
A Multi-Compartment Hybrid Computational Model Predicts Key Roles for Dendritic Cells in Tuberculosis Infection


Abstract
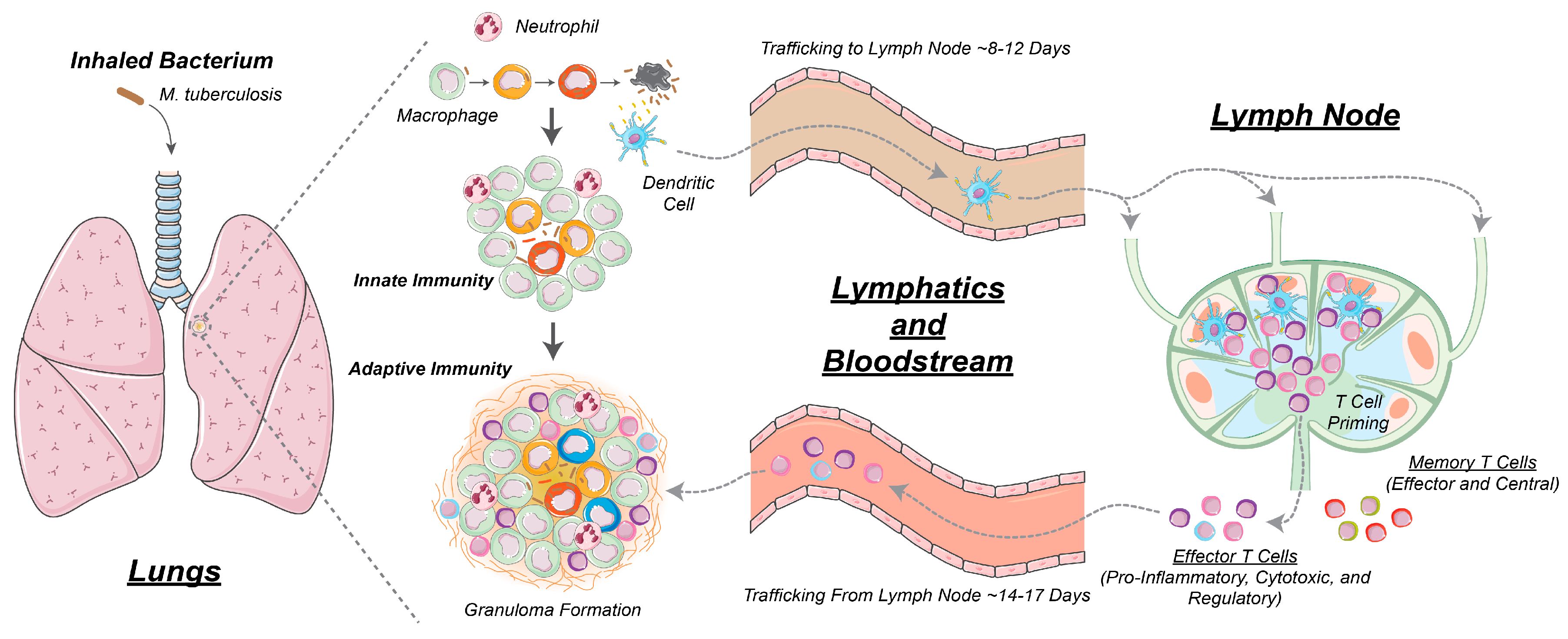
