1. The Foundation of PCCA+
Molecular simulation is used as a tool to estimate thermodynamical properties. Moreover, it could be used for investigation of molecular processes and their time scales. In computational drug design, e.g., binding affinities of ligand-receptor systems are estimated. In this case, trajectories in a
-dimensional space
of molecular conformations (
N is the number of atoms) are generated. They will be denoted as micro-states
in the following. After simulation the micro-states are classified as “bound” or “unbound”. This classification can be achieved by projecting the state space
onto a low number
n of macro-states of the system. Spectral clustering algorithms are one possible method in this context. Robust Perron Cluster Analysis (PCCA+) turned out to be a commonly used practical tool for this algorithmic step [
1].
Conformation Dynamics has been established in the late 1990s. From molecular simulations [
2] transition matrices
were derived. The entries of
P represent conditional transition probabilities between pre-defined
m subsets of the state space
of a molecular process. Today
P would be denoted as
Markov State Model. According to the transfer operator theory and PCCA [
3] the eigenvectors of these matrices were studied in the early 2000s leading to an empirical observation [
4] and
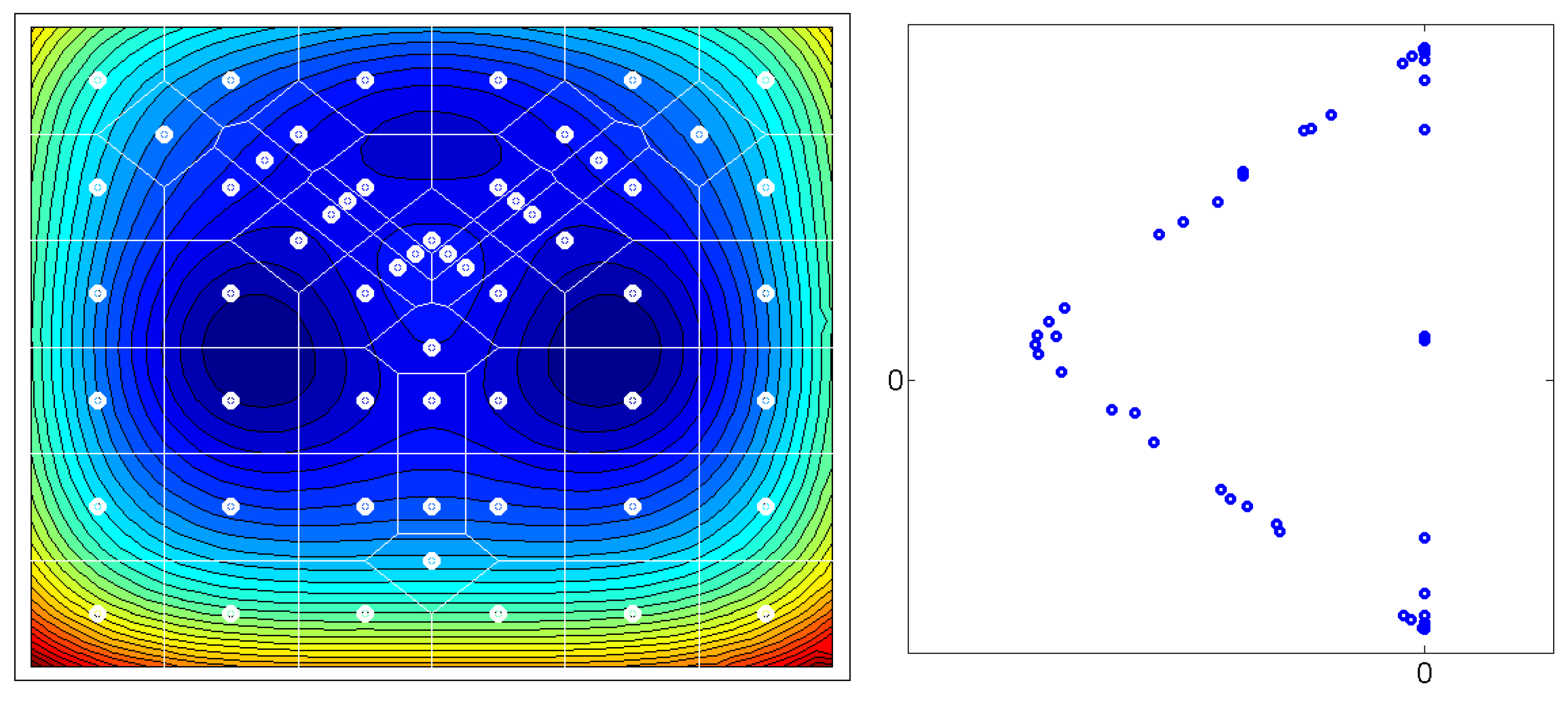
Figure 1: Given a matrix
of the
leading eigenvectors of the
-transition matrix
P, the
m rows of
X – plotted as
n-dimensional points – seem to always form an
-simplex. Some of those
m points are located at the vertices of the simplices, other points are located on the edges. The points which are located at the
n vertices of these simplices represent the thermodynamically stable conformations of the molecular systems. Whereas, the points which are located on the edges of the simplices represent transition regions. In order to transform the
-simplex
into the
-standard simplex
(spanned by the unit vectors), the
inner simplex algorithm (ISA) [
4] has been applied. ISA identifies those
n points out of the
m rows of
X which are supposed to be the vertices of the simplex
. These points are transformed into the unit vectors in
spanning
. An easy introduction to ISA can be found online [
5].
