The Role of the Reduced Laplacian Renormalization in the Kinetic Energy Functional Development



Abstract
1. Introduction
2. Theory
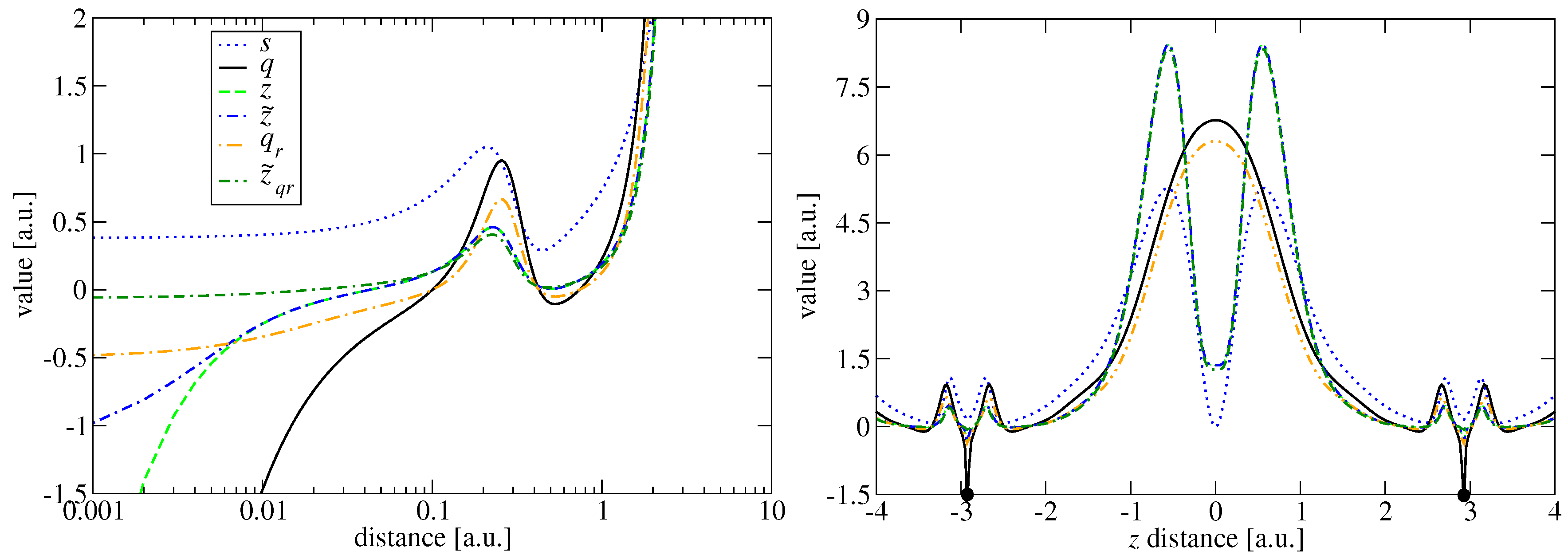
2.1. ModAPBEz
- (i)
- the value of must be of the order a.u.;
- (ii)
- the sum of the absolute differences between the calculated exact KE values () and the approximated ones () satisfies
2.2. ModAPBEq
- (i)
- when ,
- (ii)
- when ,
- (iii)
- in slowly varying region .
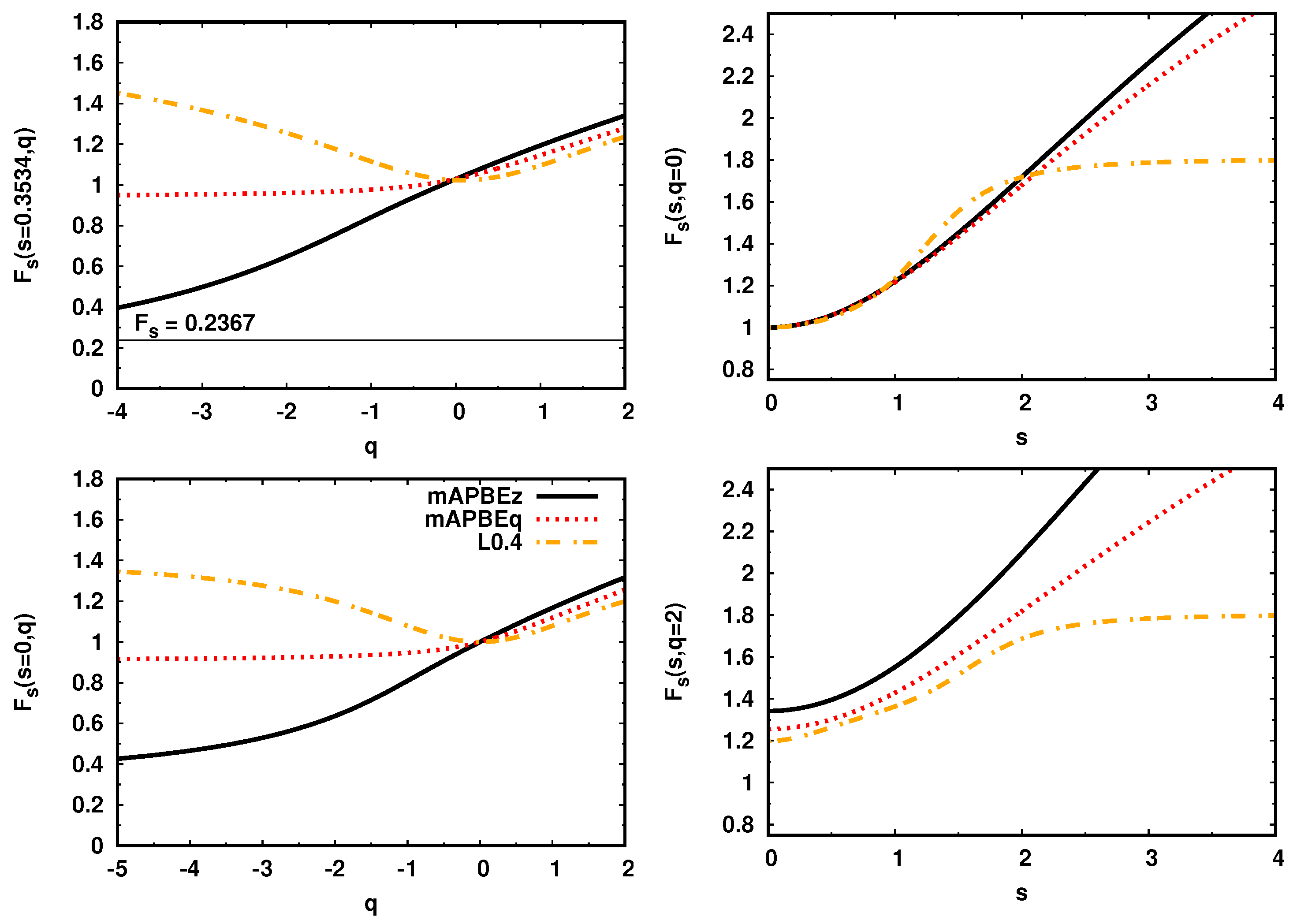
2.3. Comparison of KE Enhancement Factors
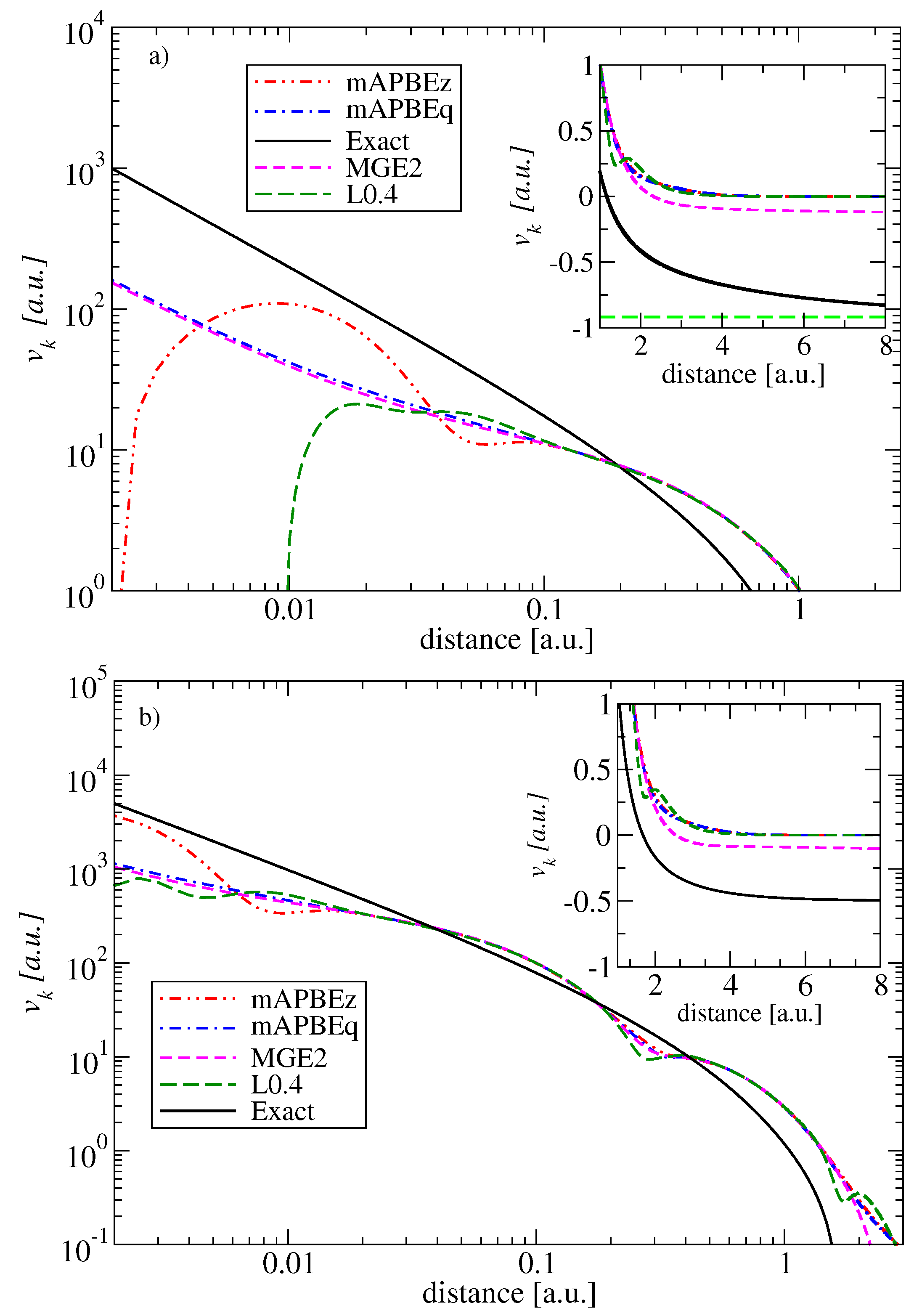
2.4. Comparison of KE Potentials
3. Computational Details
- Subsystem DFT calculations: We considered a partition of the total density into two fragments , where and are densities corresponding to subsystems A and B, respectively. The full relaxation of embedded ground-state electron densities was obtained using the freeze-and-thaw cycles [42,43] and considering convergence when the difference of dipole moments of the embedded subsystems is below a.u. In case of KS-DFT calculation, the maximum deviations in density matrix elements of a.u. were considered as convergence criteria. The benchmark set consists of five weakly interacting groups of molecular complexes used in our previous studies [21,45,46].We considered the embedding density error () and the total embedding energy error (). The first is defined aswhere N is the total number of electrons and is the deformation density calculated as the difference between the converged subsystem densities (, ) and the conventional ground state KS density (). In Equation (18), only the valence electron density was considered.
- Atoms: We considered the total KE of few small atoms (aKE test): H, N, C, O, F, Si, P, S, and Cl.
- Molecules: We considered the total KE (mKE test) and the atomization KE (atKE test), of the following small molecules: H, NH, CH, HO, FH, HCN, N, CH, HCO, HOOH, F, SIH, PH, PH, PH, SO, ClF, HCl, SH, Cl, OH, O.
- KE Ionization Potentials (IP) and Electron Affinities (EA): We considered the IP13 and EA13 tests [68], consisting of the following small atoms/molecules C, S, SH, Cl, Cl, OH, O, O, P, PH, PH, S, Si. The KE IP/KE EA has been calculated as the difference between KE of a neutral/changed system minus KE of ionized/neutral species. The errors have been calculated with respect to the values obtained from full KS-DFT calculations.
4. Results
4.1. Subsystem DFT Calculations
4.2. Results for Atoms and Molecules
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wesolowski, T.A.; Wang, Y.A. Recent Progress in Orbital-Free Density Functional Theory; World Scientific: Singapore, 2013. [Google Scholar]
- Wang, Y.A.; Carter, E.A. Orbital-Free Kinetic-Energy Density Functional Theory. In Theoretical Methods in Condensed Phase Chemistry; Springer: Dordrecht, The Netherlands, 2002; pp. 117–184. [Google Scholar] [CrossRef]