:1. Introduction
2. Materials and Methods
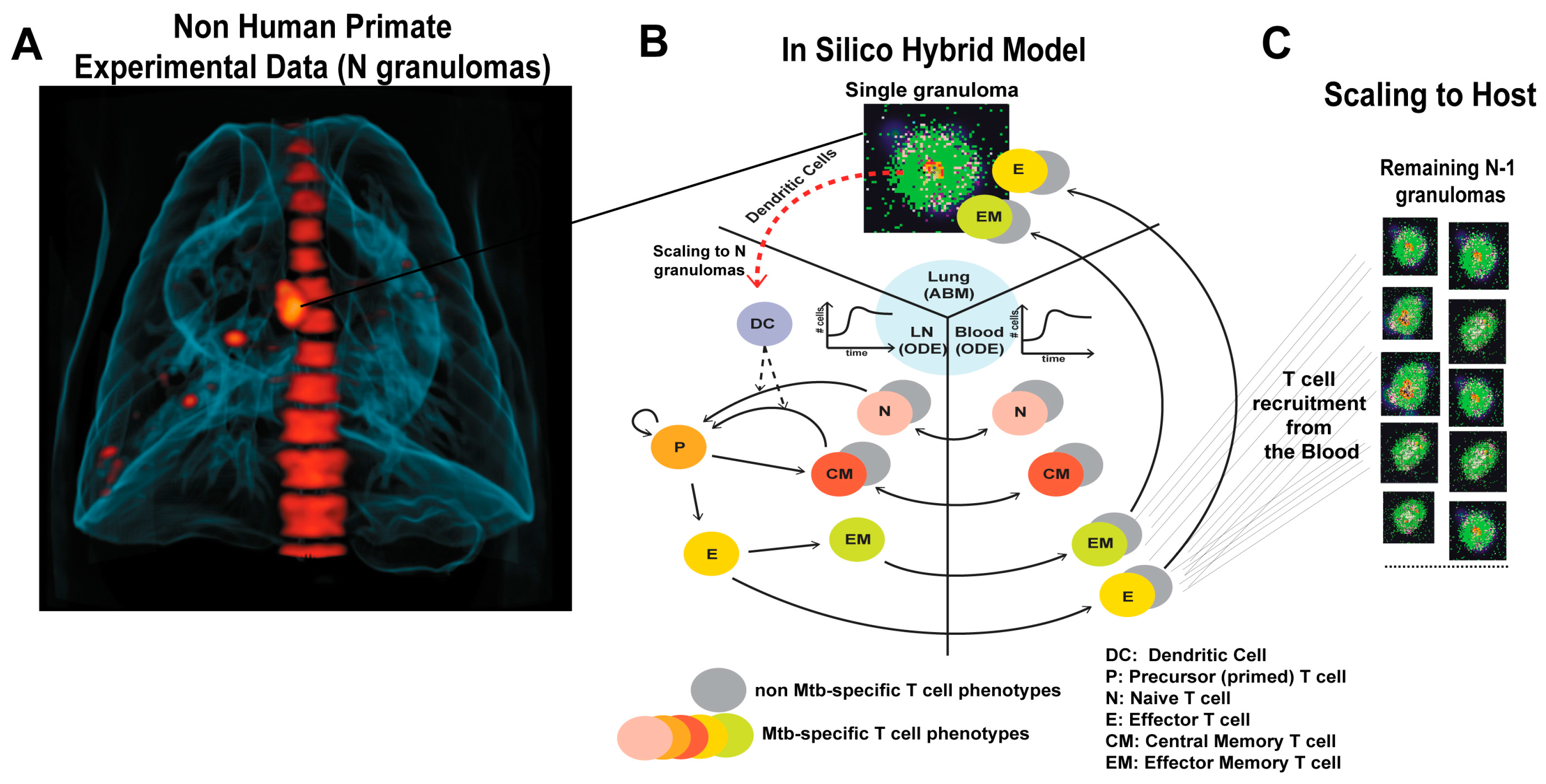
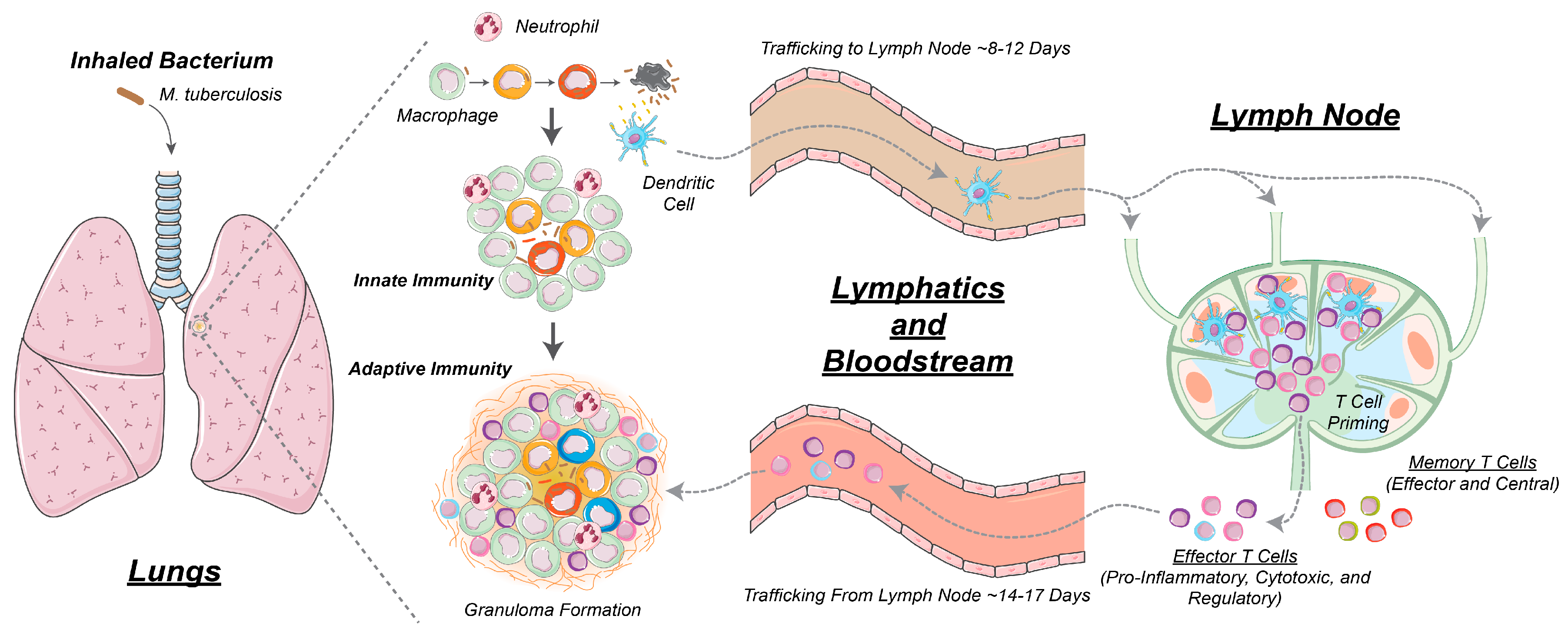
