cues. Based on the adhesion difference, the cancer stem cell-like population in MCF7, HMEC and IDC cells was separated and enriched from the mono-culture or the co-culture of HMEC and MCF7 using nanotopography and CSC surface markers. The results have shown a trend of preferential cell adhesion in both MCF7 and primary IDC cells cultured on different nanotopography after 4 and 24 h. Among the topographies tested, the nanoscale features of 350-nm gratings are consistently the most effective in enriching the CD44
+CD24
−/lowESA
+ breast CSCs. Interestingly, the total cell population has preferential adhesion to 1-μm gratings and 250-nm wells for MCF7 and primary IDC cells, respectively, suggesting that sub-populations of cancer cells exhibit different adhesion characterization to topographical structures.
Dick and colleagues’ studies in leukemic cells in the early 1990s gave one of the first descriptions of cancer stem cells [
19]. They were able to isolate a subpopulation of leukemic cells via biomarkers CD34
+/CD38
− and initiated acute myeloid leukemia in NOD/SCID mice using these leukemic cells [
22,
24]. CSCs were also found in the solid tumors of breast cancer [
4,
22,
25,
26]. Breast cancer cells have demonstrated not only self-renewal capability, but also a wide spectrum of progeny [
25]. The ESA
+CD44
+CD24
−/low lineage cells isolated from human breast tumors were able to form tumors in eight-week-old NOD/SCID mice [
4]. These cells formed a very small proportion of the total cell population, and as few as 200 cells were able to consistently form tumors in the mice and, thus, were identified as breast CSCs [
4,
25].
Although researchers were able to isolate these putative CSCs, a principle to isolate stem cells from any kind of cancer has yet to be established. In the case of breast cancer, there remains a need to determine whether CD44
+CD24
−ESA
+ cells are true breast CSCs across various breast cancer subtypes. In addition, reports have confirmed that in both breast cancer-derived cell lines and breast tumors, CD44
+CD24
− phenotypes were not necessarily associated with patient outcome or the ability to metastasize [
7,
27]. Therefore, further research is required to identify and separate CSCs in breast cancer. Meanwhile, current therapies fail to account for potential differences in drug sensitivity or target expression between the CSCs and the more frequent non-tumorigenic cells [
28]. The ability to isolate breast CSCs enables the testing of the effectiveness of cancer drugs that are currently used and will also enable the development of therapeutic strategies aimed at selectively targeting breast CSCs [
22].
Current methods for detecting and separating CSCs from solid tumors have been based on the observation of morphological features or labeling with specific markers [
25]. The labeling-assisted method is more commonly used and is highly specific. However, it requires laborious procedures prior to separation and time-consuming post-processing, such as the removal of labels [
29]. Label-free methods have been studied to separate cancer cells, which utilize differences in physical properties, such as cell size, density [
20], cell adhesion [
20,
30] and dielectric properties [
20], but the physical distribution is not high enough for efficient and accurate separation of cell populations, which limits their use. Kwon and co-workers used nanotopographic substrates to separate and enrich human breast cancer cells [
19]. They demonstrated that normal human breast epithelial cells (MCF10A) have stronger adhesion to the substratum than MCF7, regardless of culture time, surface topography and flow rates. This separation technique was reported to have 78% efficiency [
19].
Kwon’s study also found that the separation of human breast cancer cells from a mixed phase of MCF7 and MCF10A cells was optimized to occur after 2 h of culture on 400-nm gratings. A shorter duration of 2 h was chosen, as the high flow rate can exert significant shear damage to the adhered cells under longer culture periods. In contrast, in our static cultures, 4-hour is a reasonable duration, as the cells might not be able to recognize the surface within a short period of time. The 4-hour time point also appears to be the optimum duration for the separation and enrichment of the breast CSCs (as denoted by the phenotype of CD44+CD24−/lowESA+) from the heterogeneous MCF7 cancer cells and the primary cells under static culture conditions. This is in line with the development of a label-free method to isolate CSCs, as a shorter culture and analysis time is more desirable for cell analysis and cell isolation.
Kwon and colleagues determined that the use of 400-nm gratings resulted in the generation of the largest shear stress on the cell surface, facilitating the detachment of the MCF7 cells. Thus, it is the preferential detachment behavior that allowed the successful separation of the breast cancer cells in Kwon’s study [
19]. On the contrary, we have focused on the preferential attachment behavior of the cells, which may account for the difference of the choice of topographical feature. Thus, future work may involve characterizing the unattached cells, as well, to establish the most effective nanotopography for the isolation of the CSCs.
The mixture of MCF7 and MCF10A used by Kwon served to mimic the primary tissue of a breast cancer tumor; it has been proven that although breast and breast cancer cell lines have adapted to tissue culture conditions, they retain many of their phenotypic and genotypic properties over many passages. Hence, despite their acquired ability to grow
in vitro, cell lines continue to share many of the molecular and genetic features of the primary breast cancers from which they were derived [
8]. This validates the use of MCF7 to elucidate the preferential adhesion behavior of tumorigenic breast CSCs.