- Lignères, V.L.; Carter, E.A. An Introduction to Orbital-Free Density Functional Theory. In Handbook of Materials Modeling: Methods; Springer: Dordrecht, The Netherlands, 2005; pp. 137–148. [Google Scholar] [CrossRef]
- Jacob, C.R.; Neugebauer, J. Subsystem density-functional theory. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 325–362. [Google Scholar] [CrossRef]
- Wesolowski, T.A.; Shedge, S.; Zhou, X. Frozen-Density Embedding Strategy for Multilevel Simulations of Electronic Structure. Chem. Rev. 2015, 115, 5891–5928. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Harbola, M.K. Hydrodynamic approach to time-dependent density functional theory; Response properties of metal clusters. J. Chem. Phys. 2000, 113, 5614–5623. [Google Scholar] [CrossRef]
- Toscano, G.; Straubel, J.; Kwiatkowski, A.; Rockstuhl, C.; Evers, F.; Xu, H.; Mortensen, N.A.; Wubs, M. Resonance shifts and spill-out effects in self-consistent hydrodynamic nanoplasmonics. Nat. Commun. 2015, 6, 7132. [Google Scholar] [CrossRef]
- Ciracì, C.; Della Sala, F. Quantum hydrodynamic theory for plasmonics: Impact of the electron density tail. Phys. Rev. B 2016, 93, 205405. [Google Scholar] [CrossRef]
- Peverati, R.; Truhlar, D.G. Quest for a universal density functional: The accuracy of density functionals across a broad spectrum of databases in chemistry and physics. Philos. Trans. R. Soc. A 2014, 372, 20120476. [Google Scholar] [CrossRef]
- Scuseria, G.E.; Staroverov, V.N. Progress in the development of exchange-correlation functionals. In Theory and Applications of Computational Chemistry; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Constantin, L.A.; Fabiano, E.; Della Sala, F. Semilocal Pauli–Gaussian Kinetic Functionals for Orbital-Free Density Functional Theory Calculations of Solids. J. Phys. Chem. Lett. 2018, 9, 4385–4390. [Google Scholar] [CrossRef]
- Tran, F.; Wesolowski, T.A. Semilocal Approximations for the Kinetic Energy. In Recent Progress in Orbital-free Density Functional Theory; World Scientific: Singapore, 2013; pp. 429–442. [Google Scholar]
- Lembarki, A.; Chermette, H. Obtaining a gradient-corrected kinetic-energy functional from the Perdew-Wang exchange functional. Phys. Rev. A 1994, 50, 5328–5331. [Google Scholar] [CrossRef]
- Tran, F.; Wesołowski, T.A. Link between the kinetic- and exchange-energy functionals in the generalized gradient approximation. Int. J. Quantum Chem. 2002, 89, 441–446. [Google Scholar] [CrossRef]
- Laricchia, S.; Fabiano, E.; Constantin, L.A.; Della Sala, F. Generalized Gradient Approximations of the Noninteracting Kinetic Energy from the Semiclassical Atom Theory: Rationalization of the Accuracy of the Frozen Density Embedding Theory for Nonbonded Interactions. J. Chem. Theory Comput. 2011, 7, 2439–2451. [Google Scholar] [CrossRef]