Mathematically, ISA transforms
X via
into an
-matrix
.
is a regular transformation matrix. The rows of
are the new coordinates of the
m points after transformation. If the simplex
was a perfect simplex, then the entries of the matrix
are non-negative and the row sums of
are one. This means, that the entries of
can be interpreted as a partition-of-unity fuzzy clustering of the
m subsets of
into
n fuzzy sets. The membership values (denoted as membership functions) of these fuzzy sets are given by the columns of
. The theoretical justification for the distinction between thermodynamical stable regions and transition regions in
, i.e., the described localization of the
n-dimensional rows of
X in an
-simplex, has been provided by Susanna Röblitz (chapter 3.1.2 in [
6]).
Since the eigenvector data usually does not provide perfect simplices in real-world applications, ISA often results in a set of membership functions
which have negative values [
7].
The larger the deviation from the simplex structure, the higher are the absolute values of these negative entries. This can be employed to determine the number
n of membership functions [
7], such that the simplex structure is achieved. Besides this minChi-criterion, there exist other criteria for determining the number
n. Of course, using a spectral gap in the sorted list of eigenvalues is one possibility. Another possibility is to construct a set-based clustering of states, such that the transition probabilities derived from the micro-process between these sets define a Markov chain [
8]. One could also ask for a projection of the micro-process onto a Markov chain, such that the projection is as set-based as possible [
6]. The next step is to compute the transformation matrix
.
PCCA+ is an extension of ISA and was published in 2005 [
9]. In some existing software codes the inner simplex algorithm (
cluster_by_isa) is implemented and incorrectly denoted as PCCA+ like in [
5]. PCCA+ assures that the transformation
is feasible in the following sense: the resulting membership functions are a partition-of-unity and the membership values are non-negative. Briefly, the linearly constrained feasible set
of transformation matrices is given by:
where
denotes a constant function.
The feasible set
is not empty and has an interior point (proof of Lemma 3.5 in [
10]). Thus, the construction of a transformation matrix
is not unique. In PCCA+, the selection of
is given by solving a convex maximization problem. This means that there is a convex objective function
to be maximized. The optimum of a convex maximization problem on a linearly bounded compact set is attained at a vertex of
. PCCA+ is formulated as an unconstrained maximization problem
, where the function
g is a projection of an (infeasible) transformation matrix
onto a superset of the vertices of
(chapter 3 in [
10]). The objective function
accounts for the “crispness” of
. The membership functions should be as close as possible to characteristic functions of subsets of
. One example for a possible objective function
is provided at the end of
Section 3.1. Solving the PCCA+ optimization problem can be very time consuming. Berg re-implemented PCCA+ with a fast Newton-type local optimization algorithm [
11]. The implementation details of PCCA+ can be found in [
12] and in Appendix D of [
13]. In principle, the optimization problem to be solved has many local maxima. Local optimization routines cannot assure for finding the global optimum. However, there are possibilities to re-formulate PCCA+ as a linear optimization problem (chapter 3.4.3 in [
10]) which has a unique global optimum. These possibilities have not been exploited in applications so far. Although PCCA+ is only one example for a spectral clustering routine, it is discussed that
, i.e., the mapping to a simplex,
is spectral clustering [
5]. It is also discussed in artificial intelligence that spectral clustering is (like PCCA+) a special way of a basis transform [
14].
Taking some of the eigenvectors of a matrix P as the basis vectors of a linear subspace V is advantageous in terms of the matrix projection. Applying P to a vector of this subspace produces a vector , which is again an element of V. Thus, V is said to be an invariant subspace with regard to P. All actions of P in the high-dimensional space can be projected onto actions of P within a low dimensional space V. This is denoted as “invariant subspace projection”.
Since its invention in 2005, a lot of scientific contributions extended the application area of PCCA+ and evolved the theory of PCCA+ into new directions. Until 2017, the original PCCA+ article has been cited more than 250 times PCCA+ is used as a spectral clustering method in various fields of application ranging from molecular simulation to psychology. Not only the areas of application changed, but also the theory of PCCA+ went into new directions:
In contrast to PCCA+, GenPCCA is based on real invariant subspaces given by Schur decompositions [
15]. This is useful, if the transition matrices have complex eigenvectors or are non-diagonalizable [
16]. This extension of PCCA+ can be applied to non-reversible Markov chains.
Treating non-autonomous processes is also possible, i.e., whenever transition probabilities of the Markov chain depend on external variables. In this case, these variables become additional coordinates of the state space of the Markov process. The analysis of metastable sets turns into a coherent sets analysis [
17].
PCCA+ has been invented for transition matrices. However, it can be applied to transfer operators or infinitesimal generators of time-continuous Markov processes as well [
18]. In this case, adaptive discretizations of the operators play an important role [
19,
20].
Recently, the interpretation of as an equation which connects stochastics and numerics turned out to be a very fruitful link for computations in molecular sciences. The theoretical background of this equation will be explained in the next section.
Changing the point of view from a set-based towards a fuzzy clustering of
has some consequences: The set-based clustering allows for precise definition of transition states between macro-states at the interfaces of the sets [
8]. In fuzzy clustering on the other hand, every micro-state in
is more or less a transition state. This type of fuzzy clustering is also known from the core set approach [
21], where the degree of membership to one of the clusters is given by the probability to reach a certain subset of
by micro-dynamics before reaching certain other subsets. The PCCA+ approach is like a core set approach without the necessity to define core sets.