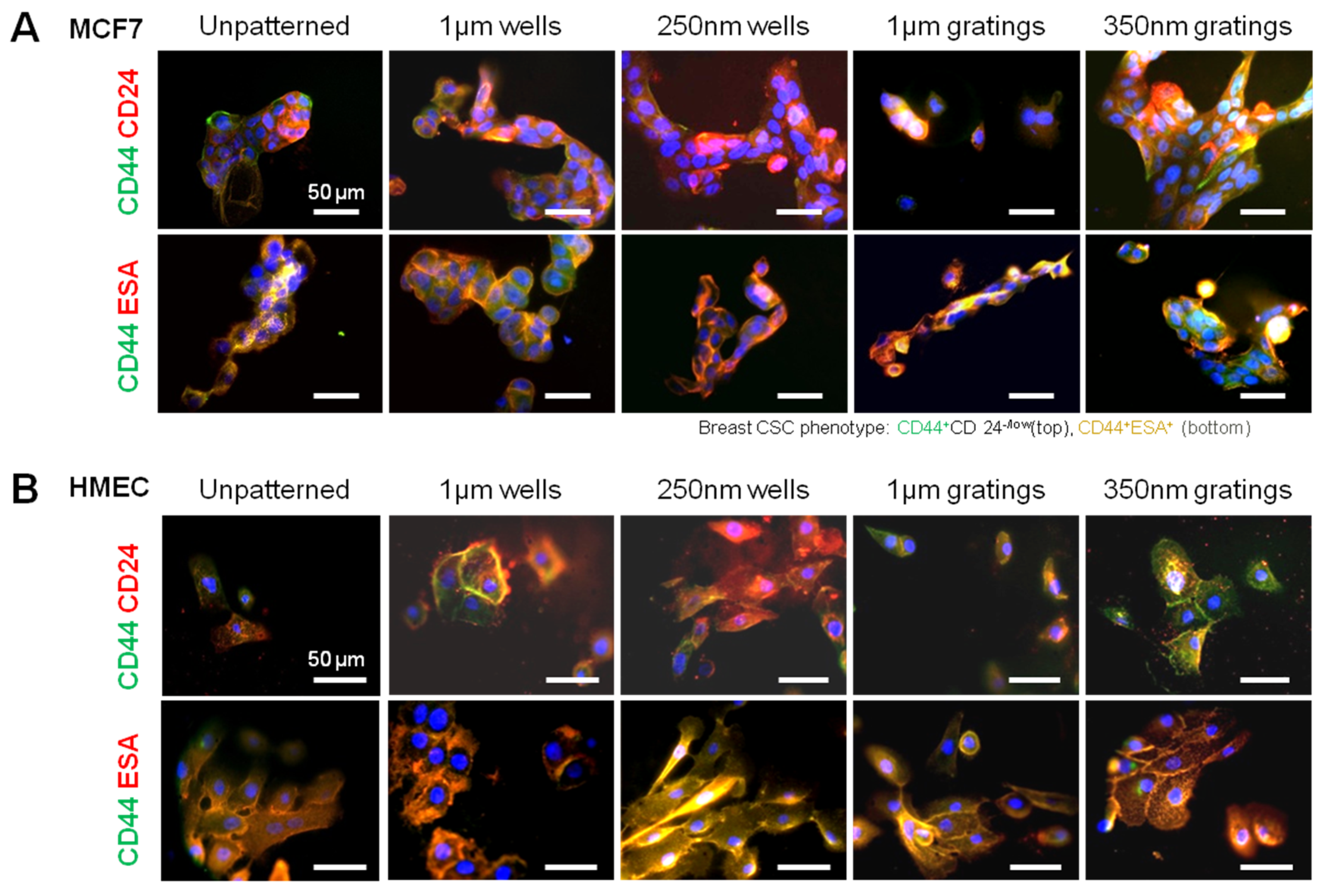
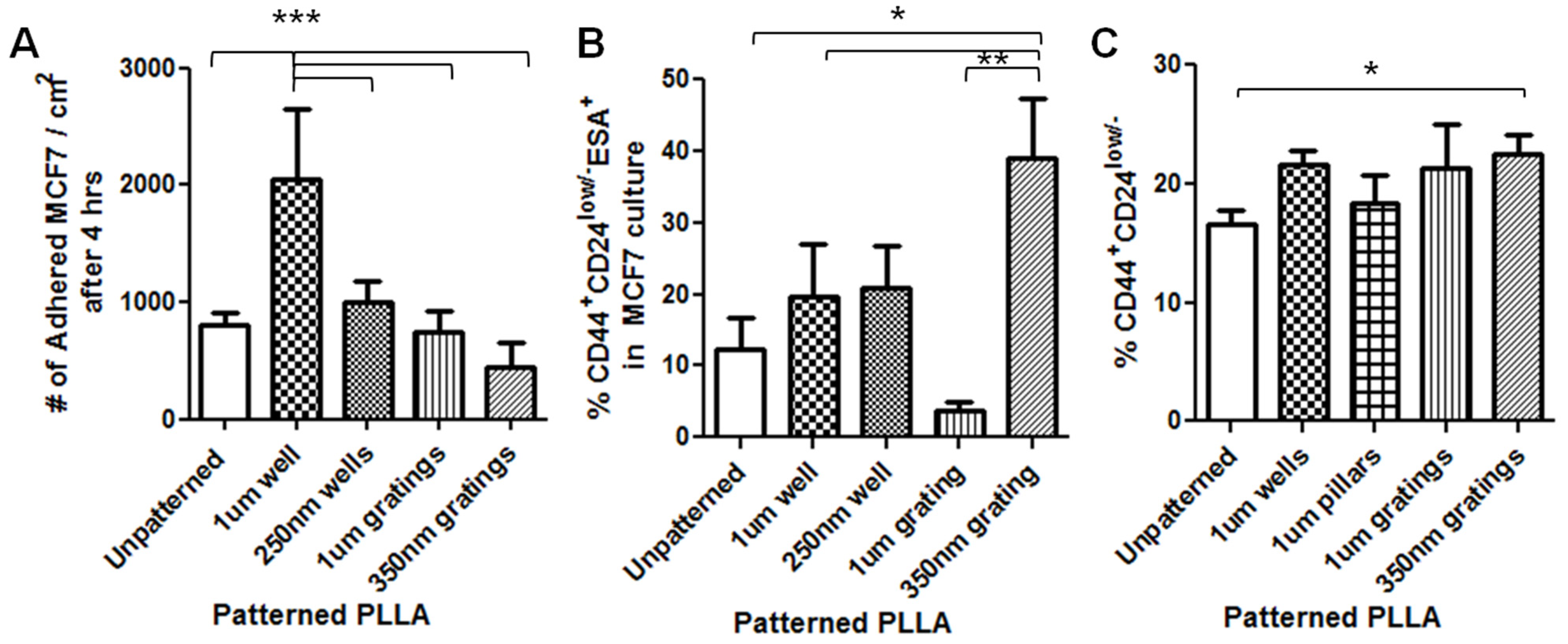
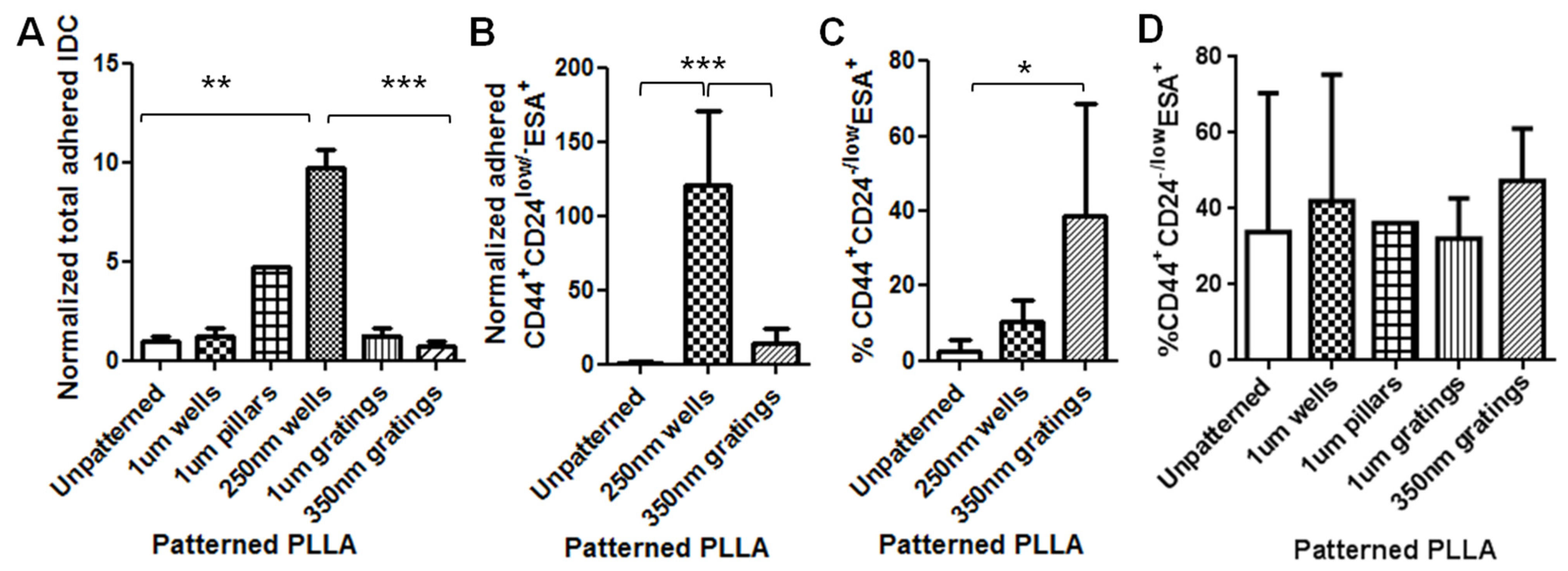
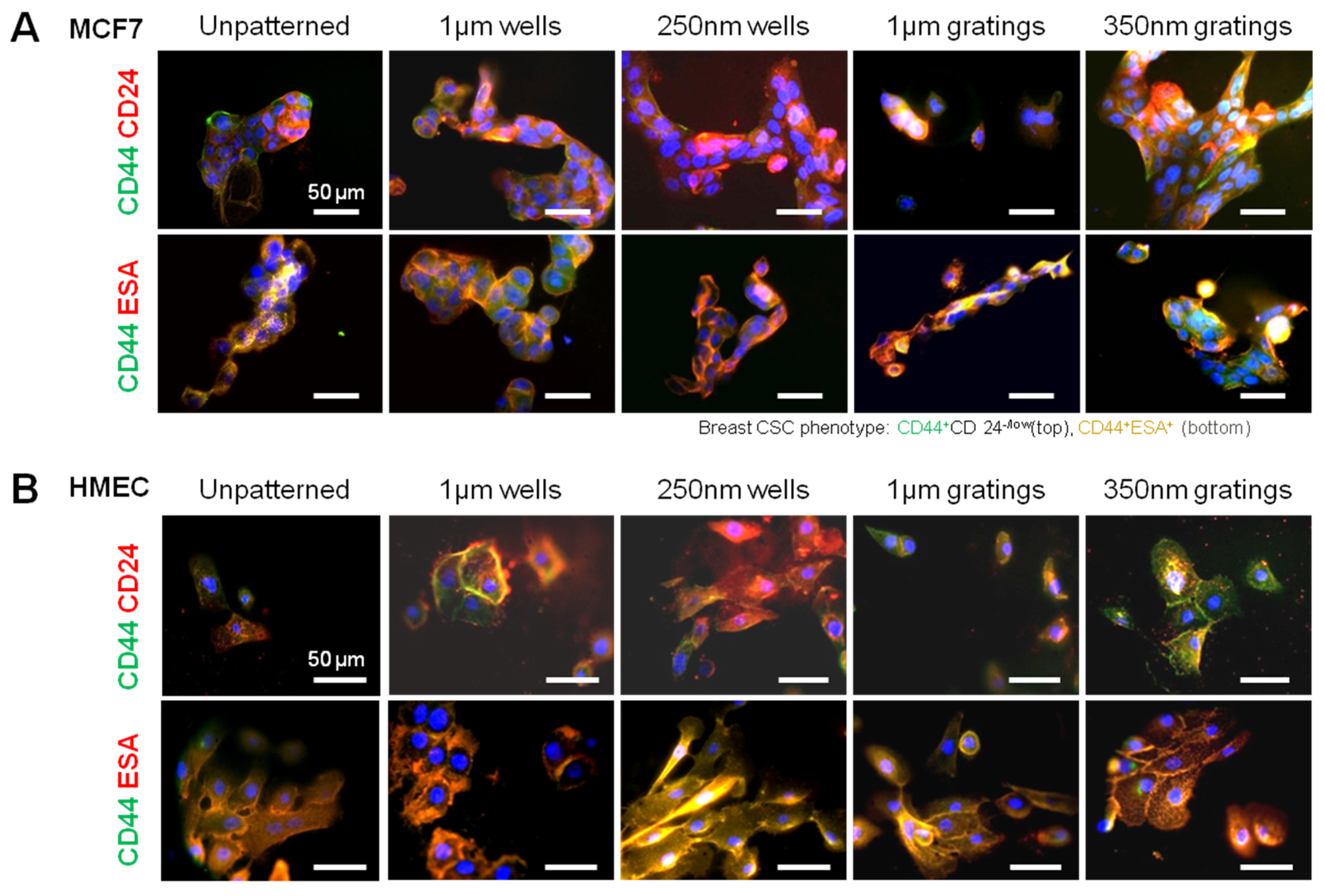
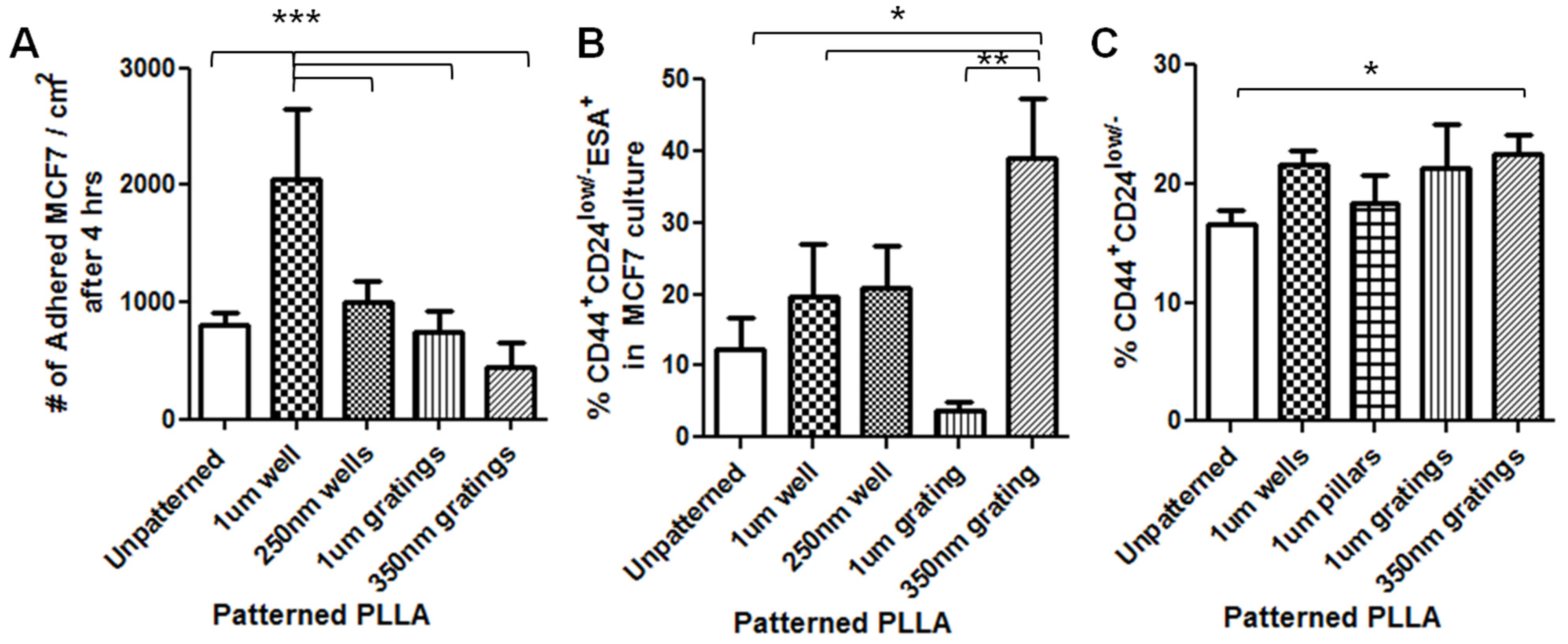
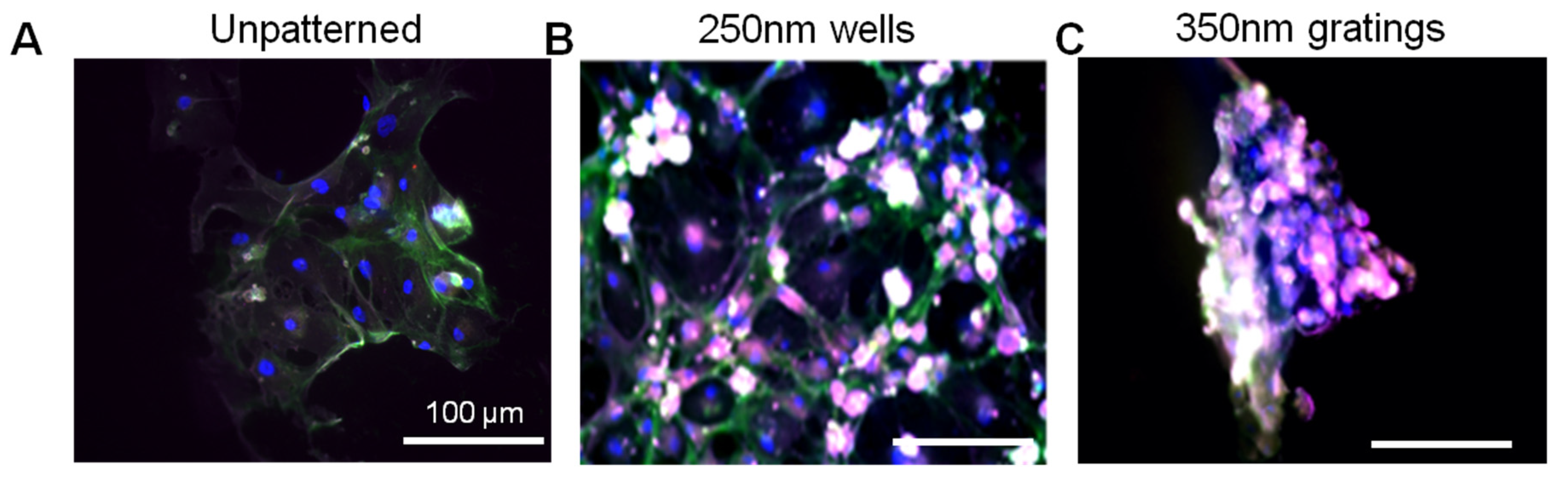
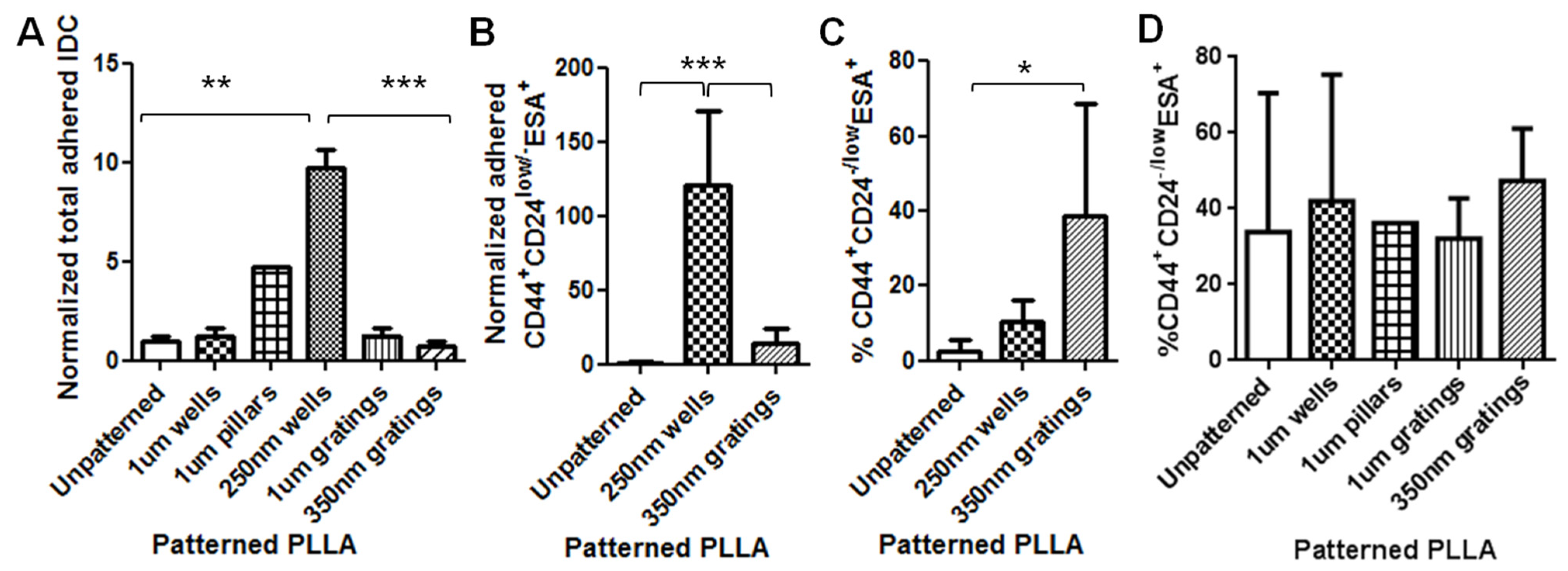
In this research, cell adhesion is evaluated based on the number of cells that adhered to the substrate and the cell morphology. From the data presented, less MCF7 and IDC cells adhered to gratings as compared to wells. From immunostaining, the MCF7 on the grating displayed higher fluorescence intensity for CD44 staining and a higher CD44+CD24−/lowESA+ population, as compared to wells and the control. By counting the number of CD44+ cells that adhered to the substrates, more than 40% of the cells that the 350-nm gratings captured were