Reduction Potential Predictions for Some 3-Aryl-Quinoxaline-2-Carbonitrile 1,4-Di-N-Oxide Derivatives with Known Anti-Tumor Properties

 and
and
Abstract
1. Introduction
2. Materials and Methods
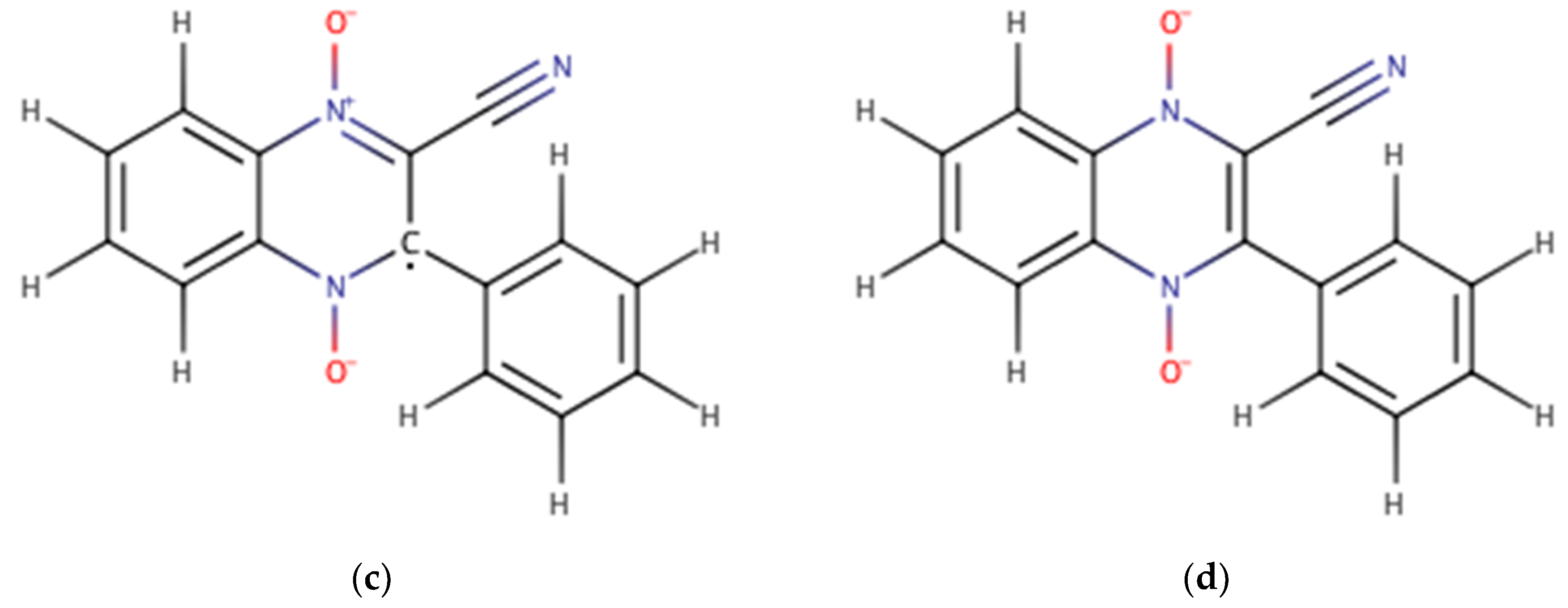
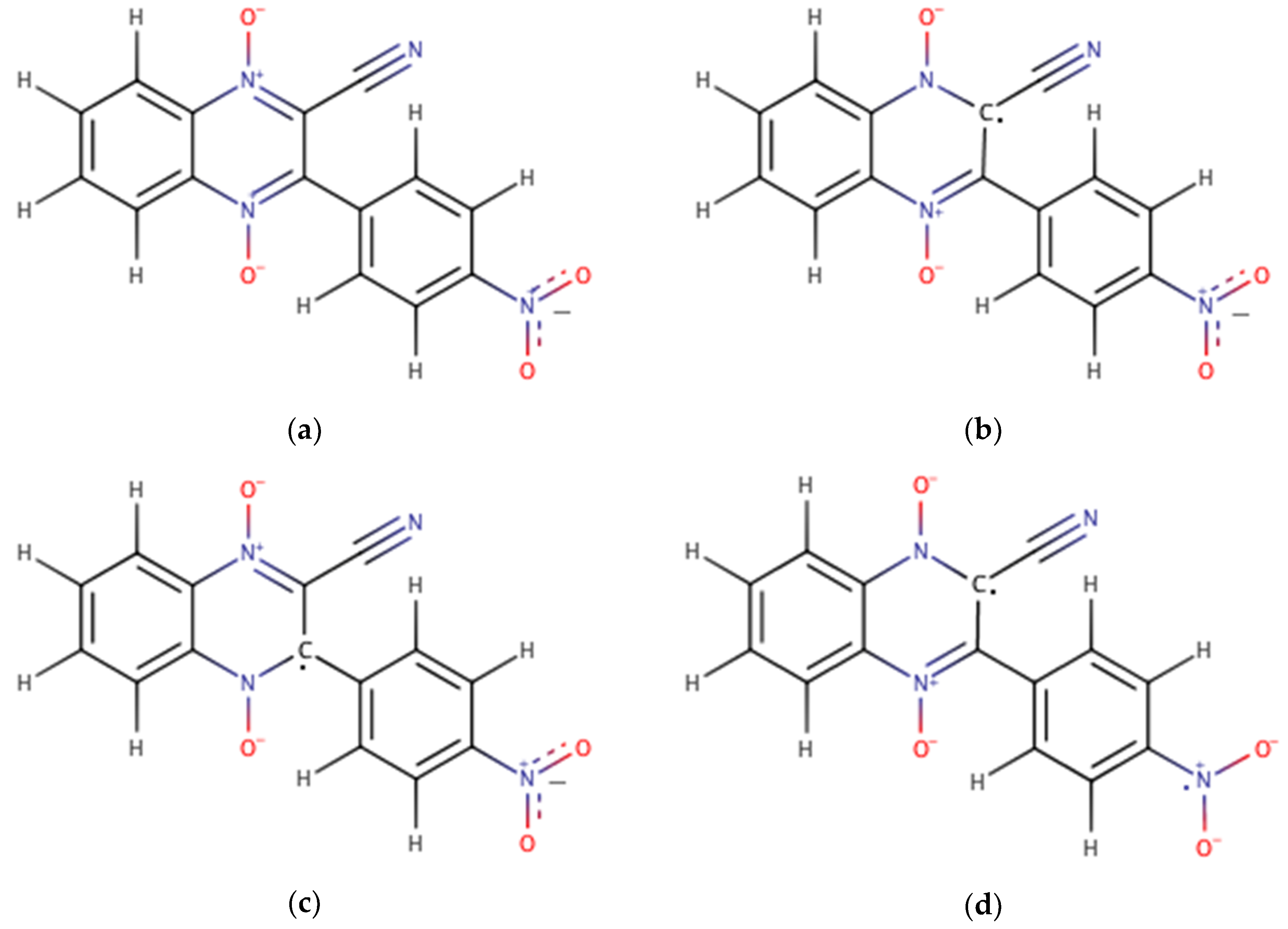
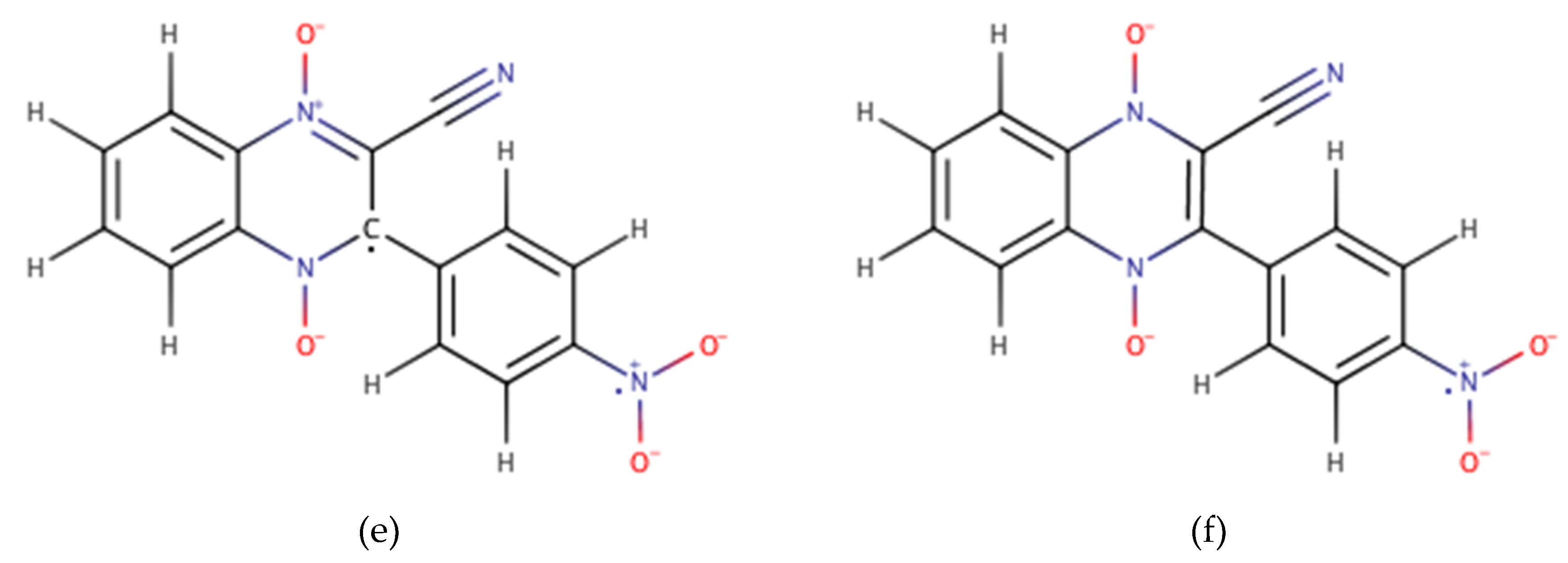
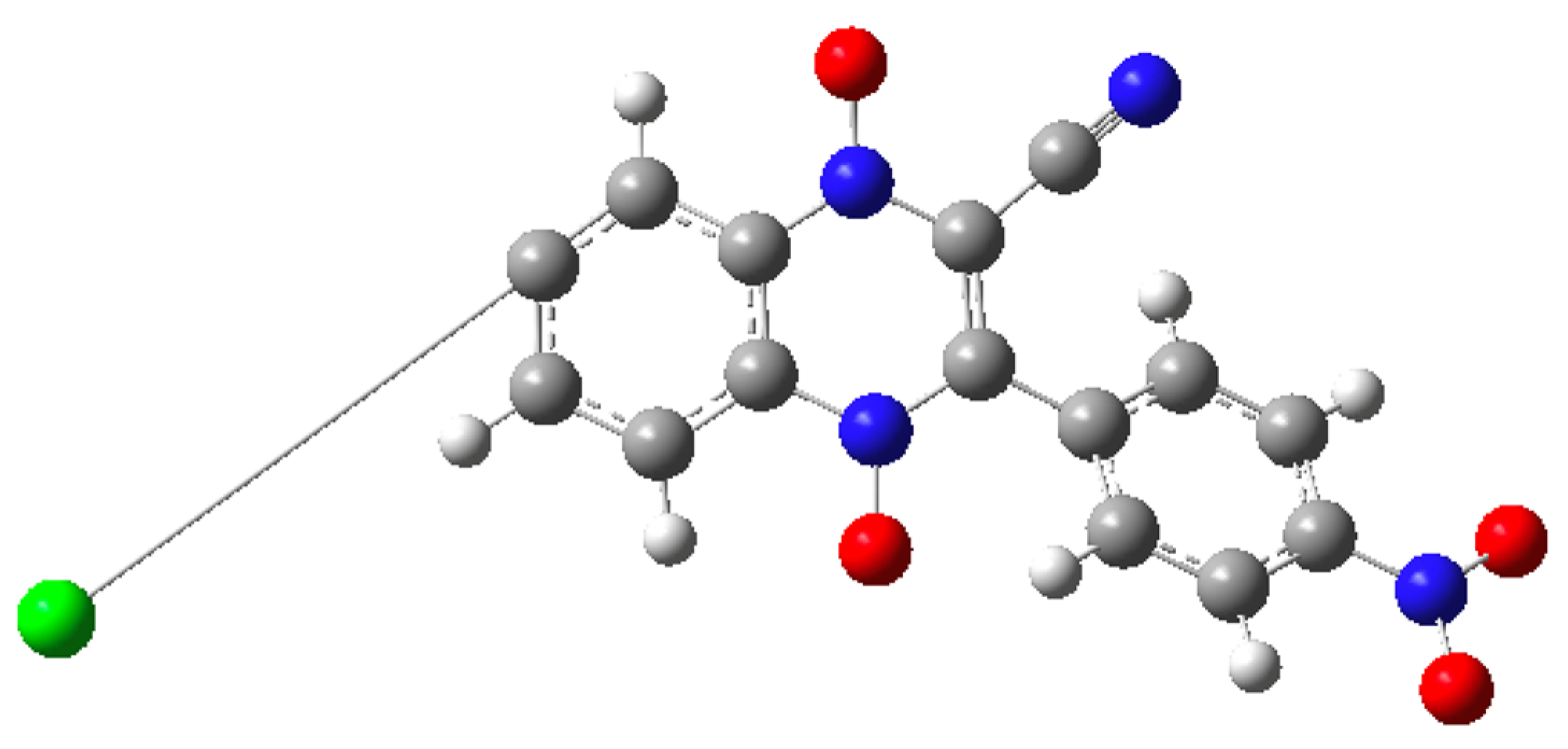
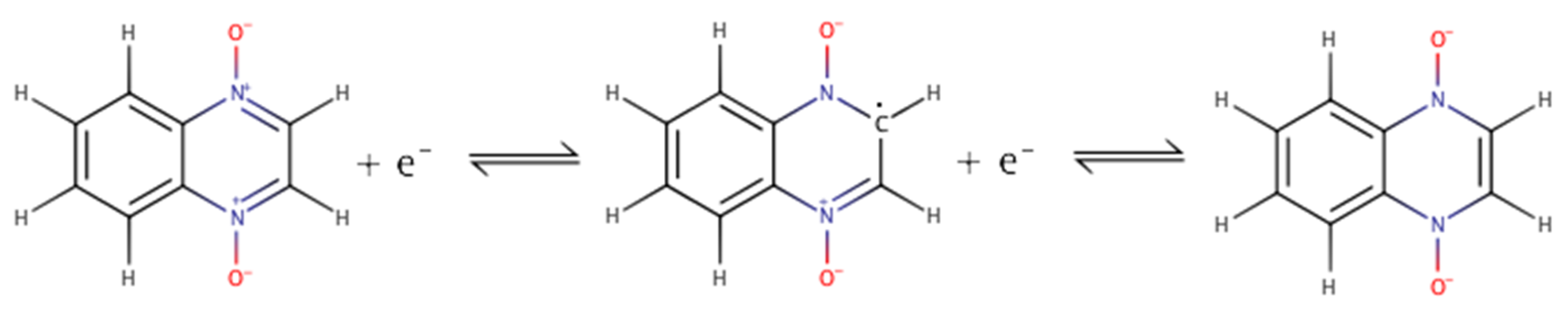
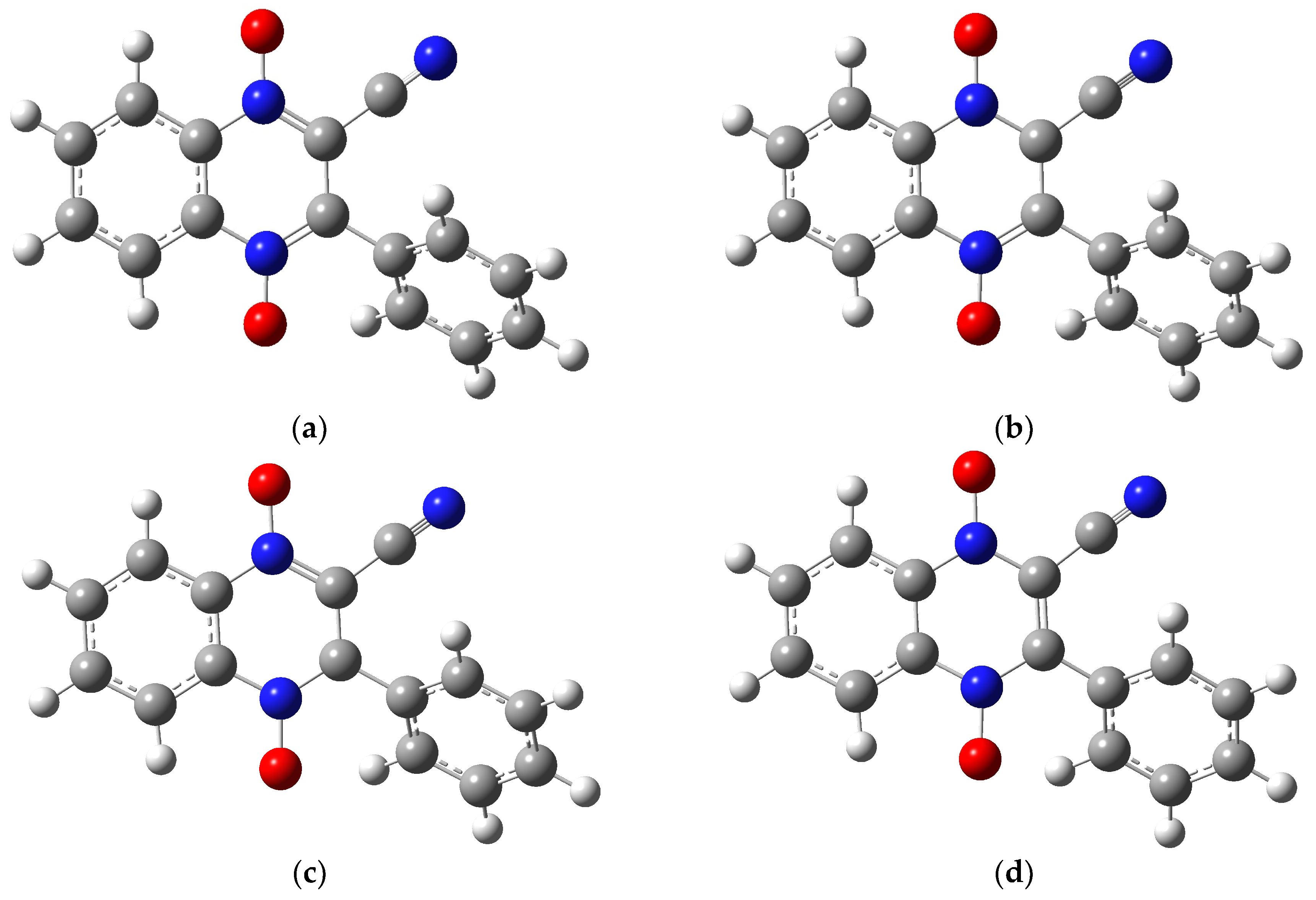
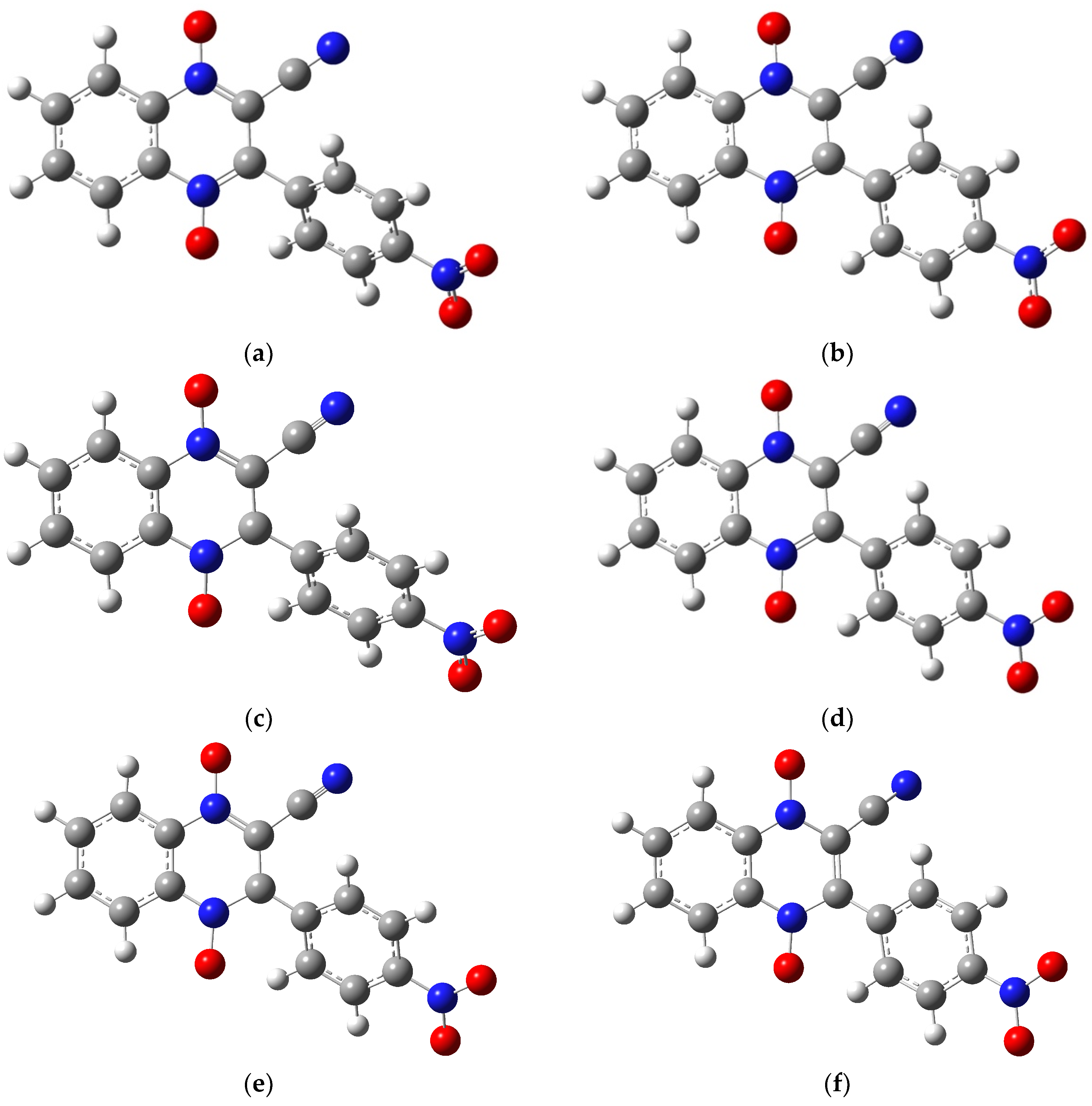
2.1. Building the Derivatives
2.2. DFT Calculations
2.3. Ferrocene/Ferrocenium Reference
3. Results and Discussion
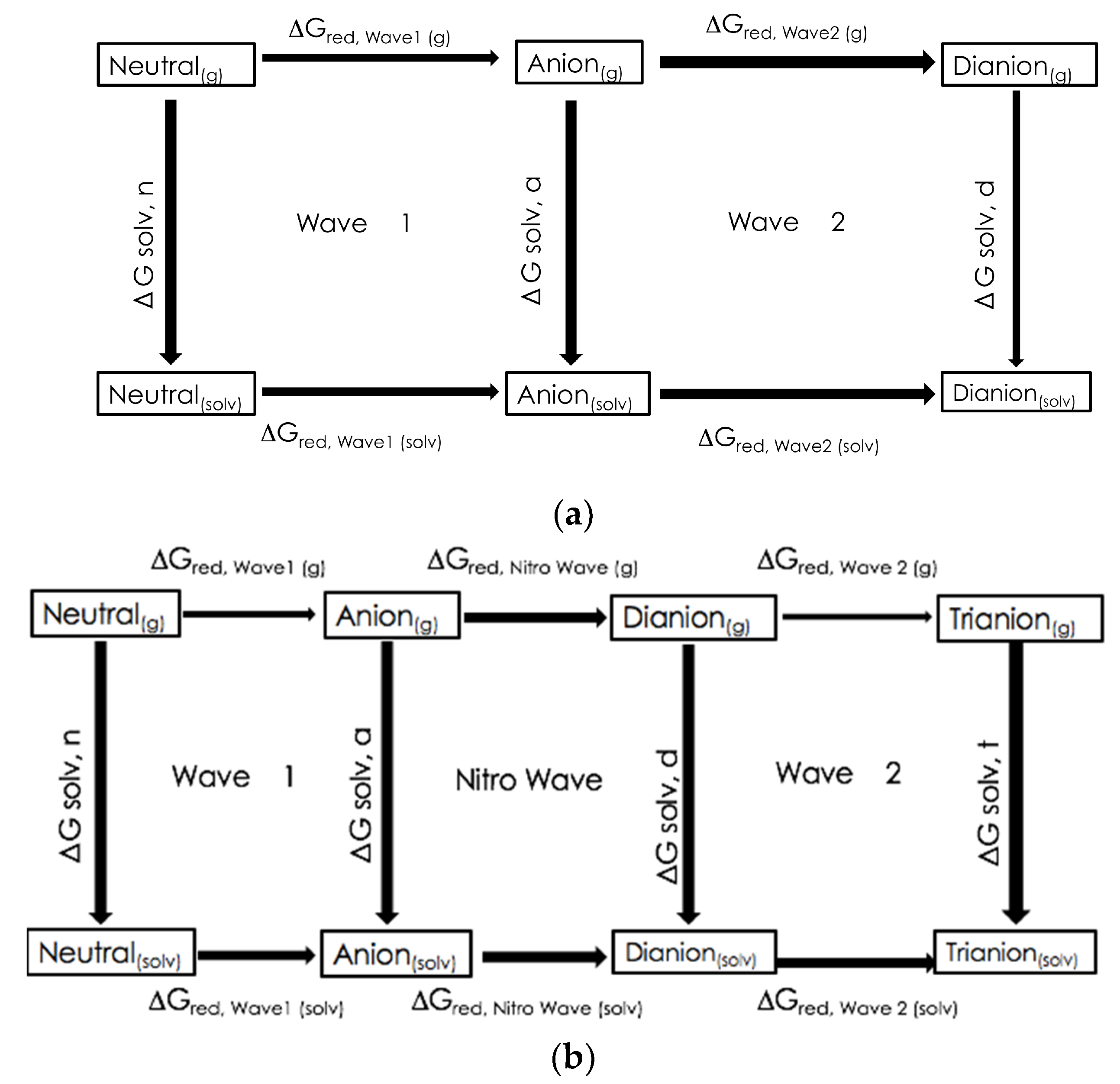
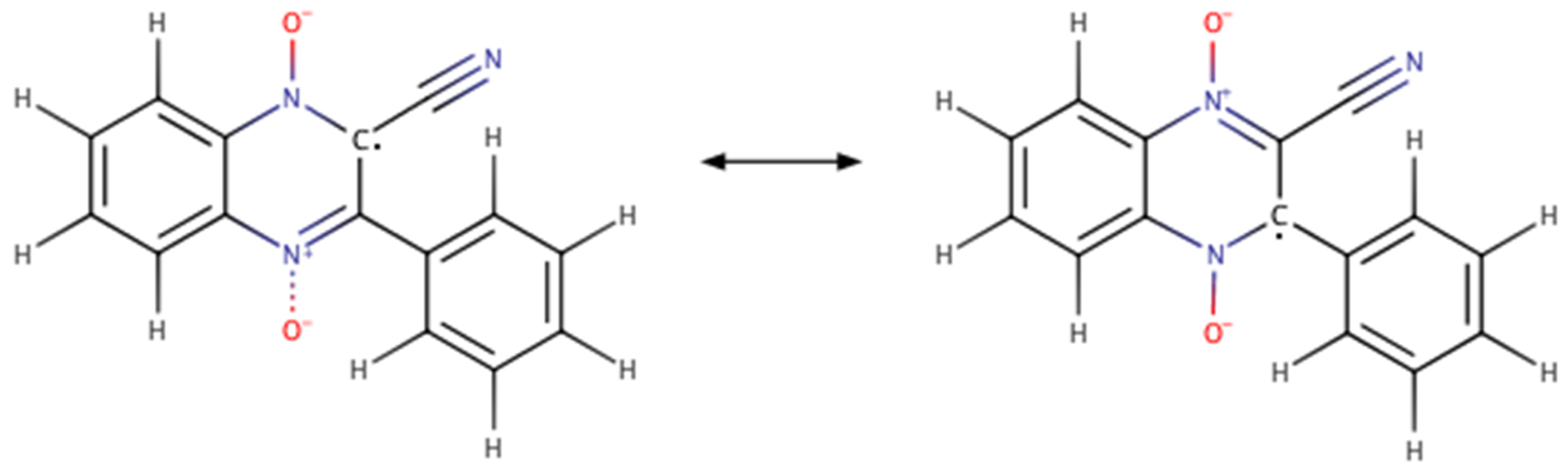
3.1. Computationally Predicted Raw Chemical Potentials
3.2. Computationally Predicted Electrochemical Cell Reaction Potentials
3.3. Comparison to Experimental Electrochemical Data
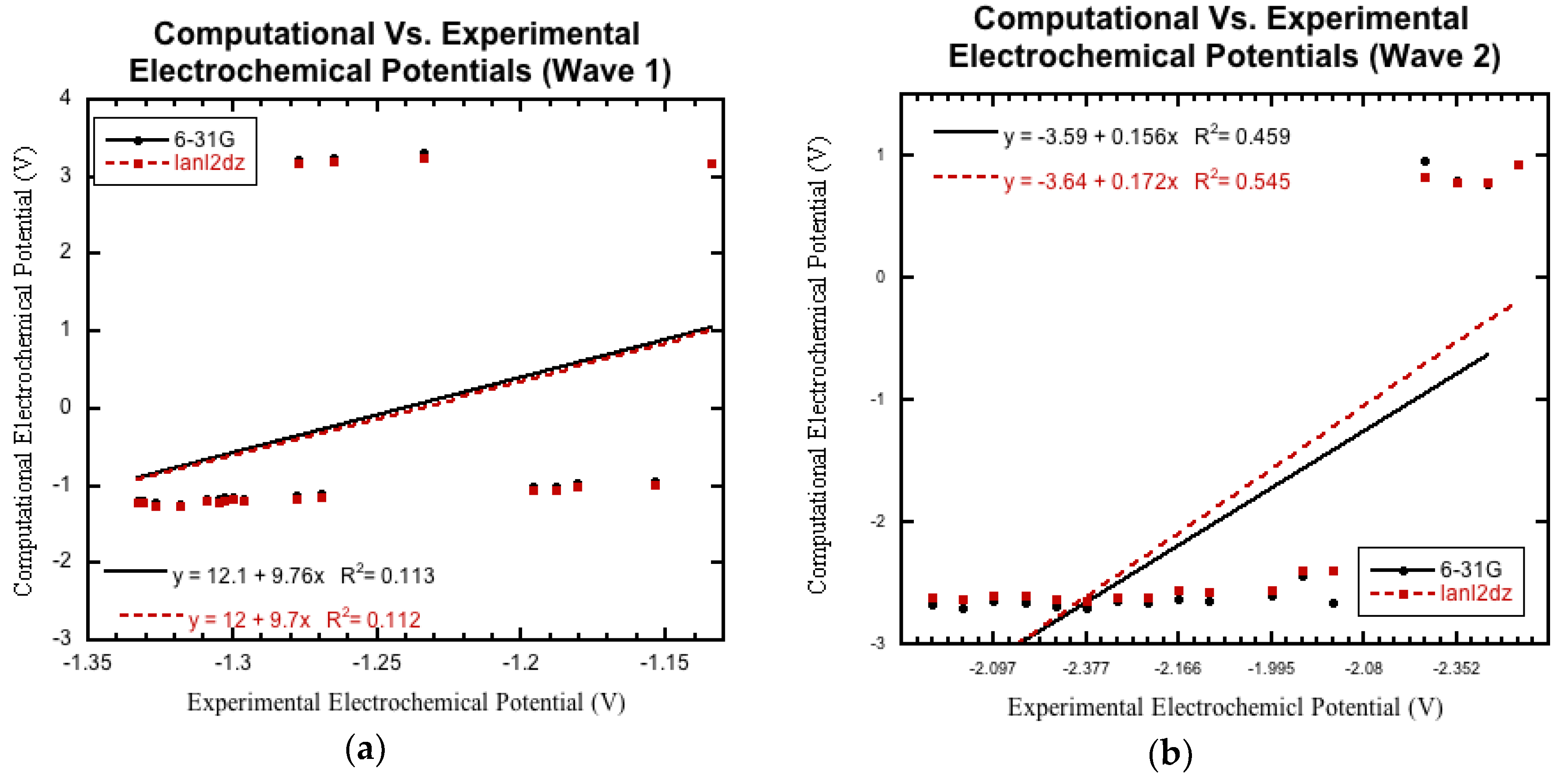
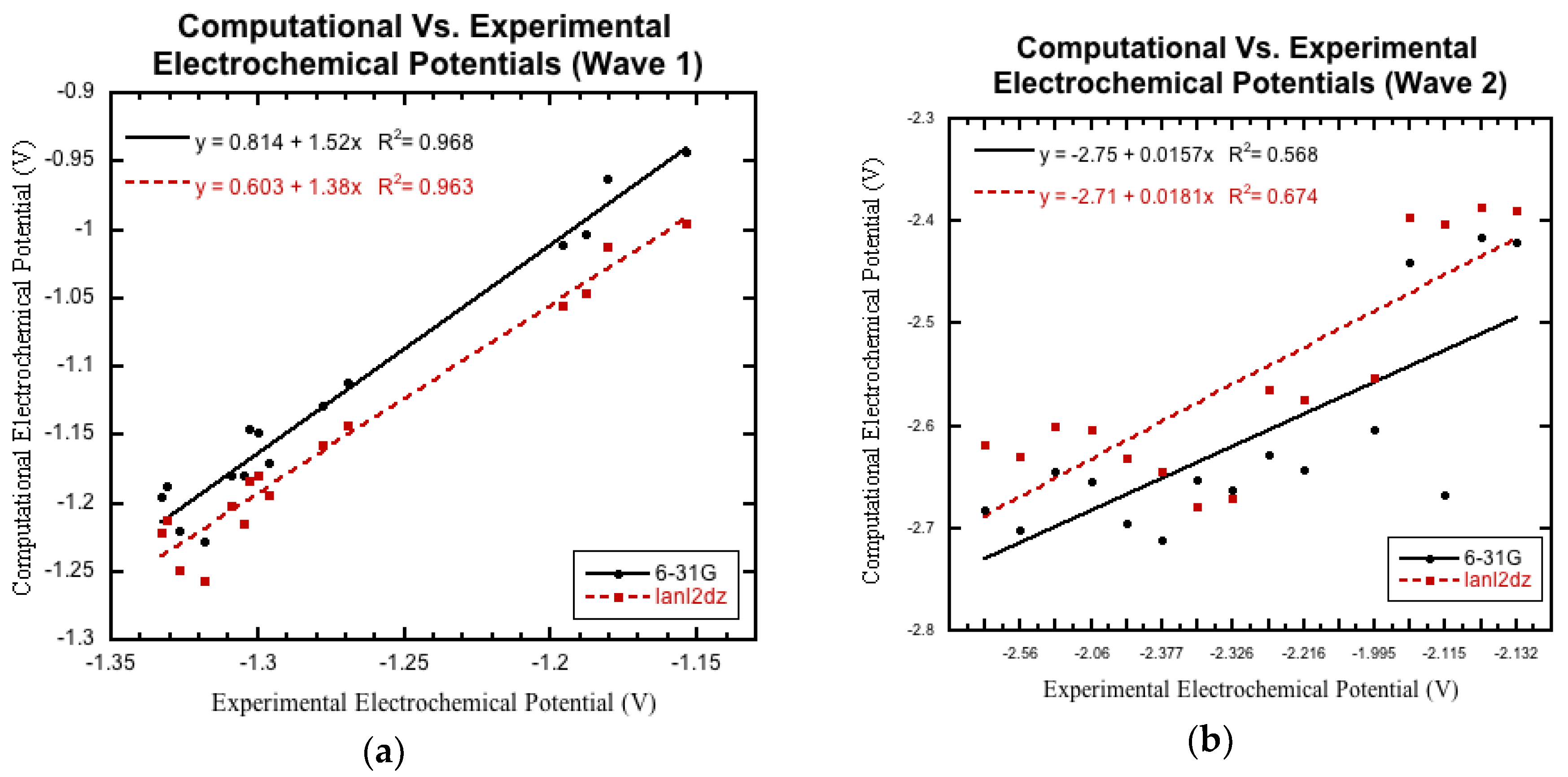
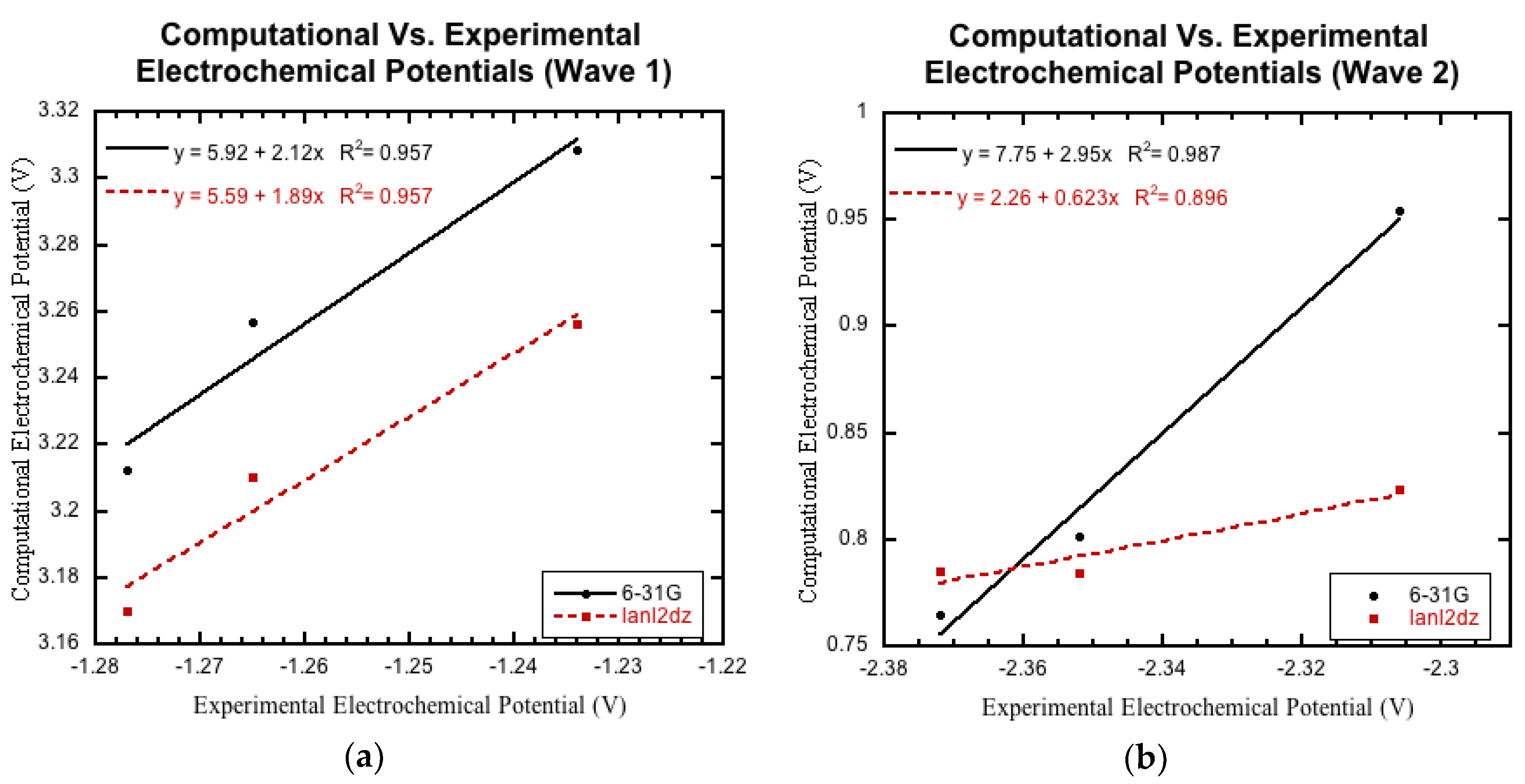
3.3.1. Initial Comparison
3.3.2. Non-Nitro Containing Derivatives