2. Interpretation of
Many mathematical models in natural sciences are based on an input-output-relation. Given a certain (input) micro-state
of a system, we want to predict its future (output) micro-states
, where
t is the time-variable and
is some state space. These input-output-relations are often formulated as initial value problems of ordinary, stochastic, or partial differential equations, where these equations are first-order with regard to time-derivatives
. Some examples are: Hamiltonian dynamics, Brownian motion, Langevin dynamics, Smoluchowski equation, Fokker-Planck equation, time-dependent Schrödinger equation, reaction kinetics, system biology models, etc. All these models can be seen as time-homogeneous (autonomous) Markov processes. All these processes are uniquely defined by their
Markov kernel. The Markov kernel is a function
The first argument of
p is a point
, which denotes the starting point of the micro-process. The second argument is a target subset
, which has a measurable size. This is denoted by
, where
defines all measurable subsets of
. The last argument is a time-span
. The function value
quantifies how likely a process starting in a micro-state
will evolve into the (target) set
at time
. A good introduction into this theory can be found in Nielsen [
22].
If we describe the properties of molecular processes, we often simplify the continuous state space
and speak of a finite set of macro-states. One example is the description of ligand-receptor-binding processes. Instead of providing all 3-dimensional coordinates
of the ligand and of the receptor to describe the state of the system, we only discriminate between a finite number of macro-states, e.g., ‘bound’ vs. ‘unbound’. For every macro-state
there is a membership function
which quantifies how much a micro-state
belongs to
i. Bujotzek established this approach for ligand-receptor binding processes [
23,
24,
25].
On the one hand we have a Markov process (
2), i.e., an input-output-relation on the level of micro-states. On the other hand, we only want to describe the process in terms of the macro-states (
3). If a Markov kernel
p is known for the micro-level it might be unknown for the macro-level. Even worse: On the macro-level the process need not be Markovian. A projected Markov process is in general not a Markov process anymore [
26].
Example 1. For example, take a particle that moves with regard to Brownian motion in a space Ω. This movement is a Markov process. Now we subdivide the space Ω into three subsets and C. Thus, the particle has 3 macro-states , and C. Once the particle crosses a barrier between two subsets, e.g., from A to B, it will more likely return to A than going to C, because of its jittering. In addition, if it crosses the barrier from C to B, it will more likely return to C than going to A. The conditional probability to move from B to one of the other sets is not independent from the past.
PCCA+ is a method for constructing the projection (3) in such a way, that the projected Markov process of micro-states is a Markov process again. The projected states are not only invariant subspaces, but they are also interpretable as macro-states (in terms of a fuzzy clustering of micro-states). Here it is explained, how this projection works. Although the dynamics of the micro-system might be highly non-linear, it is possible to fully represent these processes by linear operators, namely by
transfer operators. Transfer operators are given by solving an integral equation. In this context we will use Lebesgue integration. These integrals are based on measuring the size of subsets
with a so-called measure function
. The set of all functions with finite Lebesque integral
is denoted as
. Given a measure
, measurable sets
, and a Markov kernel
p, the transfer operator
is defined by solving the integral equation
for all
and all
chapter 1.2 in [
22]. What is a transfer operator? In order to understand the meaning of transfer operators consider the following: If the Markov process is a Markov chain on a finite set of
m states and
is the stationary distribution of this chain, then the transfer operator is simply given by the transition matrix
of the Markov chain.
is a generalization of a transition matrix for continuous spaces
.
Propagation of the micro-system is completely represented by the transfer operator
, i.e., by a linear mapping. If
has an
n-dimensional invariant real subspace
X, then propagation in the micro-system (starting with an element of that invariant subspace) can be projected onto a finite-dimensional linear mapping
within an
n-dimensional vector space, i.e., onto an
-matrix
T representing the dynamics of the macro-system, consider
Figure 2.
Invariant subspace projection of is a suitable model reduction, but it is not the desired -subset projection of . Equation provides now the recipe for constructing the functions such that the basis of the invariant subspace can be interpreted as membership functions of (fuzzy) subsets of . This is a three-step procedure:
One has to find an
n-dimensional invariant subspace
X (real vector space) of the transfer operator of the micro-system. Real eigenspaces and real Schur spaces are suitable examples for invariant subspaces [
6,
15,
18].
Then one has to linearly transform the basis of this invariant subspace X in such a way, that all basis vectors form a non-negative partition of unity. This means, one has to search for a suitable transformation matrix which provides .
With the aid of
it is possible to construct the projected transfer operator. This will be exemplified later in Equation (
5).
This invariant subspace projection assures, that propagation with the linear transfer operator commutes with the projection from micro- to macro-systems [
15,
27]. The systematic error (described in [
26]) of a set-based projection of
vanishes. The discussion of problematic recrossing events (see [
28]) for the calculation of transition rates is not needed, if
is computed via the described subspace projection.
3. Consequences of
The invariant subspace projection of some transfer operator connects micro-systems to their macro-systems. The interpretation of as a state space clustering has interesting consequences for the modeling of molecular processes. In the followings, five consequences in different fields of molecular simulation and molecular modeling are sketched.