CD44+CD24−/lowESA+ MCF7 cells, suggesting that the 350-nm grating is most effective in the enrichment of CD44+CD24−/lowESA+ MCF7 in a monoculture.
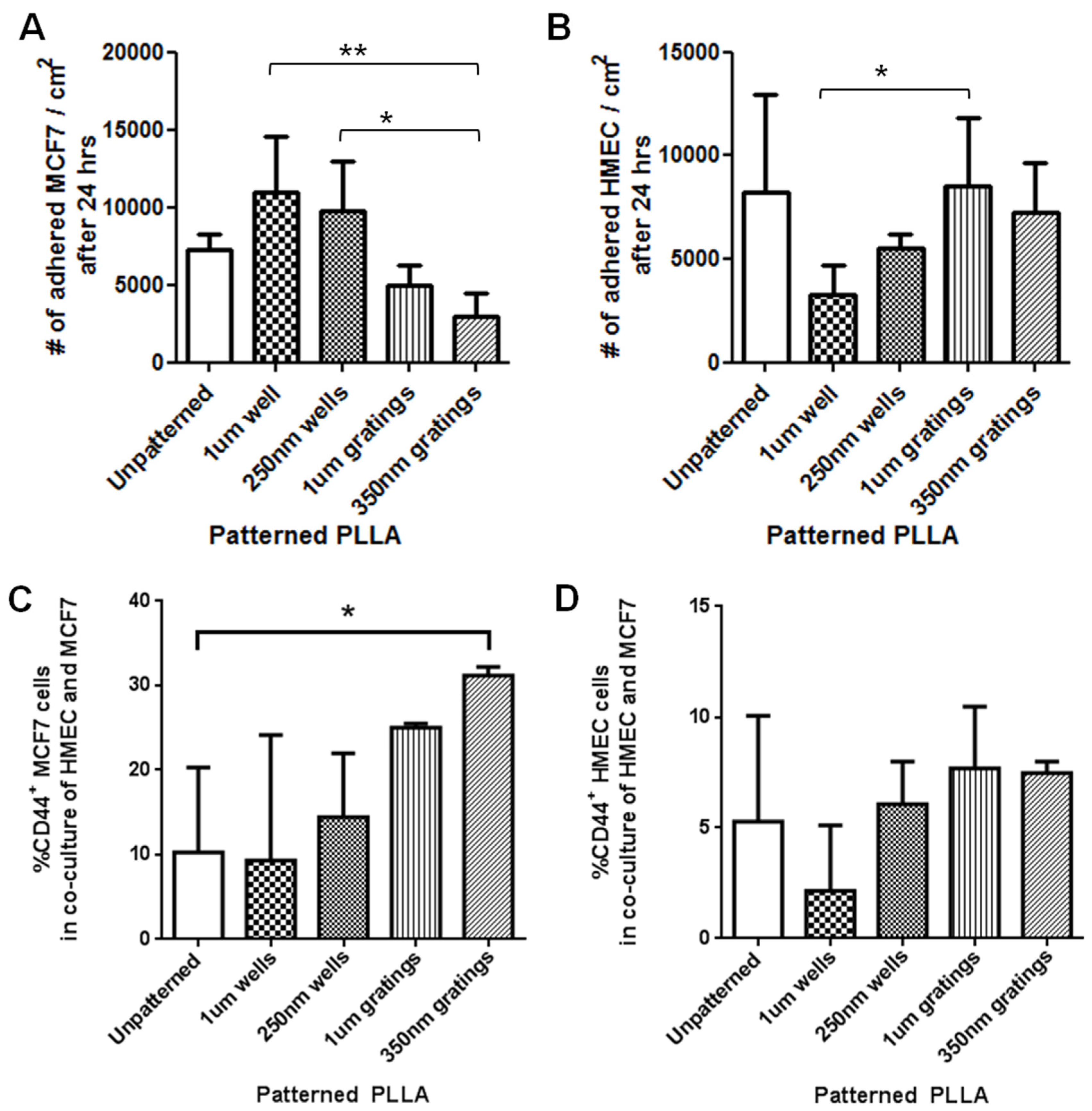
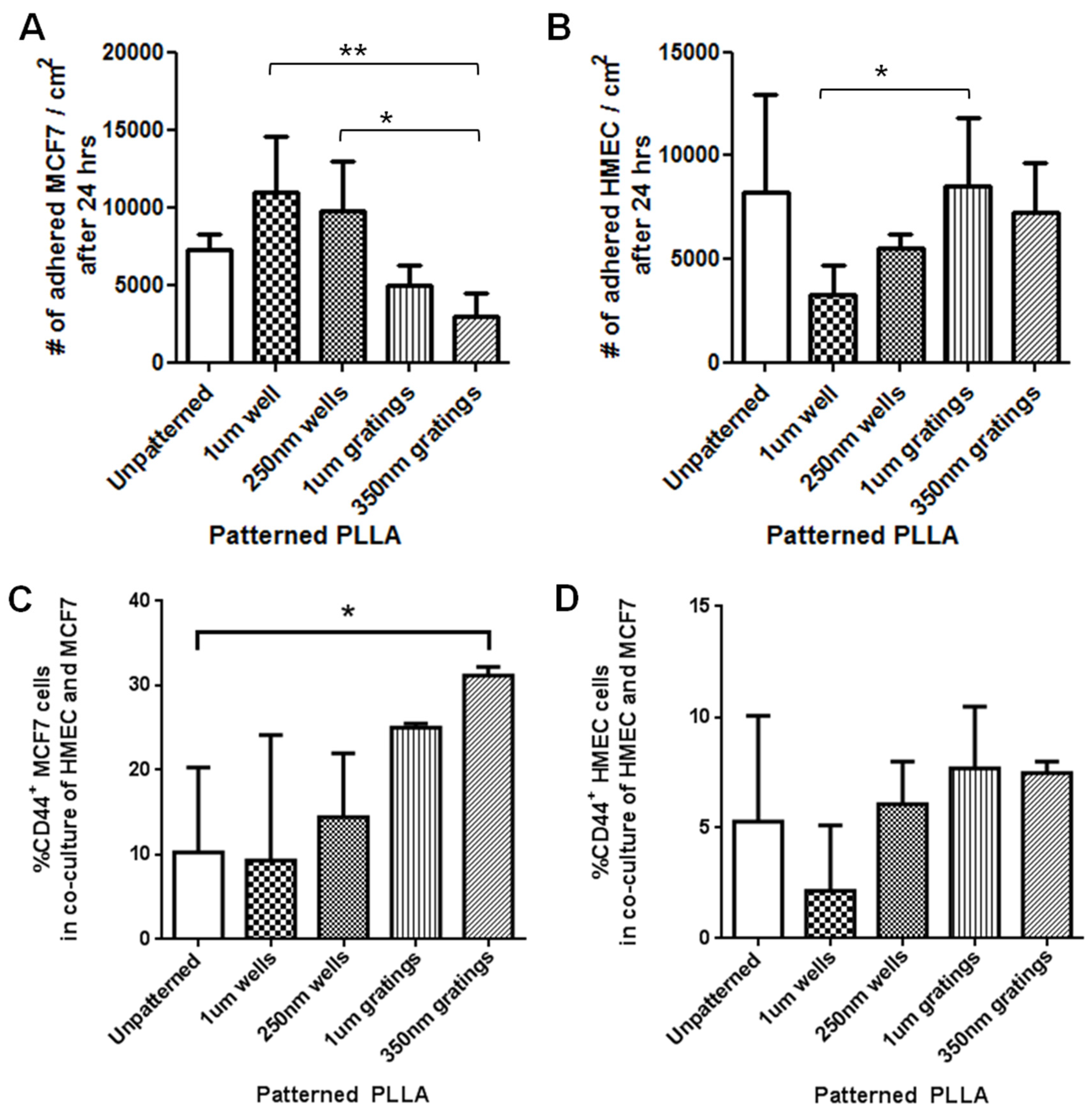
The difference in cell adhesion is a key parameter to enable isolation of breast CSCs from the heterogeneous cell suspension. Cell adhesion is affected by surface chemistry and the topography of the polymer substrate, as well as depending on the geometric parameters of the nanotopographic substrate. To mimic the heterogeneous cell population of a breast cancer tumor, the differential adhesion behavior of the co-culture of MCF7 and HMEC was studied. The nanotopography that would be the most suitable for the intended purpose would be one that captures the most CD44+ cells in the heterogeneous