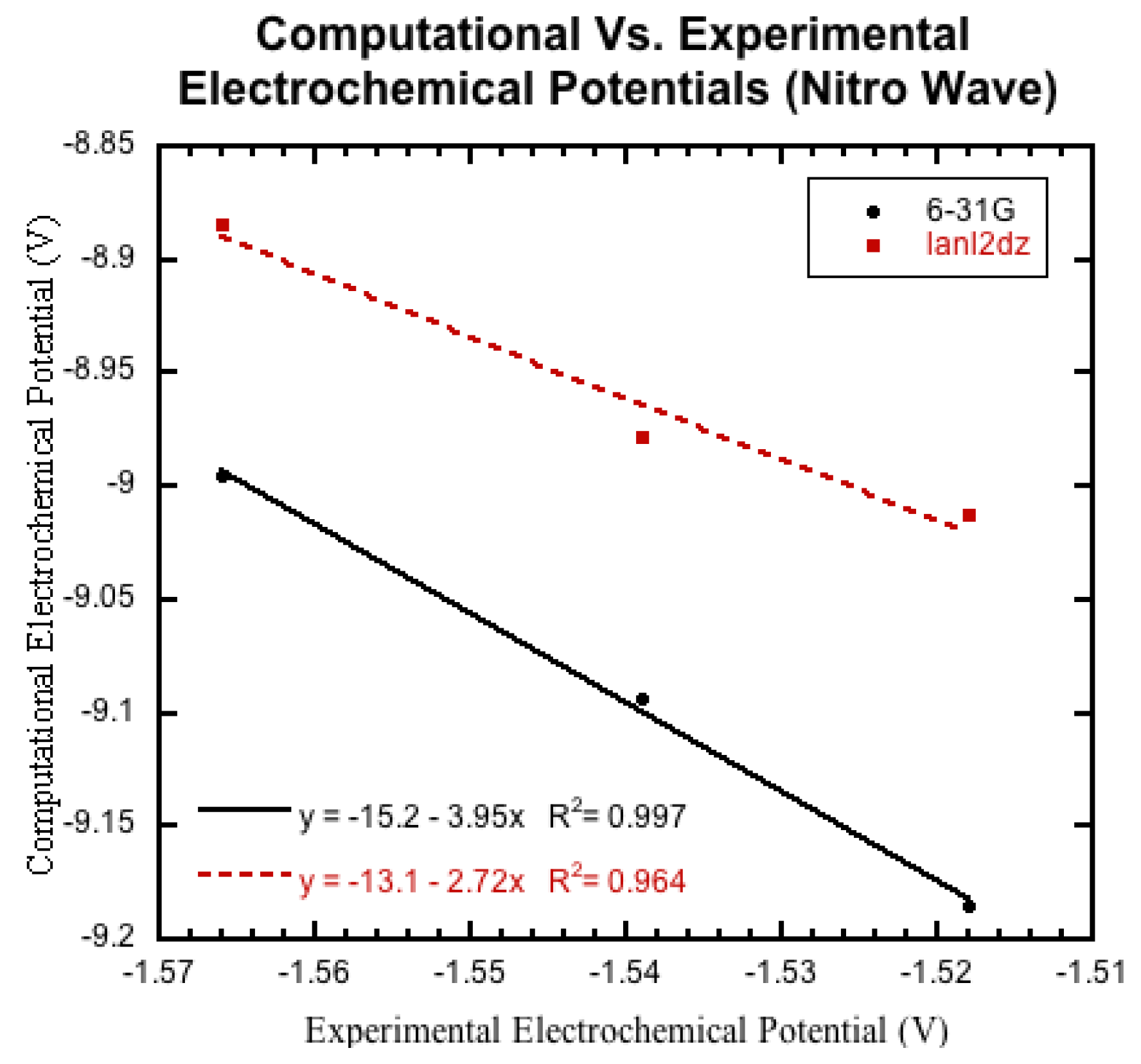
3.3.3. Nitro Containing Derivatives
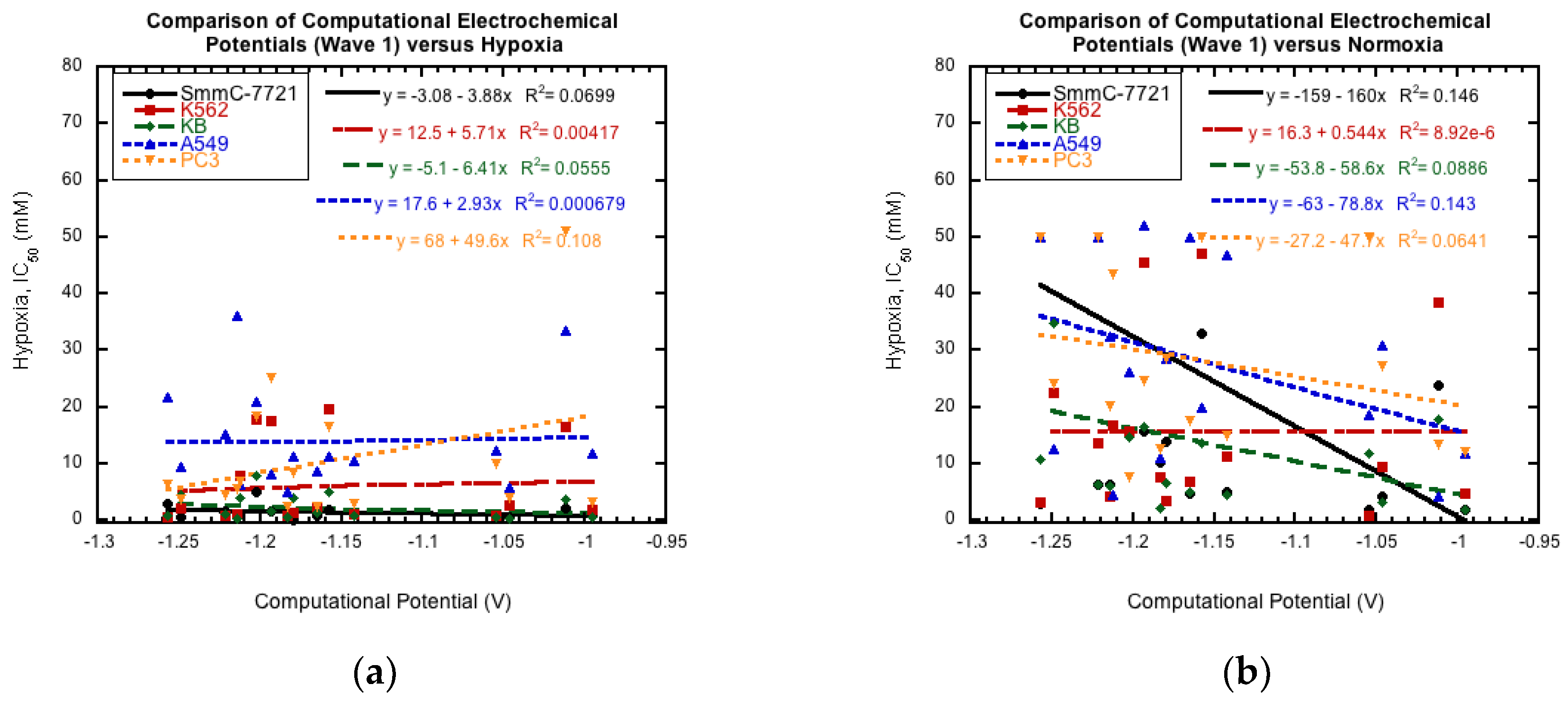
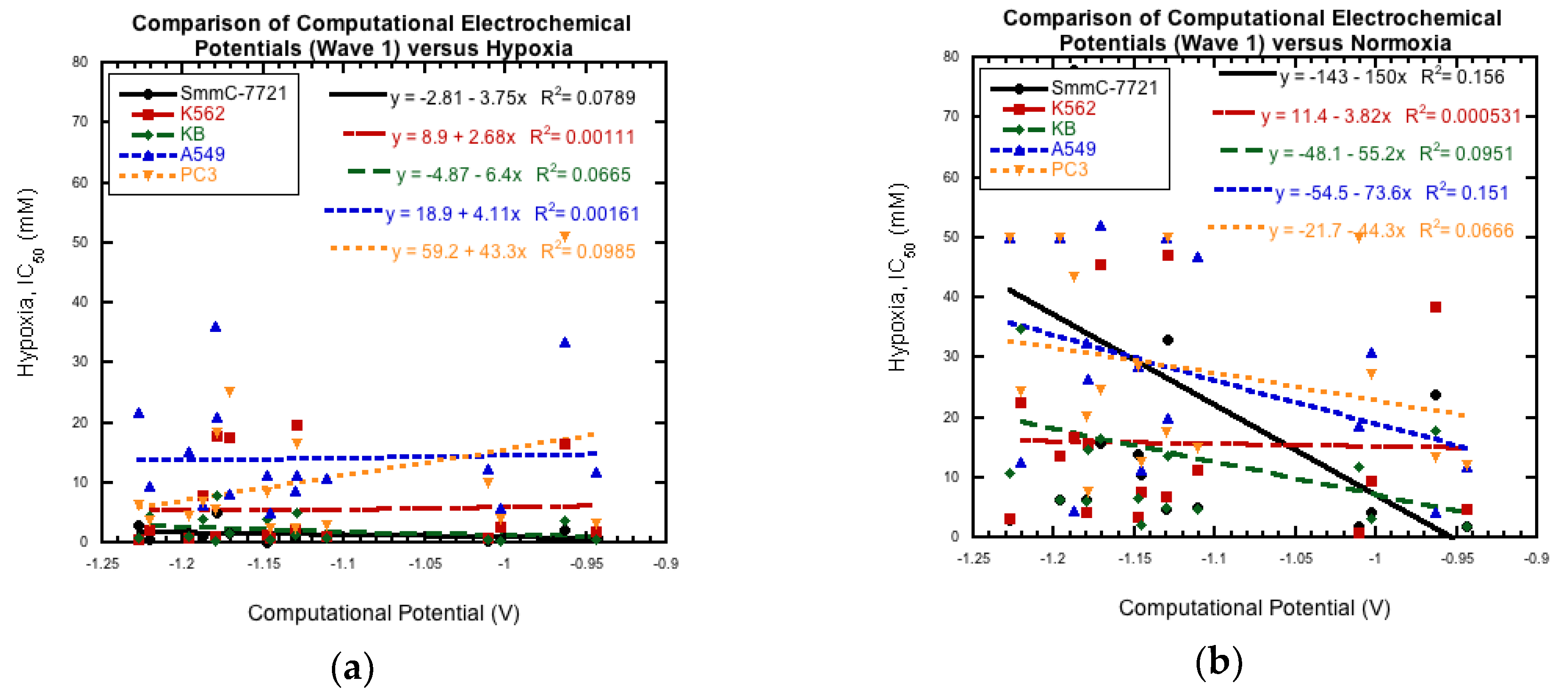
3.4. Comparison to Cytotoxicity
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A


References
- Anderson, R.F.; Yadav, P.; Shinde, S.S.; Hong, C.R.; Pullen, S.M.; Reynisson, J.; Wilson, W.R.; Hay, M.P. Radical Chemistry and Cytotoxicity of Bioreductive 3-Substituted Quinoxaline Di-N-Oxides. Chem. Res. Toxicol. 2016, 29, 1310–1324. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, B.; Jaso, A.; Aldana, I.; Monge, A. Synthesis and Anticancer Activity Evaluation of New 2-Alkylcarbonyl and 2-Benzoyl-3-trifluoromethyl-quinoxaline 1,4-Di-N-oxide Derivatives. Bioorg. Med. Chem. 2004, 12, 3711–3721. [Google Scholar] [CrossRef] [PubMed]
- Amin, K.M.; Ismail, M.M.F.; Noaman, E.; Soliman, D.H.; Ammar, Y.A. New quinoxaline 1,4-di-N-oxides. Part 1: Hypoxia-selective cytotoxins and anticancer agents derived from quinoxaline 1,4-di-N-oxides. Bioorg. Med. Chem. 2006, 14, 6917–6923. [Google Scholar] [CrossRef] [PubMed]