3.1. Rebinding Effects
Consider again the above example of a Brownian motion in
Section 2, i.e., the particle that moves in between three subsets
and
C of a (big) state space
. Once the particle crosses a barrier between two sets, e.g., from
A to
B, it will probably recross that barrier
. These recrossing events destroy the Markov property of the projected process [
28]. In the field of molecular simulation, we know this recrossing event as well. Here it is known as
rebinding [
29]. Once a ligand molecule unbinds from its receptor, it is likely to observe a rebinding event because of the still close vicinity of ligand and receptor. A good mathematical review on this subject has been written by Röhl [
30].
The rebinding phenomenon is especially relevant for multivalent ligand-receptor-binding [
25,
31]. In this case, the ligand has many “arms” that can individually bind to the receptor like in
Figure 3. Although there is a highly frequent binding and unbinding of single arms, the
total unbinding process of the multivalent ligand can be rather slow, because all arms have to unbind before one of the arms bind again. This complex multiscale binding process comprises of a lot of rebinding events and can be projected to a Markov chain on a small number
n of macro-states by the methods explained above.
It has been shown that the representation of the projected Markov chain is given by a matrix
. The projection theory of transfer operators has been worked out by Sarich [
26]. The elements of the
stochastic matrices
S and
T are computed as follows
where
denotes the
-weighted scalar product and
is a constant function.
are the membership functions of the macro-states. There is a relation between these matrices and the transformation matrix
. If
is self-adjoint (reversible Markov process) and
is the stationary density of the micro-process, then
,
, and
, where
is the diagonal matrix of the
n leading eigenvalues of
and
the diagonal matrix of the stationary distribution of the macro-process [
32].
In the reversible case, a matrix exists – – where is the diagonal matrix of the logarithm of the eigenvalues. This matrix generates P, i.e, , where denotes the matrix-exponential.
Let us assume that
P is generated by a rate matrix
Q. Rate matrices have non-negative off-diagonal elements, these are the rates between the macro-states (chapter 4 in [
34]). The diagonal elements of
Q are negative real numbers, such that the row sums of
Q are zero. Thus, the sum of all rates of the system is given by the negative trace of the matrix
Q (Equation (3.1) in [
32]):
Due to the fact that
S and
T are stochastic matrices, their determinants cannot be larger than 1. The case
only occurs, if
T and
S are unit matrices. If there are frequent transitions on the micro-level of the process (binding events of the “arms”), the stochastic matrix
T will have a low-valued determinant (Theorem 4.4 in [
10]), i.e., the negative logarithm will be a high number. Thus, the sum of the rates of the macro-processes tends to be high. However, the overlap of the membership functions, represented by the matrix
S can compensate for that: If the membership functions have a significant overlap, then also the determinant of
S is small which leads to a decreased sum of rates. The determinant of
S, therefore, is used as a measure of the stabilization of a process on the macro-level due to rebinding effects. For reversible micro-processes the matrix
directly depends on the transformation matrix
, if we take into account that the diagonal elements of
D are given by the first row of
(Lemma 3.6 in [
10]). It turns out, that the minimal rebinding effect that is included in a measured (binding) kinetics can be quantified and estimated [
30]. According to a suggestion of Röblitz, PCCA+ searches for a transformation matrix
such that the trace of the matrix
S is maximized [
6], which will also lead to high values of
. This means, that PCCA+ tries to minimize the rebinding effect (i.e, tries to minimize re-crossings).
3.2. Exit Rates
Equation
can be used to compute important dynamical properties from molecular simulations. A full decomposition of the state space
into macro-states is often not mandatory. For exit rate computations it is only needed to know how long it would take until a molecular process starting in a point
(of some subset
) leaves this subset. In particular it is asked for the exit rate, the holding probability, and the mean holding time of the set
M. The connection between these values and operator theory is very well explained by Pavliotis in [
35]. As an example,
could define the ‘bound’ macro-state of a ligand-receptor-system. The holding probability is defined by the following expectation value:
is the characteristic function of the set M. is a discontinuous function with and elsewhere. The expectation value is computed over an ensemble of trajectories with for realizations of a suitable stochastic process starting in . Only trajectories which start in M and never leave M in the interval are counted in (6).
We want to extend this approach towards continuous membership functions
instead of sets. In PCCA+, these membership functions
are computed from an invariant subspace of some transfer operator
. Here, we assume that the transfer operator is generated by an infinitesimal generator
with
. In this case,
can equivalently be computed from a real invariant subspace of
, i.e., for a 2-macro-state example there exist via PCCA+ real numbers
, such that
By multiplying this equation with the expression
, and by defining a function
, we equivalently get:
for all
with
. The definition of
can be expressed by an ordinary differential equation:
Combining Equations (8) and (9) leads to the following differential equation:
More details can be found online [
36]. For a special choice of the infinitesimal generator
, Equation (10) is the solution of the following conditional expectation value problem according to the Feynman-Kac formula (Equations III.1 and III.2 in [
37]):
In this expression, are realizations of molecular processes starting in and generated by (more precisely, by the adjoint of ).
Equation (11) is like the relaxed version of Equation (6) replacing sets by membership functions. This means, that given a membership function
with the PCCA+ property of Equation (7), the holding probability of the corresponding fuzzy set is given by Equation (11) which decreases exponentially with
and with exit rate
. Given the eigenfunction
f of
with
and the PCCA+ construction
, then
can be computed by
. The mean holding time for the membership function
and for a starting point
is therefore:
In this way, Equation can be used for finding simple analytical relations between fuzzy sets in state space and special dynamical properties of the molecular systems. In the above case, the computations of exit rates , holding probabilities , and mean holding times are directly connected to the PCCA+ construction of .