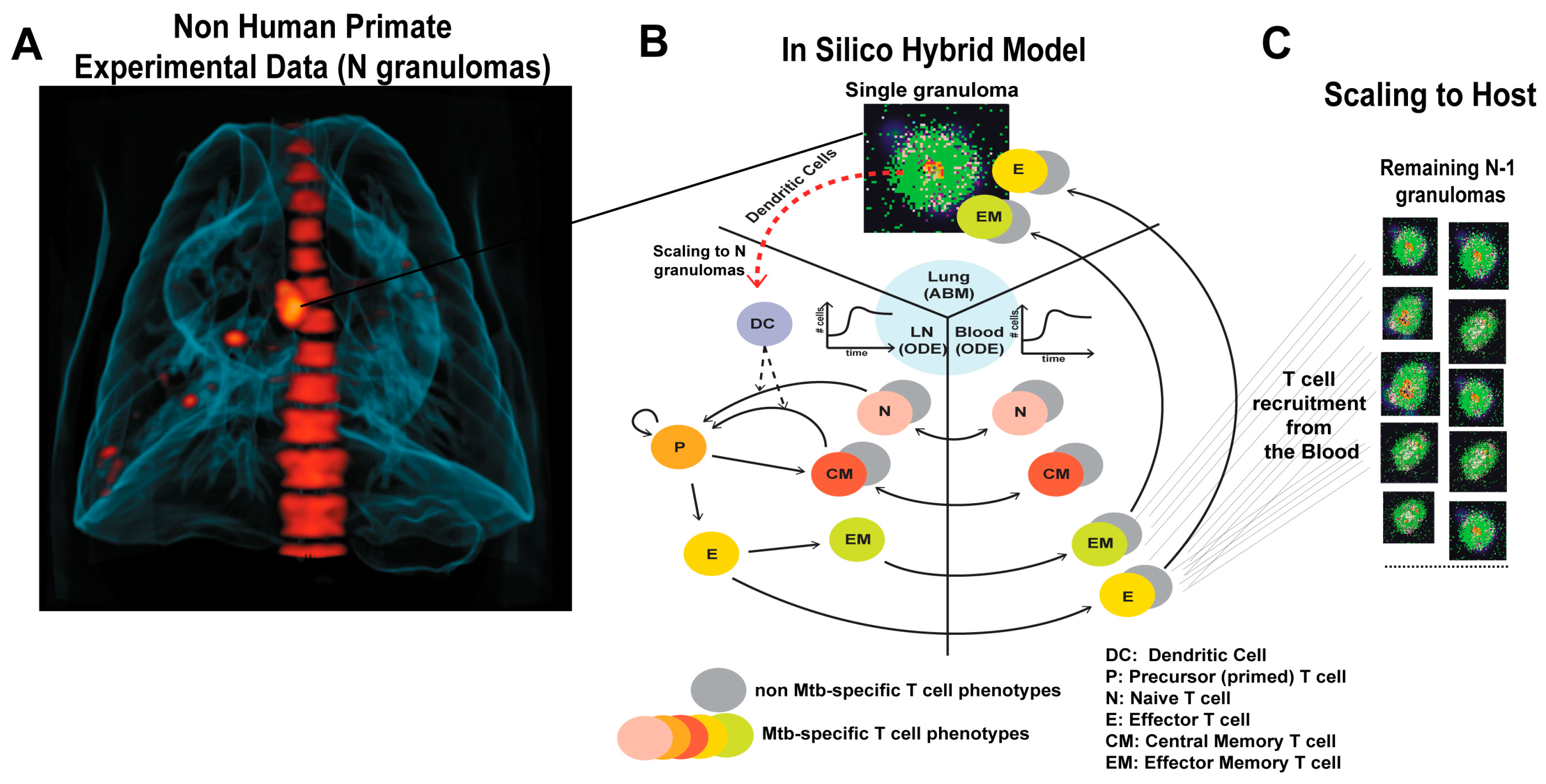
2.1. GranSim: Computational Model of Granuloma Formation and Function in the Lung
2.2. Multi-Compartment Gransim: Tracking