- Estevez, T.; Quilano, M.; Burguete, A.; Cabanillas, B.; Zimic, M.; Malaga, E.; Verastegui, M.; Pérez-Silanes, S.; Aldana, I.; Monge, A. Trypanocidal Properties, Structure-activity Relationship and Computational Studies of Quinoxaline 1,4-di-N-Oxide Derivatives. Exp. Parasitol. 2011, 127, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Ancizu, S.; Moreno, E.; Torres, E.; Burguete, A.; Pérez-Silanes, S.; Benítez, D.; Villar, R.; Solano, B.; Marín, A.; Aldana, I.; et al. Heterocyclic-2-carboxylic Acid (3-Cyano-1,4-di-N-oxidequinoxalin-2-yl)amide Derivatives as Hits for the Development of Neglected Disease Drugs. Molecules 2009, 14, 2256–2272. [Google Scholar] [CrossRef] [PubMed]
- Torres, E.; Moreno-Viguri, E.; Galiano, S.; Devarapally, G.; Crawford, P.W.; Azqueta, A.; Arbillaga, L.; Varela, J.; Birriel, E.; Di Maio, R.; et al. Novel Quinoxaline 1,4-Di-N-Oxide Derivatives as New Potential Antichagasic Agents. Eur. J. Med. Chem. 2013, 66, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Soliman, D.H. Anti-bacterial and Anti-fungal Activities of New Quinoxaline 1,4-di-N-Oxide Derivatives. Int. J. Org. Chem. 2013, 3, 65–72. [Google Scholar] [CrossRef]