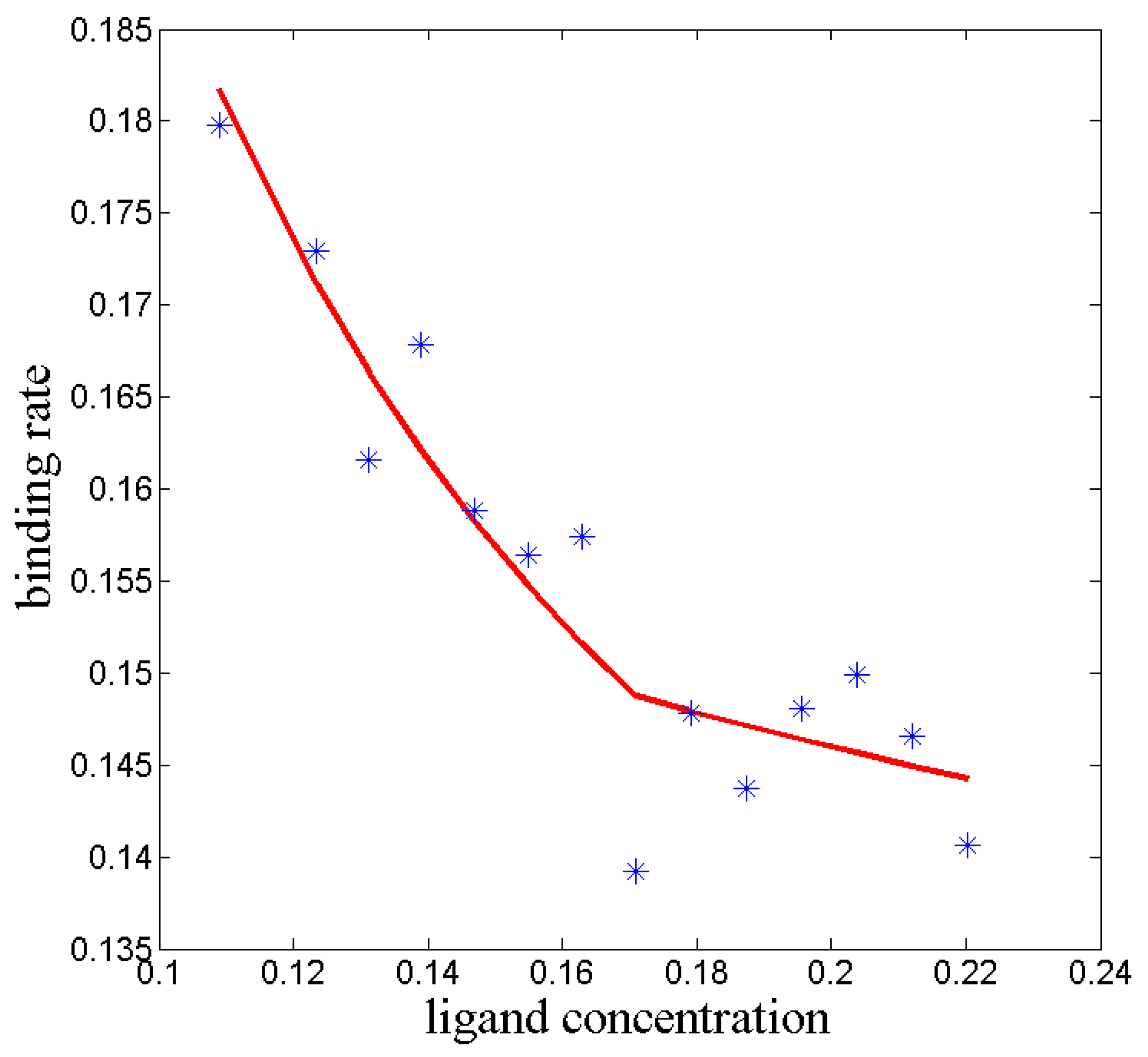
3.3. Kinetic ITC
Isothermal titration calorimetry (ITC) is an experimental approach to measure thermodynamical quantities of chemical processes [
38]. Ligand-receptor binding processes are often analyzed with this tool. One possible experimental setup can look like this: To an isolated reaction tube with a certain resolved concentration of receptors a certain amount of ligands is added successively in each step of the ITC measurement (titration). By adding the ligand, the temperature of the reaction tube changes. Assuming an endothermic reaction (i.e, the temperature tends to decrease), we have to warm up the reaction tube in order to keep its temperature constant [
39]. If the ligand-receptor-binding is a fast process, then heat must be added quickly. For a slow ligand-receptor-binding, the external heating is also slow to always assure constant temperature. This means, that the electric power used for heating is a monitor for the rate of the binding process. The binding rate can be calculated from the power profile. This calculation is named
kinetic ITC (kinITC) [
40].
The kinITC experiment provides the rate of a process which starts with a certain concentration of receptors, ligands, and (at consecutive titration steps) already formed ligand-receptor-complexes. It ends with a thermodynamically equilibrated mixture of those components in each titration step. The micro-processes taking place in the reaction tube are, thus, projected to these two macro-states, ‘unbound’ and ‘bound’. The macro-process is assumed to be Markovian: Only the initial concentrations of the components have to be known in order to explain the experimental outcome. Thus, according to
Section 2 and
Section 3.1 we assume the existence of a
-matrix
P which is the invariant subspace projection of the Markovian micro-process.