co-culture experiment. The MCF7 that adhered on 350-nm gratings continue to give high fluorescence intensity when stained for CD44, as compared to the well pattern, and have the highest percentage of cells that are CD44+ when analyzed by flow cytometry. This most likely indicates that in the co-culture, the nanotopography continues to affect the CD44+ population in MCF7 and HMEC by the same mechanism as when MCF7 was cultured alone, thus displaying a similar behavior.
Although the three different cell populations, MCF7, HMEC and IDC, showed different preferences in terms of the total cell adhesion on the 1-µm well, 1-µm gratings and 250-nm wells, respectively, the highest proportion of CD44+CD24−/lowESA+ was consistently captured on the 350-nm gratings. The observations supported our hypothesis that the different cell populations in the heterogeneous breast cancer tissue showed different preferential adhesion to various topographical patterns. In particular, the CD44+CD24−/lowESA+ CSC population showed a preferential retention and enrichment on 350-nm gratings, suggesting that this tumorigenic CSC population could be enriched by using nanoscaled gratings. However, it is uncertain whether the 350-nm grating induces the expression of CD44 or if it is able to isolate the cancer stem cell-like population in MCF7. This could be verified by studying whether the side population isolated by 350-nm gratings is indeed CSCs.
Cell adhesion is directly affected by cell-extracellular substrate interaction; however, the mechanism of the topographical regulation of cell adhesion remains unclear. One of the suggested mechanisms, known as “self-induced mechanotransduction” by Dalby
et al., is based on the hypothesis that surface topography alters nuclear morphology and chromosome position in adherent cells, leading to changes in gene transcription [
31,
32]. Another mechanism suggested by Ingber
et al. is the “direct mechanotransduction” mechanism, whereby forces encountered by cells during cell adhesion are directly transmitted to nucleus via cytoskeleton, and altered cytoskeletal tension then feeds back to induce local changes in focal adhesion assembly [
33,
34]. Our group has demonstrated that topography regulates cell morphology by modulating the physical force-sensing of the cells [
35,
36]. Moreover, the focal adhesion signaling and cytoskeletal contractility are essential for nanotopographical regulation in stem cell differentiation [
35,
37].
