
A Hybrid Computation Model to Describe the Progression of Multiple Myeloma and Its Intra-Clonal Heterogeneity


 and
and
Abstract
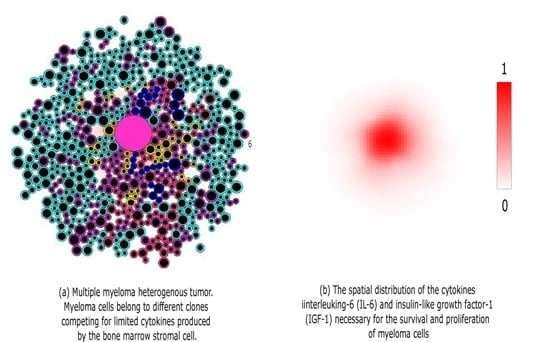
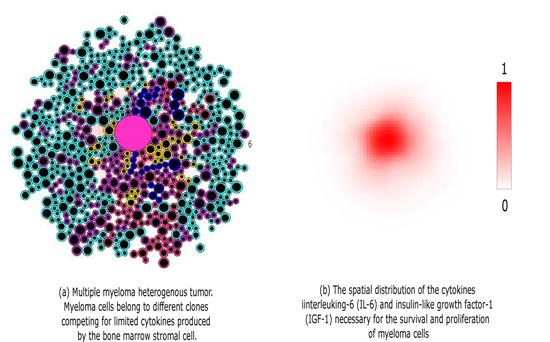
:
1. Introduction
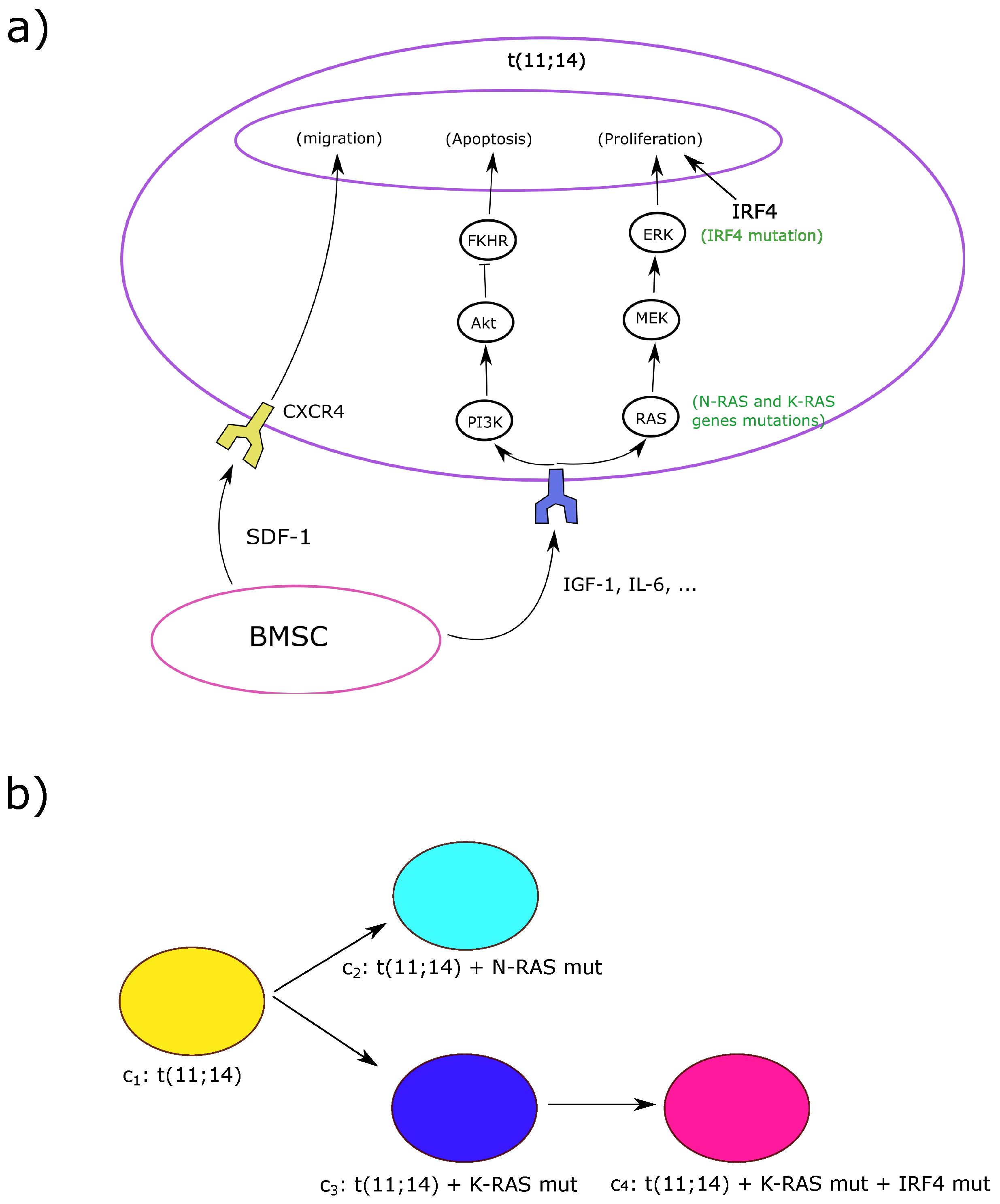
2. Hybrid Model of MM Development
2.1. Cell Motion
2.2. Extracellular Regulation
2.3. Intracellular Regulation
Myeloma Cells Division and Mutations
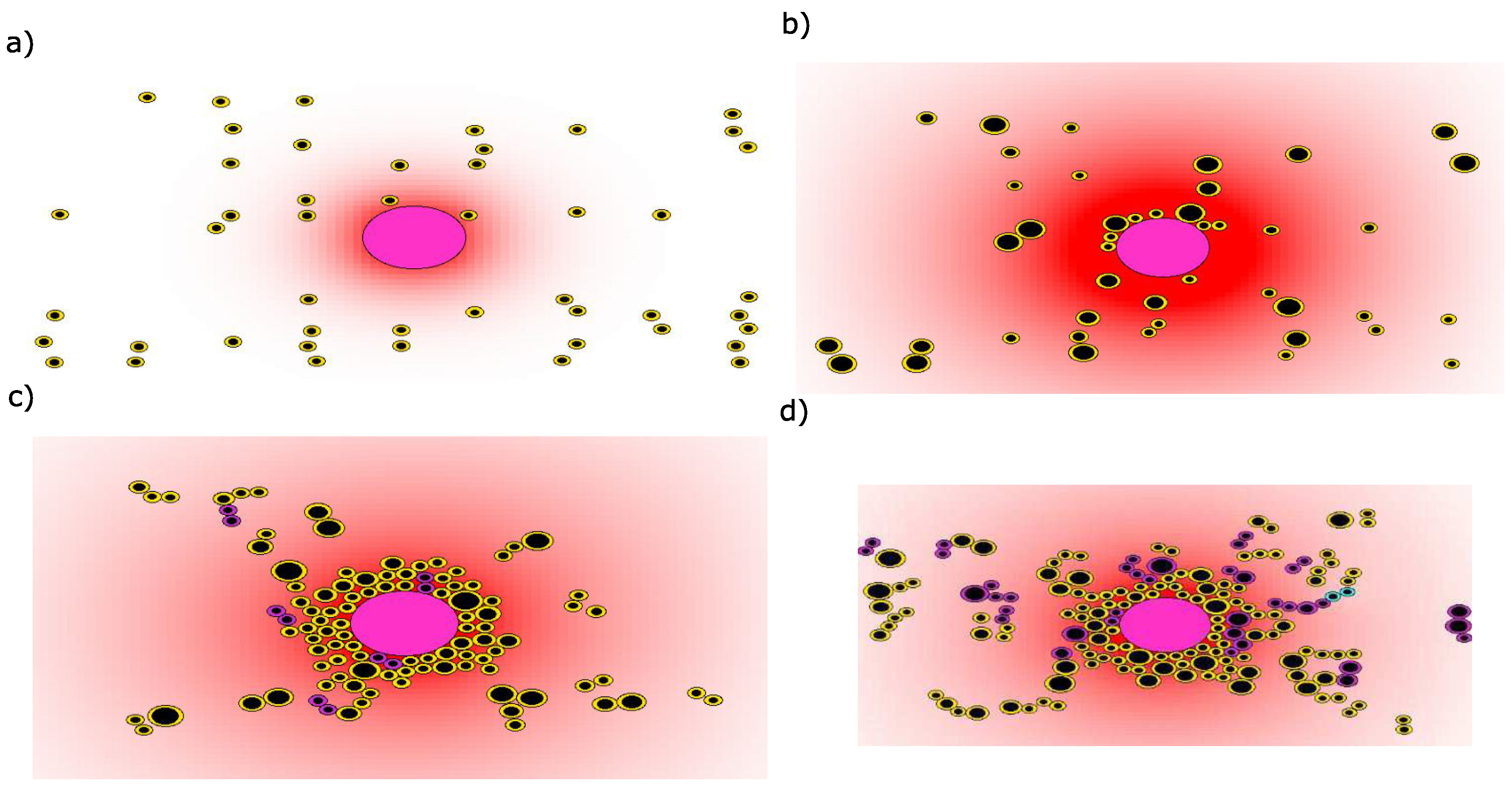
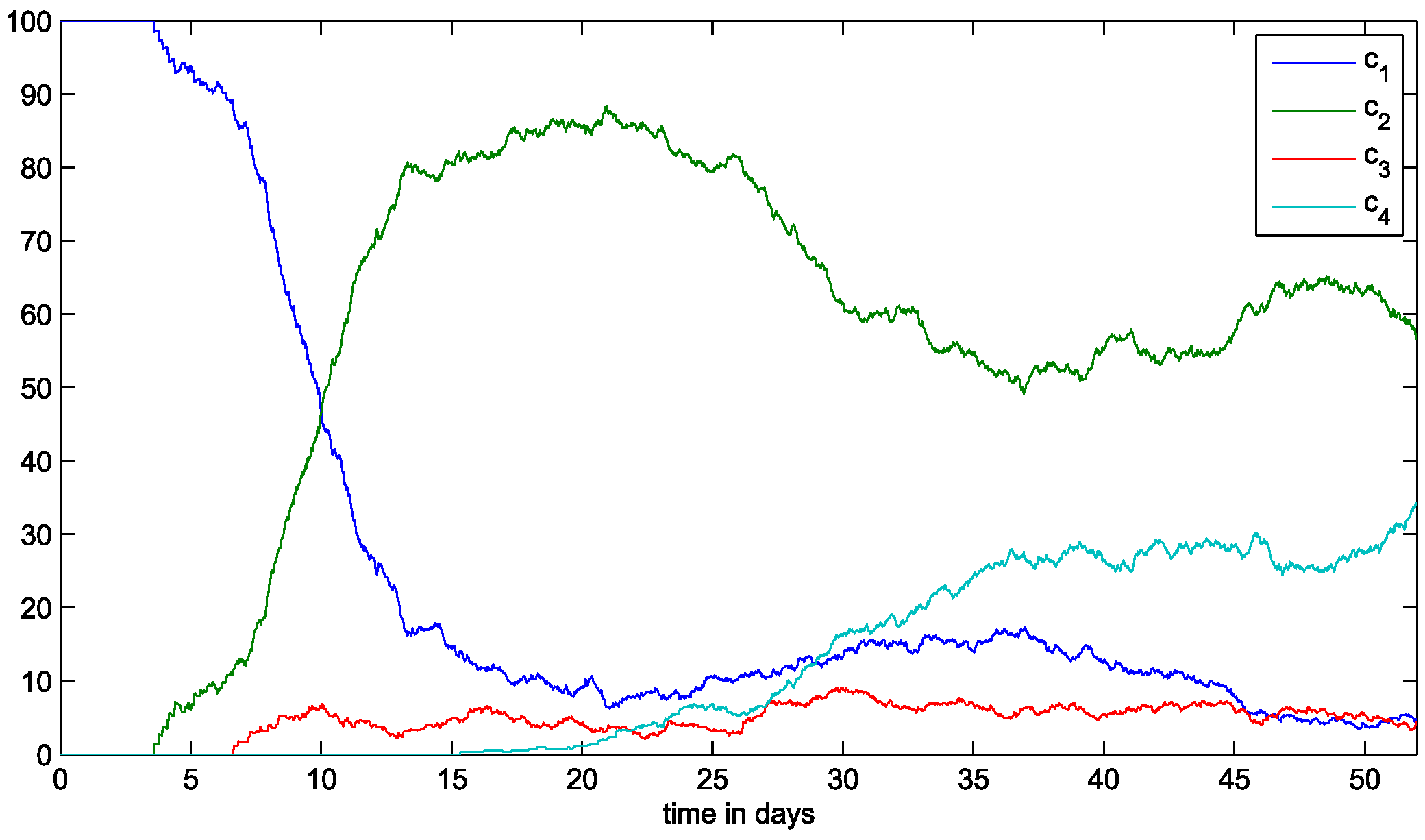
3. Results
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A. Numerical Implementation
Appendix A.1. Equations for Cell Motion and Intracellular Regulation
Appendix A.2. Reaction-Diffusion Equations for Extracellular Cytokines Concentration
Appendix B. Parameter Values

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value | Unit |
|---|---|---|
| Myeloma cell cycle length | 26 | h |
| Cell cycle variation | 13 | h |
| space variable and step | 1 | δ |
| time variable | 1 | min |
| time step | 0.01 | min |
| 0.001 | min | |
| 0.03 | min | |
| 0.002 | min | |
| 0.001 | min | |
| 0.01 | min | |
| 0.00083 | min |
| Parameter | Value | Unit |
|---|---|---|
| D | ·min | |
| W | 0.0003 | molecules··min |
| λ | 0.1 | NU |
| σ | min |
References
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Adam, J.A. A simplified mathematical model of tumor growth. Math. Biosci. 1986, 81, 229–244. [Google Scholar] [CrossRef]