- Carta, A.; Paglietta, G.; Nikookar, M.E.R.; Sanna, P.; Sechi, L.; Zanetti, S. Novel Substituted Quinoxaline 1,4-Dioxides with in vitro Antimycobacterial and Anticandida Activity. Eur. J. Med. Chem. 2002, 37, 355–366. [Google Scholar] [CrossRef]
- Vicente, E.; Pérez-Silanes, S.; Lima, L.M.; Ancizu, S.; Burguete, A.; Solano, B.; Villar, R.; Aldana, I.; Monge, A. Against Mycobacterium tuberculosis of New Quinoxaline 1,4-di-N-Oxides. Biorgan. Med. Chem. 2009, 17, 385–389. [Google Scholar] [CrossRef]
- Crawford, P.W.; Scamehorn, R.G.; Hollstein, U.; Ryan, M.D.; Kovacic, P. Cyclic Voltammetry of Phenazines and Quinoxalines Including Mono- and Di-N-Oxides. Relation to Structure and Antimicrobial Activity. Chem.-Biol. Interact. 1986, 60, 67–84. [Google Scholar] [CrossRef]
- Perez-Silanes, S.; Devarapally, G.; Torres, E.; Moreno, E.; Aldana, I.; Monge, A.; Crawford, P.W. Cyclic Voltammetric Study of Some Anti-Chagas Active Quinoxaline 1,4-Di-N-Oxide-2-Ketone Derivatives. Helv. Chim. Acta 2013, 96, 217–227. [Google Scholar] [CrossRef]