The experimental setup of kinITC does not allow for a direct calculation of the matrix
P. Instead of
P, kinITC provides its infinitesimal generator
with
and
being the time-interval of each titration step.
Q is assumed to be a rate matrix which depends on the initial concentration of the unbound ligand
to be measured directly after titration:
Using the measured ITC experimental data, the binding rate
and the dissociation rate
can be estimated for each step of the titration [
41]. Usually, these rates are assumed to be constants. In real experiments, they systematically vary in each titration step.
These variations can be explained by the formula
. In some cases
Q can be calculated theoretically by the subspace projection. If we can model the micro-processes taking place in the reaction tube, e.g., by knowing the multi-step reaction kinetics network of the binding process, we can directly access the infinitesimal generator
of
in each step of the titration. Let us assume, e.g., a two step reaction. After binding of the ligand (micro-state 1 to micro-state 2), the ligand-receptor-complex undergoes a conformational change (micro-state 2 to micro-state 3). In this case:
with suitable reaction rates
which do not depend on
.
We can (analogously to Equation (
5)) compute the invariant subspace projection of
onto
Q by applying PCCA+ to the respective eigenvectors of the “operator”
. In each step of the titration, the infinitesimal generator
(i.e., its eigenvectors) depend on the initial concentration of the unbound ligands. Hence, the projection
Q, i.e., the rates
and
, depend on
as well. A parameter fitting of the multi-step reaction network can, thus, explain the systematic changes of the rates in
Q during the kinITC experiment, see
Figure 4.
3.4. Pericyclic Reactions
PCCA+ is usually applied to eigendecompositions of finite transition matrices of reversible Markov chains. Discrete approximations
of transfer operators
are estimated by generating a sample of molecular dynamics trajectories of length
and counting transitions between (pre-defined) subsets
M of
. From a mathematical point of view, the Markov kernel
in Equation (
2) is, thus, computed for only a few initial points
and a few disjoint sets
. Equation (
4) is only solved for those functions
f which are the characteristic functions of the given subsets. In molecular simulation, these approximations of
are denoted as Markov State Models generated by an analysis of short- or fragmented long-term molecular trajectories. A very good introduction into Markov State Modeling can be found in [
42]. Another method to construct transfer operators is called Dynamic Mode Decomposition which will be subject of
Section 3.5 below.
In
Section 3.3 it has already been indicated that the estimation of the infinitesimal generator
can be an alternative approach to receive the desired invariant subspace information
X without computing
explicitly. The square root approximation (SQRT-A) is a computational method for estimating a discrete version of
, i.e., a rate matrix
which represents a suitable approximation of the dynamics of the
micro-system [
43,
44]. The matrix
L is used to approximate the invariant subspace of
and to find the desired set of
membership functions for projection of the micro-process onto an
n-states macro-process.
Consider a decomposition of into m disjoint subsets and to be the stationary distribution of the Markov process restricted to these m subsets. SQRT-A defines the transition rate between neighboring subsets i and j as . All other off-diagonal elements of L are zero, because only between neighboring subsets instantaneous transitions are possible. The diagonal elements of L are constructed such that the row sums of L are zero, i.e., L becomes a non-dissipative rate matrix.
SQRT-A can easily be extended towards the case of a time-dependent density with . In this case the matrix L is generated in the already described way, but instead of only decomposing into m subsets the larger space is decomposed and the neighborhood relation is extended to consecutive time steps as well.
This procedure is especially very useful, if solely the time-dependent evolution of a density function
of states in
at time
t is known. In other words: Whenever the individual trajectory of each “particle” of that moving density is unknown we can only exploit the information about
. One example is given by an electron density
that is propagated via the time-dependent Schrödinger equation in the 3-dimensional space
, where the “movement” of every single electron is not known [
45].