- Glass, L. Instability and mitotic patterns in tissue growth. J. Dyn. Syst. Meas. Control 1973, 95, 324–327. [Google Scholar] [CrossRef]
- McElwain, D.L.S.; Morris, L.E. Apoptosis as a volume loss mechanism in mathematical models of solid tumor growth. Math. Biosci. 1978, 39, 147–157. [Google Scholar] [CrossRef]
- Byrne, H.M.; Chaplain, M.A.J. Growth of non-necrotic tumours in the presence and absence of inhibitors. Math. Biosci. 1995, 130, 151–181. [Google Scholar] [CrossRef]
- Byrne, H.M.; Chaplain, M.A.J. Growth of Necrotic Tumours in the Presence and Absence of Inhibitors. Math. Biosci. 1996, 135, 187–216. [Google Scholar] [CrossRef]
- Wise, S.M.; Lowengrub, J.S.; Frieboes, H.B.; Cristini, V. Three-dimensional multispecies nonlinear tumor growth—I: Model and numerical method. J. Theor. Biol. 2008, 253, 524–543. [Google Scholar] [CrossRef] [PubMed]
- Stiehl, T.; Baran, N.; Ho, A.D.; Marciniak-Czohra, A. Clonal selection and therapy resistance in acute leukaemias: Mathematical modelling explains different proliferation patterns at diagnosis and relapse. J. R. Soc. Interface 2014, 11, 20140079. [Google Scholar] [CrossRef] [PubMed]
- Walenda, T.; Stiehl, T.; Braun, H.; Fröbel, J.; Ho, A.D.; Schroeder, T.; Goecke, T.W.; Rath, B.; Germing, U.; Marciniak-Czohra, A.; et al. Feedback Signals in Myelodysplastic Syndromes: Increased Self-Renewal of the Malignant Clone Suppresses Normal Hematopoiesis. PLoS Comp. Biol. 2014, 10, e1003599. [Google Scholar] [CrossRef] [PubMed]