- Constantin, L.A.; Fabiano, E.; Laricchia, S.; Della Sala, F. Semiclassical Neutral Atom as a Reference System in Density Functional Theory. Phys. Rev. Lett. 2011, 106, 186406. [Google Scholar] [CrossRef] [PubMed]
- Karasiev, V.V.; Chakraborty, D.; Shukruto, O.A.; Trickey, S. Nonempirical generalized gradient approximation free-energy functional for orbital-free simulations. Phys. Rev. B 2013, 88, 161108. [Google Scholar] [CrossRef]
- Borgoo, A.; Tozer, D.J. Density scaling of noninteracting kinetic energy functionals. J. Chem. Theory Comput. 2013, 9, 2250–2255. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Carter, E.A. Single-point kinetic energy density functionals: A pointwise kinetic energy density analysis and numerical convergence investigation. Phys. Rev. B 2015, 91, 045124. [Google Scholar] [CrossRef]
- Della Sala, F.; Fabiano, E.; Constantin, L.A. Kohn-Sham kinetic energy density in the nuclear and asymptotic regions: Deviations from the von Weizsäcker behavior and applications to density functionals. Phys. Rev. B 2015, 91, 035126. [Google Scholar] [CrossRef]
- Constantin, L.A.; Fabiano, E.; Śmiga, S.; Della Sala, F. Jellium-with-gap model applied to semilocal kinetic functionals. Phys. Rev. B 2017, 95, 115153. [Google Scholar] [CrossRef]
- Lehtomäki, J.; Lopez-Acevedo, O. Semilocal kinetic energy functionals with parameters from neutral atoms. Phys. Rev. B 2019, 100, 165111. [Google Scholar] [CrossRef]
- Perdew, J.P.; Constantin, L.A. Laplacian-level density functionals for the kinetic energy density and exchange-correlation energy. Phys. Rev. B 2007, 75, 155109. [Google Scholar] [CrossRef]
- Karasiev, V.V.; Jones, R.S.; Trickey, S.B.; Harris, F.E. Properties of constraint-based single-point approximate kinetic energy functionals. Phys. Rev. B 2009, 80, 245120. [Google Scholar] [CrossRef]
- Laricchia, S.; Constantin, L.A.; Fabiano, E.; Della Sala, F. Laplacian-Level Kinetic Energy Approximations Based on the Fourth-Order Gradient Expansion: Global Assessment and Application to the Subsystem Formulation of Density Functional Theory. J. Chem. Theory Comput. 2014, 10, 164–179. [Google Scholar] [CrossRef]
- Cancio, A.C.; Stewart, D.; Kuna, A. Visualization and analysis of the Kohn-Sham kinetic energy density and its orbital-free description in molecules. J. Chem. Phys. 2016, 144, 084107. [Google Scholar] [CrossRef]
- Constantin, L.A.; Fabiano, E.; Della Sala, F. Performance of Semilocal Kinetic Energy Functionals for Orbital-Free Density Functional Theory. J. Chem. Theory Comput. 2019, 15, 3044–3055. [Google Scholar] [CrossRef] [PubMed]