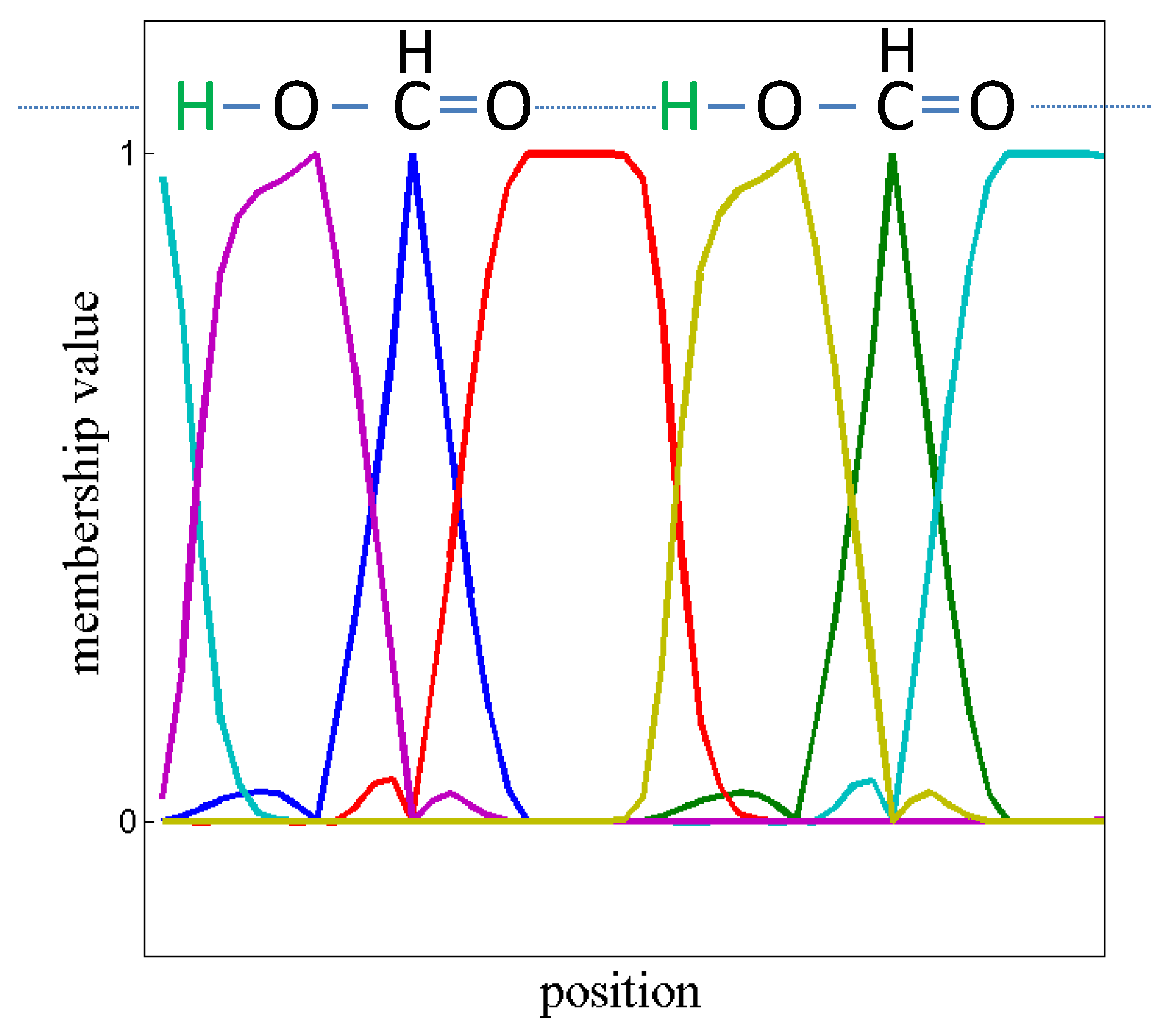
Whereas the SQRT-A matrix
L stemming from a time-independent density
represents a reversible Markov process by construction, in the time-dependent
-case the SQRT-A is usually non-reversible. Eigenvalues and eigenvectors may be complex-valued. Real invariant subspaces have to be constructed via Schur decomposition, i.e., for Schur values close to the value 0 (see also [
17]). After constructing an invariant subspace
X and after applying
, the membership functions are approximately spanning the kernel space of
L, i.e.,
. This means, that the “particles” which generate the time-dependent density will rarely cross the boundaries of the fuzzy sets
. This information can be used to reconstruct electron movement during pericyclic reactions (see chapter 4.3.7 in [
45] and
Figure 5 and
Figure 6).
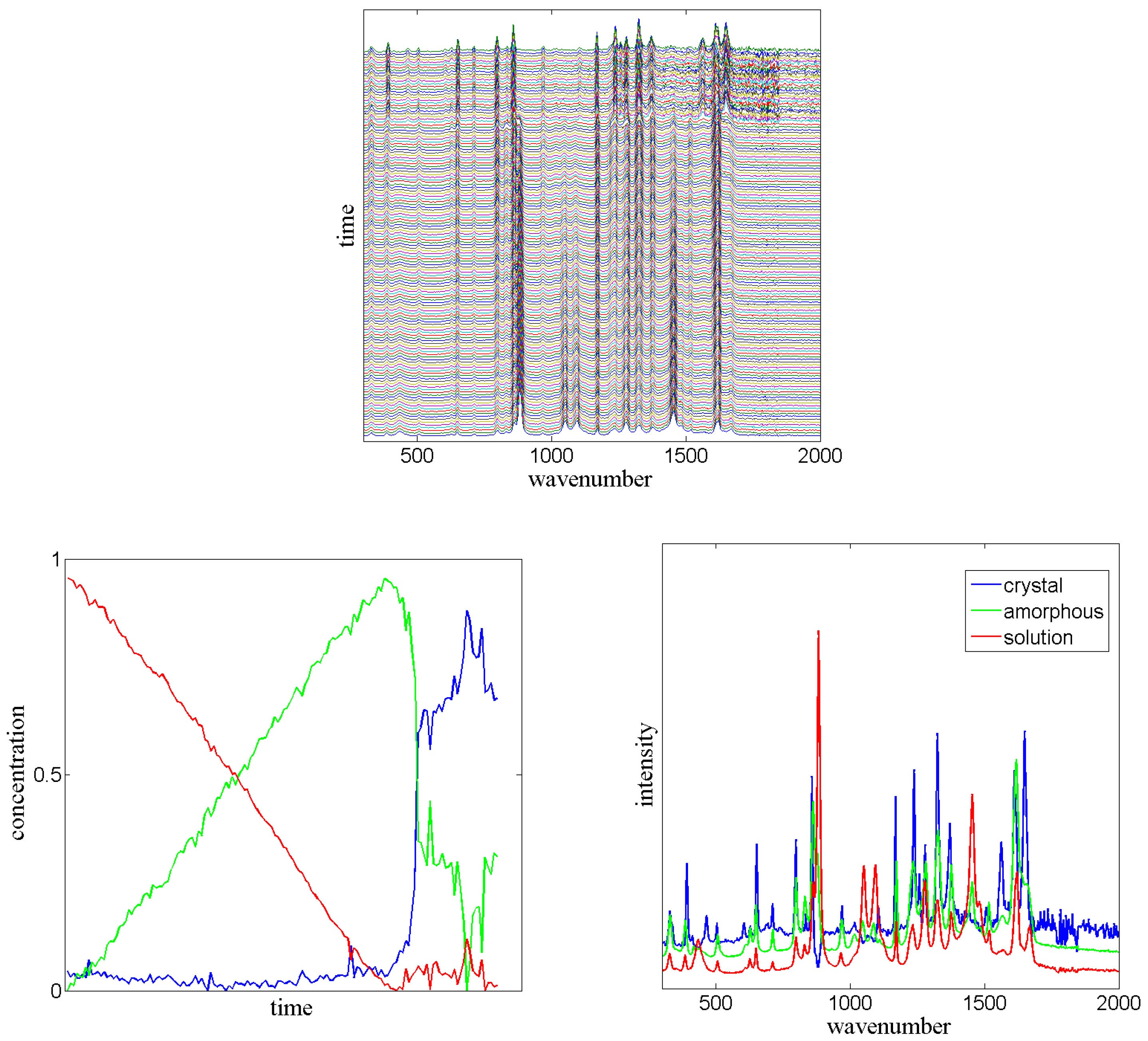
3.5. Sequential Spectroscopy
In the field of spectroscopy, several spectra (
d-dimensional non-negative vectors) are often measured sequentially in given time intervals. In most cases, closed molecular systems are observed, e.g., by sequential Raman spectroscopy. Each individual measured spectrum (also called “signal”) is the average of many ensemble signals of the components of the mixture. If we assume that each molecule (or atom, or electron) of the mixture can be assigned to a small number of
n macro-states, which can interconvert into each other by micro-processes, then the measured signal is always a convex combination of
n corresponding high-dimensional vectors
. Depending on the ratio (concentration) of components of the mixture in each of the
n states, the weights of this convex combination are changing over time [
46]. The time-dependent measured signal is:
where
is the convex combination factor of the signal
corresponding to state
i at time
t. “Convex combination” means that all these factors are non-negative and that the sum
is the unity. If we assume that the measurements are carried out at
m equally spaced points in time
with
for a constant interval
, then the convex combination factors can be arranged in a matrix
, which is a row stochastic matrix, i.e., it obeys Equation (
1). The measurements can be arranged in a matrix
. This matrix
V is usually non-negative due to the physics of spectroscopy. The
n “pure”
d-dimensional spectra
can be arranged in an
non-negative matrix
W.
The mathematical problem to be solved in sequential spectroscopy is to find the dimension n and the matrices and W given the matrix V, such that is a non-negative matrix factorization of V, whereas is a row-stochastic matrix.
PCCA+ can be used to solve this problem [
47]. Note that all convex combinations of
n vectors in a
d-dimensional space with