- Panetta, J.C. A mathematical model of drug resistance: Heterogeneous tumors. Math. Biosci. 1998, 147, 42–61. [Google Scholar] [CrossRef]
- Basanta, D.; Haralambos, H.; Deutsch, A. Studying the emergence of invasiveness in tumours using game theory. Eur. Phys. J. B 2008, 63, 393–397. [Google Scholar] [CrossRef]
- Enderling, H.; Hlatky, L.; Hahnfeldt, P. Migration rules: Tumours are conglomerates of self-metastases. Br. J. Cancer 2009, 100, 1917–1925. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, M.J.; Angus, S.D. A quantitative cellular automaton model of in vitro multicellular spheroid tumour growth. J. Theor. Biol. 2009, 258, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Drasdo, D.; Hoehme, S. A single-cell-based model of tumor growth in vitro: Monolayers and spheroids. Phys. Biol. 2005, 2, 133. [Google Scholar] [CrossRef] [PubMed]
- Shirinifard, A.; Gens, J.S.; Zaitlen, B.L.; Poplawski, N.J.; Swat, M.; Glazier, J.A. 3D Multi-Cell Simulation of Tumor Growth and Angiogenesis. PLoS ONE 2009, 4, e7190. [Google Scholar] [CrossRef] [PubMed]
- Swat, M.H.; Thomas, G.L.; Shirinifard, A.; Clandenon, S.G.; Glazier, J.A. Emergent Stratification in Solid Tumors Selects for Reduced Cohesion of Tumor Cells: A Multi-Cell, Virtual-Tissue Model of Tumor Evolution Using CompuCell3D. PLoS ONE 2015, 10, e0127972. [Google Scholar] [CrossRef] [PubMed]