Given the short time points (4 and 24 h) used in this research, it is unlikely that cell behavior would be affected by gene transcription. Preliminary data from cell adhesion counting and immunostaining support the concept that nanotopography affects cell behavior via a direct mechanism. Moreover, the morphology difference between the attached MCF7, HMEC and primary cells might provide a mechanistic explanation of the preferential cell adhesion. MCF7, HMEC and IDC cells showed elongated morphology on gratings patterns (
Figure 2 and
Figure 5); however, MCF7 and HMECs showed more pronounced cell spreading and elongation on gratings. Immunofluorescence staining of vinculin (
Supplementary Figure S4) illustrated the focal adhesion of the MCF7 and HMECs on the patterns at 24 h. MCF7 on micro-sized patterns showed fewer focal adhesions, as compared to the nano-sized patterns, while HMECs showed well-established focal adhesion on all patterns. Further studies in cell morphology with SEM could be conducted to examine cell-topography interactions to provide a better understanding of the observed morphology differences. This will also illuminate the mechanism by which nanotopography influences cell adhesion. The cell morphology of the CD44
+CD24
−/lowESA
+ cell should be further analyzed to provide a better basis for comparison of the cancer stem cell preferential adhesion among the different topographies.
In conclusion, between the two time points of four and 24 h, 4 h appear to be the optimum time mark for isolating the CD44+CD24−/lowESA+ tumorigenic cell population from both the MCF7 and the primary cell cultures. In the MCF7 and IDC and MCF-7-HMEC co-culture cell experiments, the trend of preferential adhesion behavior of these CD44+CD24−/lowESA+ cells on the nanoscale features was observed. Among the topographies tested, the 350-nm gratings are consistently the most efficient in enriching the CD44+CD24−/lowESA+ or breast CSCs from the MCF7 and primary IDC cell populations. Moving forward, to develop a biomaterial substrate for effective cancer stem cell isolation, the isolated cells can be further characterized and their cancer stem cell identity further studied, for instance, utilizing the ALDH1 enzyme as a potential marker for CSCs. Scaling up of the isolation and expansion of the isolated cells can also be further investigated.
5. Materials and Methods
5.1. Fabrication of Micro- and Nano-Patterned Poly-L-Lactic Acid Films
A poly(dimethylsiloxane) (PDMS) soft mold was made from a micro- or nano-patterned template by soft lithography, as previously described [
38]. Briefly, the PDMS 10:1 elastomer and curing agent mixture was degassed, dispensed onto the masters and cured at 65 °C.
PLLA films were made by solvent casting using 3% w/v PLLA (Sigma Aldrich, St. Louis, MO, USA) dissolved in chloroform on the PDMS soft mold. The films were dried at room temperature covered with a glass dish. PLLA films were vacuum dried in a desiccator for 5 min to remove any residual solvent before demolding. The PLLA films were trimmed to 1.0-cm2 squares and sterilized with 70% ethanol (v/v) and ultraviolet irradiation.
To verify the surface morphology and fidelity of the micro- and nano-topography replication process, PLLA films were sputter coated with gold (JEOL JFC 1600 Fine Gold Coater, JEOL Ltd., Tokyo, Japan) and examined by scanning electron microscopy (SEM, Quanta FEG 200 and JEOL JSM-5600LV Scanning Microscope, JEOL Ltd., Tokyo, Japan).
5.2. Cell Culture of Mammary Epithelial Cells, MCF7 and Primary Invasive Ductal Carcinoma Cells
HMEC (Lonza, Basel, Switzerland) were maintained using mammary epithelial basal medium (MEBM, Lonza) supplemented with bovine pituitary extract (BPE), human epithelial growth factor (hEGF), hydrocortisone, GA-1000 (Gentamicin/Amphotericin) and insulin. The medium was changed on alternate days. Cells were subcultured by washing with HEPES-buffered saline solution (HEPES-BSS), dissociated with 0.25× trypsin-EDTA and neutralized with trypsin-neutralizing solution (TNS) (Lonza). The human breast cancer cell line MCF7 was obtained from American Type Culture Collection (ATCC) and maintained using Dulbecco’s Modified Eagle’s Medium (DMEM, Sigma Aldrich, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS), 0.01 mg/mL bovine insulin, 1% penicillin streptomycin and 1% non-essential amino acid solution. The medium was changed every three days. Invasive ductal carcinoma (IDC) cells (Passage 3–5) from breast tumor tissues of patient donors were obtained from the NUH-NUS (Singapore) Tissue Repository with NUS Institutional Review Board approval (08-090). They were maintained using the mammary epithelial growth medium (MEGM, Lonza) supplemented with 10% FBS, amphotericin B and gentamicin.