- Moreno, E.; Pérez-Silanes, S.; Gouravaram, S.; Macharam, A.; Ancizu, S.; Torres, E.; Aldana, I.; Monge, A.; Crawford, P.W. 1,4-Di-N-oxide quinoxaline-2-carboxamide: Cyclic voltammetry and relationship between electrochemical behavior, structure and anti-tuberculosis activity. Electrochim. Acta 2011, 56, 3270–3275. [Google Scholar] [CrossRef]
- Ryan, M.D.; Scamehorn, R.G.; Kovacic, P. Charge Transfer in the Mechanism of Drug Action Involving Quinoxaline Di-N-Oxides. J. Pharm. Sci. 1985, 74, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Carta, A.; Corona, P.; Loriga, M. Quinoxaline 1,4-Dioxide: A Versatile Scaffold Endowed with Manifold Activities. Curr. Med. Chem. 2005, 12, 2259–2272. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Sa, W.; Cao, C.; Guo, L.; Hao, H.; Liu, Z.; Wang, X.; Yuan, Z. Quinoxaline 1,4-di-N-Oxides: Biological Activities and Mechanisms of Action. Front. Pharmacol. 2016, 7, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Tocher, J.H.; Edwards, D. I Electrochemical Characteristics of NitroHeterocyclic Compounds of Biological Interest VIII. Stability of Nitro Radical Anions from Cyclic Voltammetric Studies. Free Radic. Res. Commun. 1992, 16, 19–25. [Google Scholar] [CrossRef]
- Tocher, J.H.; Edwards, D. I Electrochemical Characteristics of Nitro-Heterocyclic Compounds of Biological Interest IV. Lifetime of the Metronidazole Radical Anion. Free Radic. Res. Commun. 1989, 6, 39–45. [Google Scholar] [CrossRef]
- Tocher, J.H.; Edwards, D. I Electrochemical Characteristics of NitroHeterocyclic Compounds of Biological Interest I. The Influence of Solvent. Free Radic. Res. Commun. 1988, 4, 269–276. [Google Scholar] [CrossRef]
- Bollo, S.; Nunez-Vergara, L.J.; Martinez, C.; Chauviere, G.; Perie, J.; Squella, J.A. Volatmmetric Study of Nitro Radical Anion Generated from Some Nitrofuran Compounds of Pharmacological Significance. Electroanalysis 2003, 15, 19–25. [Google Scholar] [CrossRef]
- MarvinSketch; ChemAxon Ltd.: Budapest, Hungary, 2018.