- Hatzikirou, H.; Brusch, L.; Schaller, C.; Simon, M.; Deutsch, A. Prediction of traveling front behavior in a lattice-gas cellular automaton model for tumor invasion. Comput. Math. Appl. 2010, 59, 2326–2339. [Google Scholar] [CrossRef]
- Aubert, M.; Badoual, M.; Fereol, S.; Christov, C.; Grammaticos, B. A cellular automaton model for the migration of glioma cells. Phys. Biol. 2006, 3, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Ramis-Conde, I.; Drasdo, D.; Chaplain, M.A.J.; Anderson, A.R.A. Modeling the influence of the E-cadherin-β-catenin pathway in cancer cell invasion: A multiscale approach. Biophys. J. 2008, 95, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Ramis-Conde, I.; Chaplain, M.A.J.; Anderson, A.R.A.; Drasdo, D. Multi-scale modelling of cancer cell intravasation: The role of cadherins in metastasis. Phys. Biol. 2009, 6, 016008. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Z.; Sagotsky, J.A.; Deisboeck, T.S. Multiscale agent-based cancer modeling. J. Math. Biol. 2009, 58, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.R.A. A hybrid mathematical model of solid tumour invasion: the importance of cell adhesion. Math. Med. Biol. 2005, 22, 163–186. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.S.; Gillies, R.D.; Gatenby, R.A. Adaptation to hypoxia and acidosis in carcinogenesis and tumor progression. Semin. Cancer Biol. 2008, 18, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Vincent, T.L.; Gatenby, R.A. An evolutionary model for initiation, promotion, and progression in carcinogenesis. Int. J. Oncol. 2008, 32, 729–737. [Google Scholar] [PubMed]