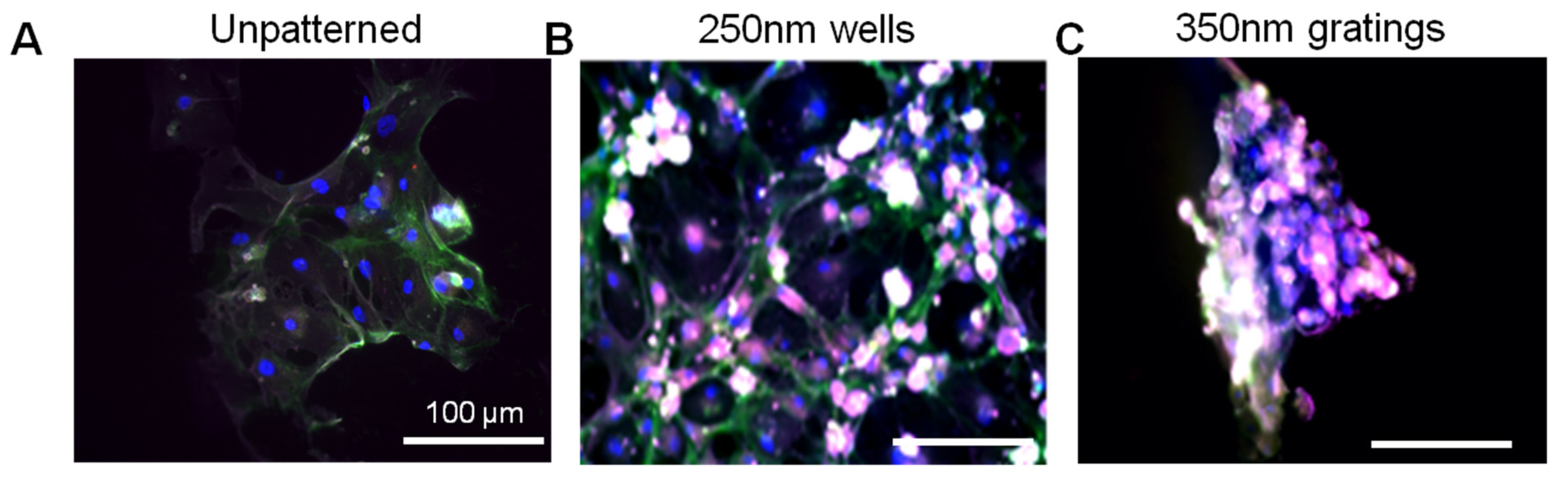
Cells were trypsinized upon reaching 80% confluence and seeded on PLLA films with a seeding density of 10,000 cells/cm2 (MCF7) or 16,000 cells/cm2 (HMEC). The substrates were then placed in an incubator for static culture. In the co-culture of MCF7 and HMEC, 16,000 cells/cm2 of each cell type (HMEC and MCF7) were seeded onto each of the samples and the control. The cell culture was maintained using MEGM. To differentiate both cell types in the co-culture system, HMEC was stained with CellTracker™ Red CMPTX (Invitrogen, Waltham, MA, USA) by incubating with 10 µM of the dye in serum-free medium (MEGM) for 30 min and subsequently replaced with MEGM for another 30 min.
5.3. Cell Adhesion Counting
The number of attached cells on nanotopographic substrates in monocultures was determined after 4 and 24 h. Unattached cells were washed twice with PBS or HBSS. Cells were then detached with trypsin from the PLLA films, and the cell number was determined by hemocytometer counting. Adhesion assays were performed with 5 replicates.
In another method, cell adhesion counting was performed after 4 h of culture. In order to identify the live cells, the attached cells were stained with the dye, CellTracker™ Red CMPTX (Invitrogen). After 3 h of culture, cells were labeled with CellTracker dye. At the 4-hour time point, the samples were gently washed to remove the unattached cells, then fixed with 4% formaldehyde solution and stained with DAPI. The samples were washed and mounted before imaging with a fluorescent microscope (Leica DC 300F, Leica, Wetzlar, Germany). The number of adhered cells was determined by counting the DAPI- and CellTracker-stained cells. For each sample, 5 random images were taken at 20× magnification. Cell adhesion counting was carried out for both HMEC and MCF7 mono-cultures.
5.4. Immunostaining of CD44, CD24 and Epithelial Surface Antigen Markers
After 4 or 24 h of culture, the cells were fixed with 4% formaldehyde and incubated in blocking buffer (1% w/v bovine serum albumin (BSA) and 10% goat serum in PBS). The cells were stained with antibodies specific for human cell surface markers: CD44-FITC (Abcam, Cambridge, UK), CD24-PE (Abcam) and ESA (Santa Cruz, Starr County, TX, USA) (
Table 1). After overnight incubation at 4 °C, the cells were washed and incubated with the corresponding secondary antibodies: Alexa Fluor 488, Alexa Fluor 546 and Alexa Fluor 647 (Invitrogen) for 1 h at room temperature. The cells were counter-stained with DAPI. For each sample, 5 random images were taken at 20× magnification. The number of cells that were of the CD44
+CD24
−ESA
+ phenotype was counted. The CD24
−/low immunophenotype was taken as CD24
− for cell counting.
Table 1.
Dilution ratio of the antibodies used in immunostaining.
Table 1.
Dilution ratio of the antibodies used in immunostaining.
| Antigen | Primary antibody | Secondary antibody |
|---|
| CD44 | Anti-CD44, rat, conjugated with FITC 1:500 | FITC, anti-rat 1:750 |
| CD24 | Anti-CD24, mouse, conjugated with PE 1:50 | Alexa Fluor 546, anti-mouse 1:750 |
| ESA | Anti-ESA, rabbit, unconjugated 1:50 | Alexa Fluor 647, anti-rabbit 1:750 |
5.5. Flow Cytometry Analysis
To analyze the expression of CD44, CD24 and ESA after 4 or 24 h of culture, flow cytometry was performed on the MCF7 mono-culture and IDC cell experiment. At the 4- or 24-hour time point, the cells attached on the films were trypsinized, centrifuged and counted. The harvested cells were incubated with 2% goat serum in staining buffer (1% goat serum, 0.5% BSA in PBS) for 10 min in an ice bath and stained with antibodies specific for human cell surface markers: CD44-FITC (Abcam), CD24-PE (Abcam) and ESA (Santa Cruz) (
Table 2). After incubation for 30 minutes at 4 °C, the suspensions were washed three times in staining buffer. Cells were then incubated with the secondary antibody, Alexa Fluor 647 for ESA, for 30 min at 4 °C. The cells were washed twice and fixed with 1% formaldehyde for 1 h before analyzing with the flow cytometer (Dako Cytomation Cyan LX, Glostrup, Denmark). The data were analyzed by FlowJo (version 10.0.7).
Table 2.
Dilution ratio of the antibodies used in flow cytometry.
Table 2.
Dilution ratio of the antibodies used in flow cytometry.
| Antigen | Primary antibody | Secondary antibody |
|---|
| CD44 | Rat anti-CD44 conjugated with FITC 1:50 | None |
| CD24 | Mouse anti-CD24 conjugated with PE 1:10 | None |
| ESA | Rabbit anti-ESA Un-conjugated 1:100 | Goat anti-rabbit conjugated with Alexa Fluor 647 1:500 |
5.6. Statistics Analysis
Data are presented as the mean ± SD unless specified otherwise. The statistical significance of the results obtained was assessed by one-way analysis of variance (ANOVA) and Tukey’s post hoc test or the Kruskal–Wallis test followed by Dunn’s post hoc test for flow cytometry analysis data. Results with P < 0.05 were considered statistically different.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}