- Seino, J.; Kageyama, R.; Fujinami, M.; Ikabata, Y.; Nakai, H. Semi-local machine-learned kinetic energy density functional with third-order gradients of electrond ensity. J. Chem. Phys. 2018, 148, 241705. [Google Scholar] [CrossRef] [PubMed]
- Golub, P.; Manzhos, S. Kinetic energy densities based on the fourth order gradient expansion: Performance in different classes of materials and improvement via machine learning. Phys. Chem. Chem. Phys. 2019, 21, 378–395. [Google Scholar] [CrossRef] [PubMed]
- Constantin, L.A.; Fabiano, E.; Della Sala, F. Kinetic and Exchange Energy Densities near the Nucleus. Computation 2016, 4, 19. [Google Scholar] [CrossRef]
- Brack, M.; Jennings, B.K.; Chu, Y.H. On the extended Thomas–Fermi approximation to the kinetic energy density. Phys. Lett. B 1976, 65, 1–4. [Google Scholar] [CrossRef]
- Hodges, C.H. Quantum corrections to the Thomas–Fermi approximation: The Kirzhnits method. Can. J. Phys. 1973, 51, 1428. [Google Scholar] [CrossRef]
- Constantin, L.A.; Fabiano, E.; Della Sala, F. Modified fourth-order kinetic energy gradient expansion with hartree potential-dependent coefficients. J. Chem. Theory Comput. 2017, 13, 4228–4239. [Google Scholar] [CrossRef]
- Zhao, Q.; Levy, M.; Parr, R.G. Applications of coordinate-scaling procedures to the exchange-correlation energy. Phys. Rev. A 1993, 47, 918–922. [Google Scholar] [CrossRef]
- Jemmer, P.; Knowles, P.J. Exchange energy in Kohn-Sham density-functional theory. Phys. Rev. A 1995, 51, 3571–3575. [Google Scholar] [CrossRef]
- Filatov, M.; Thiel, W. Exchange-correlation density functional beyond the gradient approximation. Phys. Rev. A 1998, 57, 189. [Google Scholar] [CrossRef]
- Springborg, M.; Dahl, J.P. On exact and approximate exchange-energy densities. J. Chem. Phys. 1999, 110, 9360–9370. [Google Scholar] [CrossRef][Green Version]
- Cancio, A.C.; Chou, M. Beyond the local approximation to exchange and correlation: The role of the Laplacian of the density in the energy density of Si. Phys. Rev. B 2006, 74, 081202. [Google Scholar] [CrossRef]
- Cancio, A.C.; Wagner, C.E.; Wood, S.A. Laplacian-based models for the exchange energy. Int. J. Quantum Chem. 2012, 112, 3796–3806. [Google Scholar] [CrossRef]
- Lee, H.; Lee, C.; Parr, R.G. Conjoint gradient correction to the Hartree-Fock kinetic- and exchange-energy density functionals. Phys. Rev. A 1991, 44, 768–771. [Google Scholar] [CrossRef] [PubMed]
- March, N.H. Electron density theory of atoms and molecules. J. Phys. Chem. 1982, 86, 2262–2267. [Google Scholar] [CrossRef]
- Wesolowski, T.A. Chemistry: Reviews of Current Trends; Leszczynski, J., Ed.; World Scientific: Singapore, 2006; Volume 10, pp. 1–82. [Google Scholar]
- Wesolowski, T.A.; Weber, J. Kohn-Sham equations with constrained electron density: An iterative evaluation of the ground-state electron density of interacting molecules. Chem. Phys. Lett. 1996, 248, 71–76. [Google Scholar] [CrossRef]