- Chisholm, R.H.; Lorenzi, T.; Lorz, A.; Larsen, A.K.; de Almeida, L.N.; Escargueil, A.; Clairambault, J. Emergence of drug tolerance in cancer cell populations: An evolutionary outcome of selection, nongenetic instability, and stress-induced adaptation. Cancer Res. 2015, 75, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Ayati, B.P.; Edwards, C.M.; Webb, G.F.; Wikswo, J.P. A mathematical model of bone remodeling dynamics for normal bone cell populations and myeloma bone disease. Biol. Direct 2010, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Bouchnita, A.; Eymard, N.; Moyo, T.K.; Koury, M.J.; Volpert, V. Bone marrow infiltration by multiple myeloma causes anemia by reversible disruption of erythropoiesis. Am. J. Hematol. 2016, 91, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Wardell, C.P.; Melchor, L.; Hulkki, S.; Potter, N.E.; Johnson, D.C.; Fenwick, K.; Kozarewa, I.; Gonzalez, D.; Lord, C.J.; et al. Intraclonal heterogeneity and distinct molecular mechanisms characterize the development of t(4;14) and t(11;14) myeloma. Blood 2012, 120, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Bouchnita, A.; Belmaati, F.E.; Aboulaich, R.; Ellaia, R.; Volpert, V. Mathematical modelling of intra-clonal heterogeneity in multiple myeloma. In Proceedings of the CARI 2016, Hammamet, Tunisia, 11–14 October 2016.