Cell Dynamics in the Lymph Node and Blood
2.3. Adding DCs to GranSim
2.4. Scaling to Host Feature
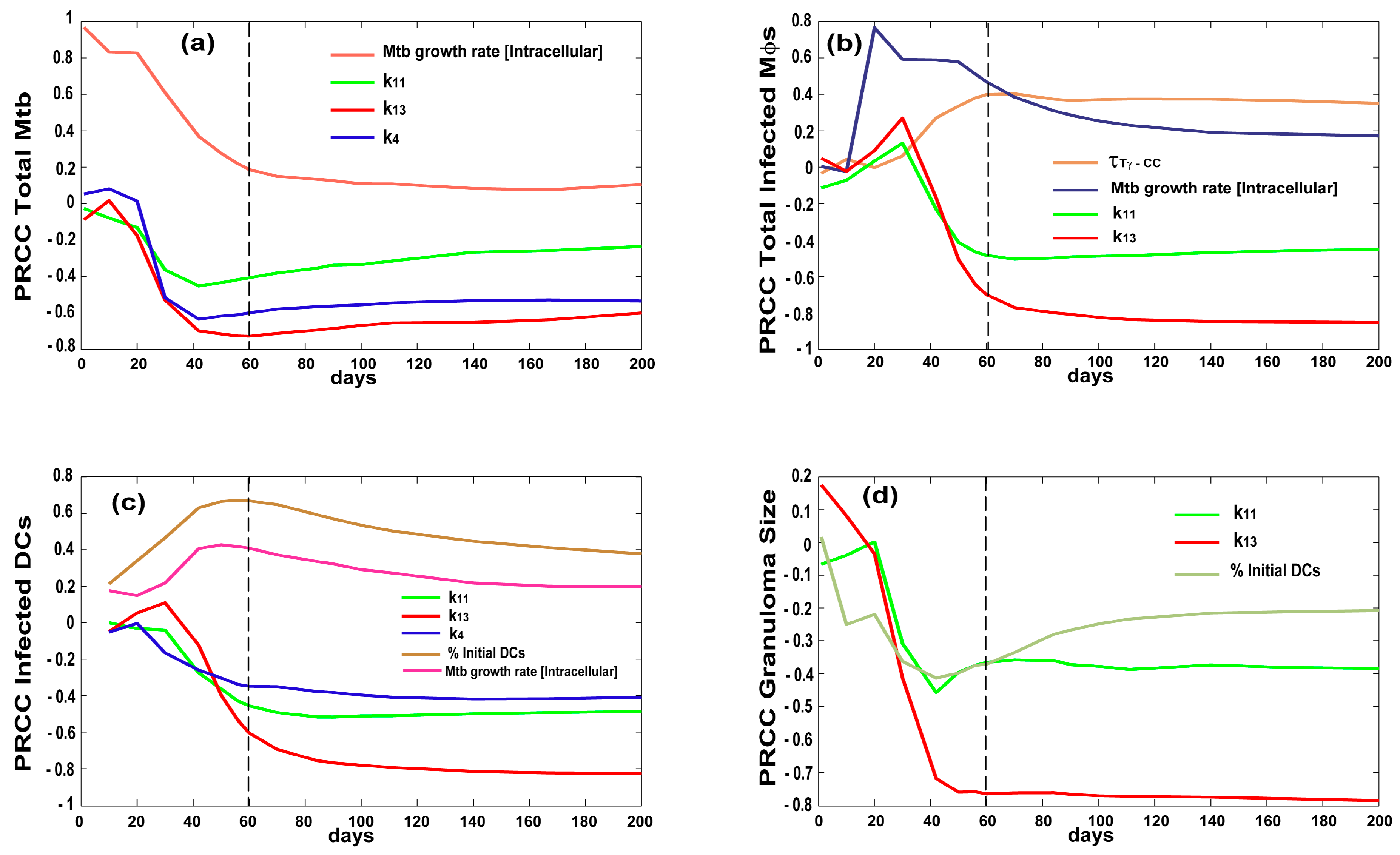