- Aguilar-Martínez, M.; Cuevas, G.; Jiménez-Estrada, M.; González, I.; Lotina-Hennsen, B.; Macías-Ruvalcaba, N. An Experimental and Theoretical Study of the Substituent Effects on the Redox Properties of 2-[(R-phenyl)amine]-1,4-naphthalenediones in Acetonitrile. J. Org. Chem. 1999, 64, 3684–3694. [Google Scholar] [CrossRef]
- Hodgson, J.L.; Namazian, M.; Bottle, S.E.; Coote, M.L. One-Electron Oxidation and Reduction Potentials of Nitroxide Antioxidants: A Theoretical Study. J. Phys. Chem. A 2007, 111, 13595–13605. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro da Silva, M.D.M.C.; Gomes, J.R.B.; Gonçalves, J.M.; Sousa, E.A.; Pandey, S.; Acree, W.E. Thermodynamic Properties of Quinoxaline-1,4-Dioxide Derivatives: A Combined Experimental and Computational Study. J. Org. Chem. 2004, 69, 2785–2792. [Google Scholar] [CrossRef] [PubMed]
- Lauria, A.; Almerico, A.M.; Barone, G. The influence of substitution in the quinoxaline nucleus on 1,3-dipolar cycloaddition reactions: A DFT study. Comput. Theor. Chem. 2013, 1013, 116–122. [Google Scholar] [CrossRef]
- Mishra, A.; Verma, C.; Srivastava, V.; Lgaz, H.; Quraishi, M.A.; Ebenso, E.E.; Chung, I.-M. Chemical, Electrochemical and Computational Studies of Newly Synthesized Novel and Environmental Friendly Heterocyclic Compounds as Corrosion Inhibitors for Mild Steel in Acidic Medium. J. Bio- Tribo-Corros. 2018, 4, 32. [Google Scholar] [CrossRef]
- Hu, Y.; Xia, Q.; Shangguan, S.; Liu, X.; Hu, Y.; Sheng, R. Synthesis and Biological Evaluation of 3-Aryl-quinoxaline-2-carbonitrile 1,4-di-N-Oxide Derivatives as Hypoxic Selective Anti-tumor Agents. Molecules 2012, 17, 9683–9696. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.E.; Xia, Q.; Cella, E.M.; Nenninger, W.A.; Mruzik, N.M.; Brillos-Monia, A.K.; Hu, Z.Y.; Sheng, R.; Ragain, M.C.; Crawford, W.P. Voltammetric Study of Some 3-Aryl-quinoxaline-2-carbonitrile 1,4-di-N-oxide Derivatives with Anti-Tumor Activities. Molecules 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Leo, A.; Hoekman, D. Exploring QSAR.; Fundamentals and Applications in Chemistry and Biology; American Chemical Society: Washington, DC, USA, 1995. [Google Scholar]
- Kunz, K.R.; Iyengar, B.S.; Dorr, R.T.; Alberts, D.S.; Remers, W.A. Structure activity relationship for mitomycin C and mitomycin A analogs. J. Med. Chem. 1991, 34, 2281–2286. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.; Millam, J. Millam GaussView, Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision 5.0.9; Gaussian, Inc.: Wallingford CT, USA, 2016. [Google Scholar]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Miertus̃, S.; Tomasi, J. Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. Chem. Phys. 1982, 65, 239–245. [Google Scholar] [CrossRef]
- Pascual-ahuir, J.L.; Silla, E.; Tuñon, I. GEPOL: An improved description of molecular surfaces. III. A new algorithm for the computation of a solvent-excluding surface. J. Comput. Chem. 1994, 15, 1127–1138. [Google Scholar] [CrossRef]
- Palma, J.L.; Batista, V.S. Tutorial on Ab Initio Redox Potential Calculations. 2016. Available online: http://ursula.chem.yale.edu/~batista/classes/tutorials/redoxpotentials.pdf (accessed on 10 January 2019).
- Karlsson, C.; Jämstorp, E.; Strømme, M.; Sjödin, M. Computational Electrochemistry Study of 16 Isoindole-4,7-diones as Candidates for Organic Cathode Materials. J. Phys. Chem. C 2012, 116, 3793–3801. [Google Scholar] [CrossRef]
- Sawyer, D.T.; Sobkowiak, A.; Roberts, J.L. Elecrochemistry for Chemists, 2nd ed.; Wiley-Interscience Publication: New York, NY, USA, 1995; ISBN 978-0-471-59468-0. [Google Scholar]
- Crawford, P.W.; Carlos, E.; Ellegood, J.C.; Cheng, C.C.; Dong, Q.; Liu, D.F.; Luo, Y.L. The electrochemistry of antineoplastic furanquinones: Electrochemical properties of benzo[b]naphtho[2,3-d]furan-6,11-dione derivatives. Electrochim. Acta 1996, 41, 2399–2403. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | R1 | R2 |
|---|---|---|
| A1 | H | H |
| A2 | CH3 | H |
| A3 | OCH3 | H |
| A4 | Cl | H |
| B1 | H | 3-CH3 |
| B2 | CH3 | 3-CH3 |
| B3 | OCH3 | 3-CH3 |
| B4 | Cl | 3-CH3 |
| C1 | H | 3-Cl |
| C2 | CH3 | 3-Cl |
| C3 | OCH3 | 3-Cl |
| C4 | Cl | 3-Cl |
| D1 | H | 4-Br |
| D2 | CH3 | 4-Br |
| D3 | OCH3 | 4-Br |
| D4 | Cl | 4-Br |
| E1 | H | 4-NO2 |
| E2 | CH3 | 4-NO2 |
| E3 | OCH3 | 4-NO2 |
| E4 | Cl | 4-NO2 |
| Wave 1 | Nitro Wave (E1–E4 Only) | Wave 2 | ||||
|---|---|---|---|---|---|---|
| Compounds | Lanl2dz | 6-31g | Lanl2dz | 6-31g | Lanl2dz | 6-31g |
| A1 | 3.6006 | 3.2891 | - - - - - | - - - - - | 2.1763 | 1.7774 |
| A2 | 3.5453 | 3.2395 | - - - - - | - - - - - | 2.1618 | 1.7652 |
| A3 | 3.5818 | 3.2723 | - - - - - | - - - - - | 2.2294 | 1.8321 |
| A4 | 3.7477 | 3.4570 | - - - - - | - - - - - | 2.3981 | 2.0192 |
| B1 | 3.5920 | 3.2807 | - - - - - | - - - - - | 2.1636 | 1.7586 |
| B2 | 3.5370 | 3.2319 | - - - - - | - - - - - | 2.0861 | 1.7480 |
| B3 | 3.5729 | 3.2639 | - - - - - | - - - - - | 2.2190 | 1.8172 |
| B4 | 3.7390 | 3.4489 | - - - - - | - - - - - | 2.3920 | 1.7917 |
| C1 | 3.6516 | 3.3487 | - - - - - | - - - - - | 2.1944 | 1.8146 |
| C2 | 3.6111 | 3.3141 | - - - - - | - - - - - | 2.1161 | 1.8077 |
| C3 | 3.6295 | 3.3292 | - - - - - | - - - - - | 2.2472 | 1.8647 |
| C4 | 3.7986 | 3.5162 | - - - - - | - - - - - | 2.4078 | 2.0440 |
| D1 | 3.6364 | 3.3307 | - - - - - | - - - - - | 2.1920 | 1.8055 |
| D2 | 3.5797 | 3.2800 | - - - - - | - - - - - | 2.1228 | 1.7977 |
| D3 | 3.6150 | 3.3119 | - - - - - | - - - - - | 2.2407 | 1.8561 |
| D4 | 3.7825 | 3.4970 | - - - - - | - - - - - | 2.4046 | 2.0388 |
| E1 | 8.0502 | 7.7682 | −4.2182 | −4.7249 | 5.6176 | 5.4141 |