- Götz, A.W.; Beyhan, S.M.; Visscher, L. Performance of Kinetic Energy Functionals for Interaction Energies in a Subsystem Formulation of Density Functional Theory. J. Chem. Theory Comput. 2009, 5, 3161–3174. [Google Scholar] [CrossRef] [PubMed]
- Śmiga, S.; Fabiano, E.; Constantin, L.A.; Della Sala, F. Laplacian-dependent models of the kinetic energy density: Applications in subsystem density functional theory with meta-generalized gradient approximation functionals. J. Chem. Phys. 2017, 146, 064105. [Google Scholar] [CrossRef]
- Śmiga, S.; Fabiano, E.; Laricchia, S.; Constantin, L.A.; Della Sala, F. Subsystem density functional theory with meta-generalized gradient approximation exchange-correlation functionals. J. Chem. Phys. 2015, 142, 154121. [Google Scholar] [CrossRef]
- Mejia-Rodriguez, D.; Trickey, S. Deorbitalization strategies for meta-generalized-gradient-approximation exchange-correlation functionals. Phys. Rev. A 2017, 96, 052512. [Google Scholar] [CrossRef]
- Thomas, L.H. The calculation of atomic fields. Proc. Camb. Philos. Soc. 1926, 23, 542–548. [Google Scholar] [CrossRef]
- Fermi, E. Un metodo statistico per la determinazione di alcune priorieta dell’atome. Rend. Accad. Naz. Lincei 1927, 6, 602–607. [Google Scholar]
- Fermi, E. Eine statistische Methode zur Bestimmung einiger Eigenschaften des Atoms und ihre Anwendung auf die Theorie des periodischen Systems der Elemente. Zeitschrift für Physik 1928, 48, 73–79. [Google Scholar] [CrossRef]
- Della Sala, F.; Fabiano, E.; Constantin, L.A. Kinetic-energy-density dependent semilocal exchange-correlation functionals. Int. J. Quantum Chem. 2016, 116, 1641–1694. [Google Scholar] [CrossRef]
- Fabiano, E.; Constantin, L.A. Relevance of coordinate and particle-number scaling in density-functional theory. Phys. Rev. A 2013, 87, 012511. [Google Scholar] [CrossRef]
- Staroverov, V.N.; Scuseria, G.E.; Tao, J.; Perdew, J.P. Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes. J. Chem. Phys. 2003, 119, 12129–12137. [Google Scholar] [CrossRef]
- Buksztel, A.; Śmiga, S.; Grabowski, I. The Correlation Effects in Density Functional Theory Along the Dissociation Path. In Electron Correlation in Molecules—ab initio Beyond Gaussian Quantum Chemistry; Hoggan, P.E., Ozdogan, T., Eds.; Academic Press: Cambridge, MA, USA, 2016. [Google Scholar] [CrossRef]
- Becke, A.D. Hartree–Fock exchange energy of an inhomogeneous electron gas. Int. J. Quantum Chem. 1983, 23, 1915–1922. [Google Scholar] [CrossRef]
- Tao, J. Exchange energy density of an atom as a functional of the electron density. J. Chem. Phys. 2001, 115, 3519–3530. [Google Scholar] [CrossRef]