- Brioli, A.; Melchor, L.; Cavo, M.; Morgan, G.J. The impact of intra-clonal heterogeneity on the treatment of multiple myeloma. Br. J. Haematol. 2014, 165, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Melchor, L.; Brioli, A.; Wardell, C.P.; Murison, A.; Potter, N.E.; Kaiser, M.F.; Fryer, R.A.; Johnson, D.C.; Begum, D.B.; Wilson, S.H.; et al. Single-cell genetic analysis reveals the composition of initiating clones and phylogenetic patterns of branching and parallel evolution in myeloma. Leukemia 2014, 28, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Chesi, M.; Bergsagel, P.L.; Brents, L.A.; Smith, C.M.; Gerhard, D.S.; Kuehl, W.M. Dysregulation of cyclin D1 by translocation into an IgH gamma switch region in two multiple myeloma cell lines. Blood 1996, 88, 674–681. [Google Scholar] [PubMed]
- Bouyssou, J.M.C.; Ghobrial, I.M.; Roccaro, A.M. Targeting SDF-1 in multiple myeloma tumor microenvironment. Cancer Lett. 2015, 380, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Vanderkerken, K.; van Camp, B.; de Greef, C.; Broek, I.V.; Asosingh, K.; van Riet, I. Homing of the myeloma cell clone. Acta Oncol. 2000, 39, 771–776. [Google Scholar] [PubMed]
- Hideshima, T.; Mitsiades, C.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 2007, 7, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Rozemuller, H.; van der Spek, E.; Bogers-Boer, L.H.; Zwart, M.C.; Verweij, V.; Emmelot, M.; Groen, R.W.; Spaapen, R.; Bloem, A.C.; Lokhorst, H.M.; et al. A bioluminescence imaging based in vivo model for preclinical testing of novel cellular immunotherapy strategies to improve the graft-versus-myeloma effect. Haematologica 2008, 93, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Manier, S.; Sacco, A.; Leleu, X.; Ghobrial, I.M.; Roccaro, A.M. Bone marrow microenvironment in multiple myeloma progression. BioMed Res. Int. 2012, 2012, 157496. [Google Scholar] [CrossRef] [PubMed]
- Kitano, H. Systems Biology: A brief overview. Science 2002, 295, 1662–1664. [Google Scholar] [CrossRef] [PubMed]







© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouchnita, A.; Belmaati, F.-E.; Aboulaich, R.; Koury, M.J.; Volpert, V. A Hybrid Computation Model to Describe the Progression of Multiple Myeloma and Its Intra-Clonal Heterogeneity. Computation 2017, 5, 16. https://doi.org/10.3390/computation5010016
Bouchnita A, Belmaati F-E, Aboulaich R, Koury MJ, Volpert V. A Hybrid Computation Model to Describe the Progression of Multiple Myeloma and Its Intra-Clonal Heterogeneity. Computation. 2017; 5(1):16. https://doi.org/10.3390/computation5010016
Chicago/Turabian StyleBouchnita, Anass, Fatima-Ezzahra Belmaati, Rajae Aboulaich, Mark J. Koury, and Vitaly Volpert. 2017. "A Hybrid Computation Model to Describe the Progression of Multiple Myeloma and Its Intra-Clonal Heterogeneity" Computation 5, no. 1: 16. https://doi.org/10.3390/computation5010016
APA StyleBouchnita, A., Belmaati, F.-E., Aboulaich, R., Koury, M. J., & Volpert, V. (2017). A Hybrid Computation Model to Describe the Progression of Multiple Myeloma and Its Intra-Clonal Heterogeneity. Computation, 5(1), 16. https://doi.org/10.3390/computation5010016