| E2 | 8.0043 | 7.7169 | −4.1840 | −4.6334 | 5.5784 | 5.2614 |
| E3 | 7.9644 | 7.6723 | −4.0895 | −4.5349 | 5.5797 | 5.2245 |
| E4 | 7.9769 | 7.6480 | −3.8809 | - - - - - | 5.7205 | - - - - - |
| Wave 1 | Nitro Wave (E1–E4 Only) | Wave 2 | ||||
|---|---|---|---|---|---|---|
| Compounds | Lanl2dz | 6-31g | Lanl2dz | 6-31g | Lanl2dz | 6-31g |
| A1 | −1.9136 | −1.8908 | - - - - - | - - - - - | −3.3379 | −3.4025 |
| A2 | −1.9690 | −1.9404 | - - - - - | - - - - - | −3.3524 | −3.4147 |
| A3 | −1.9325 | −1.9076 | - - - - - | - - - - - | −3.2848 | −3.3479 |
| A4 | −1.7666 | −1.7230 | - - - - - | - - - - - | −3.1161 | −3.1607 |
| B1 | −1.9223 | −1.8993 | - - - - - | - - - - - | −3.3507 | −3.4213 |
| B2 | −1.9773 | −1.9480 | - - - - - | - - - - - | −3.4282 | −3.4319 |
| B3 | −1.9413 | −1.9160 | - - - - - | - - - - - | −3.2952 | −3.3627 |
| B4 | −1.7752 | −1.7310 | - - - - - | - - - - - | −3.1222 | −3.3882 |
| C1 | −1.8626 | −1.8312 | - - - - - | - - - - - | −3.3199 | −3.3654 |
| C2 | −1.9032 | −1.8658 | - - - - - | - - - - - | −3.3982 | −3.3722 |
| C3 | −1.8847 | −1.8507 | - - - - - | - - - - - | −3.2671 | −3.3152 |
| C4 | −1.7156 | −1.6637 | - - - - - | - - - - - | −3.1064 | −3.1359 |
| D1 | −1.8778 | −1.8492 | - - - - - | - - - - - | −3.3233 | −3.3744 |
| D2 | −1.9346 | −1.8999 | - - - - - | - - - - - | −3.3915 | −3.3822 |
| D3 | −1.8993 | −1.8680 | - - - - - | - - - - - | −3.2735 | −3.3238 |
| D4 | −1.7318 | −1.6829 | - - - - - | - - - - - | −3.1097 | −3.1411 |
| E1 | 2.5360 | 2.5883 | −9.7324 | −9.9049 | 0.10341 | 0.23426 |
| E2 | 2.4901 | 2.5369 | −9.6982 | −9.8133 | 0.06423 | 0.08157 |
| E3 | 2.4502 | 2.4924 | −9.6038 | −9.7148 | 0.06548 | 0.04466 |
| E4 | 2.4627 | 2.4681 | −9.3952 | - - - - - | 0.20624 | - - - - - |
| Wave 1 | Nitro Wave (E1–E4 Only) | Wave 2 | ||||
|---|---|---|---|---|---|---|
| Compounds | Lanl2dz | 6-31g | Lanl2dz | 6-31g | Lanl2dz | 6-31g |
| A1 | −1.1936 | −1.1708 | - - - - - | - - - - - | −2.6179 | −2.6825 |
| A2 | −1.2490 | −1.2204 | - - - - - | - - - - - | −2.6324 | −2.6947 |
| A3 | −1.2125 | −1.1876 | - - - - - | - - - - - | −2.5648 | −2.6279 |
| A4 | −1.0466 | −1.0030 | - - - - - | - - - - - | −2.3961 | −2.4407 |
| B1 | −1.2023 | −1.1793 | - - - - - | - - - - - | −2.6307 | −2.7013 |
| B2 | −1.2573 | −1.2280 | - - - - - | - - - - - | −2.7082 | −2.7119 |
| B3 | −1.2213 | −1.1960 | - - - - - | - - - - - | −2.5752 | −2.6427 |
| B4 | −1.0552 | −1.0110 | - - - - - | - - - - - | −2.4022 | −2.6682 |
| C1 | −1.1426 | −1.1112 | - - - - - | - - - - - | −2.5999 | −2.6454 |
| C2 | −1.1832 | −1.1458 | - - - - - | - - - - - | −2.6782 | −2.6522 |
| C3 | −1.1647 | −1.1307 | - - - - - | - - - - - | −2.5471 | −2.5952 |
| C4 | −0.9956 | −0.9437 | - - - - - | - - - - - | −2.3864 | −2.4159 |
| D1 | −1.1578 | −1.1292 | - - - - - | - - - - - | −2.6033 | −2.6544 |
| D2 | −1.2146 | −1.1799 | - - - - - | - - - - - | −2.6715 | −2.6622 |
| D3 | −1.1793 | −1.1480 | - - - - - | - - - - - | −2.5535 | −2.6038 |
| D4 | −1.0118 | −0.9629 | - - - - - | - - - - - | −2.3897 | −2.4211 |
| E1 | 3.2560 | 3.3083 | −9.0124 | −9.1849 | 0.82341 | 0.95426 |
| E2 | 3.2101 | 3.2569 | −8.9782 | −9.0933 | 0.78423 | 0.80157 |
| E3 | 3.1702 | 3.2124 | −8.8838 | −8.9948 | 0.78548 | 0.76466 |
| E4 | 3.1827 | 3.1881 | −8.6752 | - - - - - | 0.92624 | - - - - - |
| Wave 1 | Nitro Wave (E1–E4 Only) | Wave 2 | |
|---|---|---|---|
| Compounds | E1/2 (V) | E1/2 (V) | Epc (V) |
| A1 | −1.296 | - - - - - | −2.163 |
| A2 | −1.327 | - - - - - | −2.310 |
| A3 | −1.331 | - - - - - | −2.166 |
| A4 | −1.188 | - - - - - | −1.973 |
| B1 | −1.309 | - - - - - | −2.56 b |
| B2 | −1.318 | - - - - - | −2.377 |
| B3 | −1.333 | - - - - - | −2.216 |
| B4 | −1.196 | - - - - - | −2.115 |
| C1 | −1.269 | - - - - - | −2.097 |
| C2 | −1.303 | - - - - - | −2.125 |
| C3 | −1.401 a | - - - - - | No Value |
| C4 | −1.154 | - - - - - | −2.080 |
| D1 | −1.278 | - - - - - | −2.06 b |
| D2 | −1.305 | - - - - - | −2.326 |
| D3 | −1.300 | - - - - - | −1.995 |
| D4 | −1.181 | - - - - - | −2.132 |
| E1 | −1.234 | −1.518 | −2.306 |
| E2 | −1.265 | −1.539 | −2.352 |
| E3 | −1.277 | −1.566 | −2.372 |
| E4 | −1.134 | −1.514 | −2.141 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miller, E.M.; Brazel, C.J.; Brillos-Monia, K.A.; Crawford, P.W.; Hufford, H.C.; Loncaric, M.R.; Mruzik, M.N.; Nenninger, A.W.; Ragain, C.M. Reduction Potential Predictions for Some 3-Aryl-Quinoxaline-2-Carbonitrile 1,4-Di-N-Oxide Derivatives with Known Anti-Tumor Properties. Computation 2019, 7, 6. https://doi.org/10.3390/computation7010006
Miller EM, Brazel CJ, Brillos-Monia KA, Crawford PW, Hufford HC, Loncaric MR, Mruzik MN, Nenninger AW, Ragain CM. Reduction Potential Predictions for Some 3-Aryl-Quinoxaline-2-Carbonitrile 1,4-Di-N-Oxide Derivatives with Known Anti-Tumor Properties. Computation. 2019; 7(1):6. https://doi.org/10.3390/computation7010006
Chicago/Turabian StyleMiller, Eric M., Cody J. Brazel, Krystina A. Brillos-Monia, Philip W. Crawford, Hannah C. Hufford, Michael R. Loncaric, Monica N. Mruzik, Austin W. Nenninger, and Christina M. Ragain. 2019. "Reduction Potential Predictions for Some 3-Aryl-Quinoxaline-2-Carbonitrile 1,4-Di-N-Oxide Derivatives with Known Anti-Tumor Properties" Computation 7, no. 1: 6. https://doi.org/10.3390/computation7010006
APA StyleMiller, E. M., Brazel, C. J., Brillos-Monia, K. A., Crawford, P. W., Hufford, H. C., Loncaric, M. R., Mruzik, M. N., Nenninger, A. W., & Ragain, C. M. (2019). Reduction Potential Predictions for Some 3-Aryl-Quinoxaline-2-Carbonitrile 1,4-Di-N-Oxide Derivatives with Known Anti-Tumor Properties. Computation, 7(1), 6. https://doi.org/10.3390/computation7010006