- Cancio, A.C.; Wagner, C.E. Laplacian-based generalized gradient approximations for the exchange energy. ArXiv e-prints 2013, arXiv:1308.3744. [Google Scholar]
- Clementi, E.; Roetti, C. Roothaan-Hartree-Fock atomic wavefunctions: Basis functions and their coefficients for ground and certain excited states of neutral and ionized atoms, Z≤54. Atomic Data Nucl. Data Tables 1974, 14, 177–478. [Google Scholar] [CrossRef]
- Ribeiro, R.F.; Burke, K. Leading corrections to local approximations. II. The case with turning points. Phys. Rev. B 2017, 95, 115115. [Google Scholar] [CrossRef]
- Constantin, L.A.; Snyder, J.C.; Perdew, J.P.; Burke, K. Communication: Ionization potentials in the limit of large atomic number. J. Chem. Phys. 2011, 133, 241103. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. A Bond Path: A Universal Indicator of Bonded Interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Essén, H. The characterization of atomic interactions. J. Chem. Phys. 1984, 80, 1943–1960. [Google Scholar] [CrossRef]
- von Weizsäcker, C.F. Zur Theorie der Kernmassen. Z. Phys. A 1935, 96, 431–458. [Google Scholar] [CrossRef]
- Siecinńska, S.; Fabiano, E.; Śmiga, S. About the methods to generate the reference total and Pauli kinetic energy potentials. Unpublished work.
- Laricchia, S.; Fabiano, E.; Della Sala, F. Frozen density embedding with hybrid functionals. J. Chem. Phys. 2010, 133, 164111. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Rappoport, D.; Furche, F. Property-optimized gaussian basis sets for molecular response calculations. J. Chem. Phys. 2010, 133, 134105. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Weigend, F.; Furche, F.; Ahlrichs, R. Gaussian basis sets of quadruple zeta valence quality for atoms H–Kr. J. Chem. Phys. 2003, 119, 12753–12762. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- TURBOMOLE V6.2, 2009, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007. Available online: http://www.turbomole.com (accessed on 10 October 2019).




{kind=link}
{kind=link}
{kind=link}
| GGAs | metaGGAs | |||||
|---|---|---|---|---|---|---|
| System | revAPBE | LGAP | L0.4 | mAPBEq | mAPBEz | |
| Weak interactions (WI) | ||||||
| He-Ne | 0.05 | 0.09 | 0.22 | 0.06 | 0.08 | |
| He-Ar | 0.06 | 0.07 | 0.22 | 0.10 | 0.12 | |
| Ne-Ne | 0.04 | 0.06 | 0.25 | 0.04 | 0.08 | |
| Ne-Ar | 0.06 | 0.07 | 0.28 | 0.11 | 0.13 | |
| CH-Ne | 0.07 | 0.08 | 0.34 | 0.11 | 0.14 | |
| CH-Ne | 0.13 | 0.15 | 0.28 | 0.19 | 0.22 | |
| CH-CH | 0.60 | 0.71 | 0.37 | 0.81 | 0.86 | |
| MAE | 0.14 | 0.18 | 0.28 | 0.20 | 0.23 | |
| Dipole interactions (DI) | ||||||
| HS-HS | 1.85 | 1.97 | 2.62 | 2.04 | 2.03 | |
| HCl-HCl | 1.87 | 1.89 | 2.22 | 1.94 | 1.97 | |
| HS-HCl | 3.70 | 3.70 | 3.59 | 3.71 | 3.79 | |
| CHCl-Hcl | 2.38 | 2.44 | 2.54 | 2.44 | 2.47 | |
| CHSH-HCN | 1.72 | 1.86 | 2.47 | 1.87 | 1.83 | |