are included in an
-dimensional affine linear subspace of
. Following steps lead to the solution:
In order to transform the affine linear space into a linear subspace, one could for example subtract the mean vector (taking the mean over all measurements) from all rows of V. This leads to a normalized matrix , where is an m-dimensional constant vector, all elements are 1.
If the assumptions for solving the problem are correct, then should have rank . Together with a singular value decomposition , this can be used to determine n.
We know that the columns of the matrix span a linear space in which the vector is included. Thus, after determining n and , we construct the n-dimensional linear space by accounting for the first column vectors in . With the aid of Gram-Schmidt orthogonalization, this basis of a linear space can be transformed into an orthogonal basis with being the first basis vector. The n orthogonalized basis vectors are organized in a matrix . This matrix provides the linear space for the convex combination factors, i.e., there must be a linear transformation , such that .
Given the matrix X constructed from the spectral data, PCCA+ applied to X will provide a row-stochastic matrix which is the proposed matrix of concentrations. Thus, PCCA+ solves the problem.
However, due to errors in measurement and maybe due to deviations from the affine linear space, the matrix
constructed from the pseudo-inverse of
need not be non-negative. Röhm [
47] analyzed in his master’s thesis, whether it is useful to include an additional term (which measures the deviation of
W from a non-negative matrix) into the objective function
of PCCA+. It turned out, that this does not improve the results of
significantly.
Dynamic Mode Decomposition [
48] can help to improve the PCCA+ result, i.e., to provide a better transformation matrix
from a physical point of view. If the measured dynamical process takes place in a closed environment, then it is assumed to be autonomous. In this case, there should be an unknown stochastic transition matrix
, i.e., according to
Section 2 there should be a projected transfer operator for the
-discretized process. If
is the
i-th row of
, then
provides the concentration vector for the next step. Thus, we construct
which includes the first
rows of
and we construct
which includes the last
rows. For these matrices we get
. By using
, we get
, where
and
are constructed from
X analogously. Thus,
can directly be constructed from the measured data. In PCCA+,
must be searched in such a way that
is a stochastic matrix. This additional criterion improves the solution of the sequential spectroscopy problem, see
Figure 7.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