| CHSH-Hcl | 4.08 | 4.11 | 3.81 | 4.10 | 4.18 | |
| MAE | 2.60 | 2.66 | 2.88 | 2.68 | 2.71 | |
| Hydrogen bonds (HB) | ||||||
| NH-NH | 1.79 | 1.97 | 2.68 | 2.05 | 2.02 | |
| HF-HF | 1.53 | 1.51 | 1.76 | 1.54 | 1.56 | |
| HO-HO | 2.01 | 2.08 | 2.53 | 2.14 | 2.14 | |
| NH-HO | 3.11 | 3.22 | 3.58 | 3.26 | 3.26 | |
| HF-HCN | 2.77 | 2.84 | 2.96 | 2.86 | 2.88 | |
| (HCONH) | 2.72 | 2.94 | 3.58 | 2.93 | 2.87 | |
| (HCOOH) | 3.35 | 3.48 | 3.73 | 3.48 | 3.47 | |
| MAE | 2.47 | 2.58 | 2.97 | 2.61 | 2.60 | |
| Dihydrogen bonds (DHB) | ||||||
| AlH-HCl | 5.81 | 5.79 | 5.86 | 5.75 | 5.87 | |
| AlH-HF | 3.82 | 3.80 | 3.29 | 3.74 | 3.83 | |
| LiH-HCl | 14.72 | 14.90 | 16.35 | 14.80 | 14.80 | |
| LiH-HF | 7.58 | 7.68 | 7.40 | 7.58 | 7.59 | |
| MgH-HCl | 4.61 | 4.66 | 4.53 | 4.63 | 4.67 | |
| MgH-HF | 3.21 | 3.23 | 2.97 | 3.18 | 3.23 | |
| BeH-HCl | 3.92 | 3.97 | 3.90 | 3.98 | 4.02 | |
| BeH-HF | 3.42 | 3.40 | 3.13 | 3.36 | 3.44 | |
| MAE | 5.89 | 5.93 | 5.93 | 5.88 | 5.93 | |
| Charge transfer (CT) | ||||||
| NF-HCN | 0.29 | 0.35 | 0.15 | 0.39 | 0.41 | |
| CH-F | 6.35 | 6.32 | 6.43 | 6.33 | 6.34 | |
| NF-HNC | 0.58 | 0.61 | 0.83 | 0.64 | 0.65 | |
| CH-Cl | 5.77 | 5.49 | nc | 5.42 | 5.76 | |
| NH-F | 9.60 | 9.53 | 9.44 | 9.55 | 9.57 | |
| NF-ClF | 1.73 | 1.62 | 1.17 | 1.60 | 1.68 | |
| NF-HF | 0.95 | 0.94 | 0.95 | 0.94 | 0.96 | |
| CH-ClF | 6.02 | 5.66 | nc | 5.56 | 5.97 | |
| HCN-ClF | 3.21 | 3.09 | 3.02 | 3.02 | 3.12 | |
| NH-Cl | 7.60 | 7.23 | nc | 7.17 | 7.60 | |
| HO-ClF | 5.06 | 4.79 | nc | 4.74 | 5.03 | |
| NH-ClF | 11.19 | 13.61 | nc | 15.78 | 12.72 | |
| MAE | 4.86 | 4.94 | - | 5.10 | 4.98 | |
| rwMAE | 0.91 | 0.97 | 1.141 | 0.99 | 1.02 | |
| GGAs | metaGGAs | |||||
|---|---|---|---|---|---|---|
| System | revAPBE | LGAP | L0.4 | mAPBEq | mAPBEz | |
| Weak interactions (WI) | ||||||
| He-Ne | 0.08 | 0.15 | 0.23 | 0.13 | 0.11 | |
| He-Ar | 0.05 | 0.13 | 0.23 | 0.10 | 0.08 | |
| Ne-Ne | 0.14 | 0.26 | 0.46 | 0.21 | 0.17 | |
| Ne-Ar | 0.11 | 0.24 | 0.52 | 0.20 | 0.15 | |
| CH-Ne | 0.12 | 0.26 | 0.57 | 0.24 | 0.19 | |
| CH-Ne | −0.03 | 0.20 | 1.45 | 0.30 | 0.17 | |
| CH-CH | −0.38 | −0.24 | 0.96 | −0.15 | −0.27 | |
| MAE | 0.13 | 0.21 | 0.63 | 0.19 | 0.16 | |
| Dipole interactions (DI) | ||||||
| HS-HS | −0.47 | −0.61 | −0.57 | −0.60 | −0.61 | |
| HCl-HCl | 0.07 | −0.10 | 0.06 | −0.07 | −0.09 | |
| HS-HCl | 0.40 | 0.08 | −0.70 | 0.06 | 0.17 | |
| CHCl-HCl | 0.02 | −0.35 | 0.10 | −0.13 | −0.12 | |
| CHSH-HCN | −1.18 | −1.35 | −0.92 | −1.23 | −1.24 | |
| CHSH-HCl | 0.73 | 0.23 | −0.47 | 0.35 | 0.55 | |
| MAE | 0.48 | 0.45 | 0.47 | 0.41 | 0.46 | |
| Hydrogen bonds (HB) | ||||||
| NH-NH | −0.95 | −1.14 | −1.01 | −1.10 | −1.13 | |
| HF-HF | 0.79 | 0.53 | 0.64 | 0.55 | 0.49 | |
| HO-HO | −0.20 | −0.52 | −0.81 | −0.51 | −0.51 | |
| NH-HO | −0.44 | −0.86 | −1.87 | −0.90 | −0.79 | |
| HF-HCN | 0.43 | −0.05 | −1.43 | −0.16 | −0.04 | |
| (HCONH) | −4.21 | −5.37 | −7.43 | −5.16 | −4.89 | |
| (HCOOH) | −1.88 | −3.33 | −7.08 | −3.29 | −2.73 | |
| MAE | 1.27 | 1.69 | 2.90 | 1.67 | 1.51 | |
| Dihydrogen bonds (DHB) | ||||||
| AlH-HCl | 2.54 | 2.01 | −1.18 | 1.71 | 2.06 | |
| AlH-HF | 3.99 | 3.54 | 1.30 | 3.32 | 3.59 | |
| LiH-HCl | 4.63 | 3.83 | −2.01 | 3.14 | 4.05 | |
| LiH-HF | 5.08 | 4.44 | 0.86 | 4.06 | 4.67 | |
| MgH-HCl | 1.79 | 1.29 | −1.36 | 1.04 | 1.34 | |
| MgH-HF | 3.65 | 3.19 | 0.92 | 2.97 | 3.26 | |
| BeH-HCl | 0.70 | 0.42 | −0.73 | 0.30 | 0.37 | |
| BeH-HF | 2.24 | 1.94 | 0.76 | 1.82 | 1.9 | |
| MAE | 3.08 | 2.58 | 1.14 | 2.30 | 2.66 | |
| Charge transfer (CT) | ||||||
| NF-HCN | −0.41 | −0.27 | 1.28 | −0.16 | −0.30 | |
| CH-F | 4.27 | 4.40 | 6.06 | 4.54 | 4.41 | |
| NF-HNC | −0.13 | −0.22 | −0.15 | −0.28 | −0.33 | |
| CH-Cl | 1.52 | 0.71 | nc | 0.94 | 1.64 | |
| NH-F | 6.90 | 6.90 | 8.60 | 7.08 | 6.96 | |
| NF-ClF | 2.14 | 1.85 | 2.21 | 2.08 | 2.12 | |
| NF-HF | 0.91 | 0.69 | 0.35 | 0.63 | 0.62 | |
| CH-ClF | 3.71 | 2.92 | nc | 3.17 | 3.84 | |
| HCN-ClF | 1.62 | 0.97 | −0.10 | 1.21 | 1.53 | |
| NH-Cl | 2.84 | 1.97 | nc | 1.94 | 2.81 | |
| HO-ClF | 2.42 | 1.67 | nc | 1.91 | 2.36 | |
| NH-ClF | 4.44 | 0.86 | nc | 0.63 | 0.87 | |
| MAE | 2.61 | 1.95 | - | 2.05 | 2.32 | |
| rwMAE | 0.94 | 0.94 | 1.37 1 | 0.90 | 0.92 | |
| aKE | mKE | atKE | IP13-KE | EA13-KE | |
|---|---|---|---|---|---|
| revAPBEk | 0.83% | 0.54% | 0.156 | 0.101 | 0.060 |
| APBEk | 0.40% | 0.33% | 0.141 | 0.101 | 0.061 |
| GE4 | 1.60% | 0.98% | 0.264 | 0.104 | 0.064 |
| L0.4 | 1.32% | 0.91% | 0.188 | 0.100 | 0.062 |
| mAPBEz | 1.02% | 0.74% | 0.154 | 0.101 | 0.060 |
| mAPBEq | 1.61% | 1.21% | 0.162 | 0.100 | 0.059 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Śmiga, S.; Constantin, L.A.; Della Sala, F.; Fabiano, E. The Role of the Reduced Laplacian Renormalization in the Kinetic Energy Functional Development. Computation 2019, 7, 65. https://doi.org/10.3390/computation7040065
Śmiga S, Constantin LA, Della Sala F, Fabiano E. The Role of the Reduced Laplacian Renormalization in the Kinetic Energy Functional Development. Computation. 2019; 7(4):65. https://doi.org/10.3390/computation7040065
Chicago/Turabian StyleŚmiga, Szymon, Lucian A. Constantin, Fabio Della Sala, and Eduardo Fabiano. 2019. "The Role of the Reduced Laplacian Renormalization in the Kinetic Energy Functional Development" Computation 7, no. 4: 65. https://doi.org/10.3390/computation7040065
APA StyleŚmiga, S., Constantin, L. A., Della Sala, F., & Fabiano, E. (2019). The Role of the Reduced Laplacian Renormalization in the Kinetic Energy Functional Development. Computation, 7(4), 65. https://doi.org/10.3390/computation7040065