2.5. Uncertainty and Sensitivity Analysis
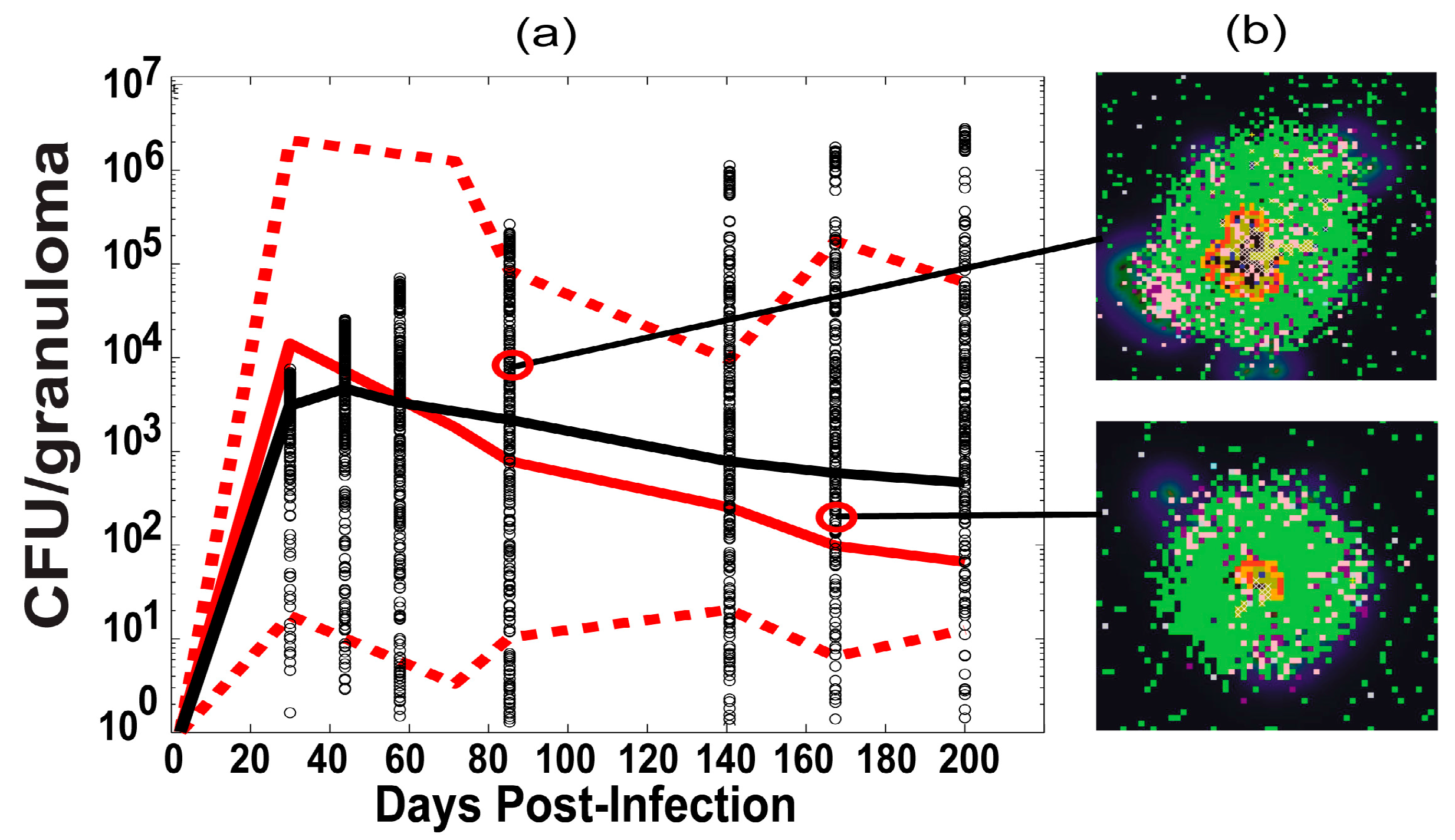
2.6. Experimental Data: Non Human Primate Lung and Blood Data
3. Results
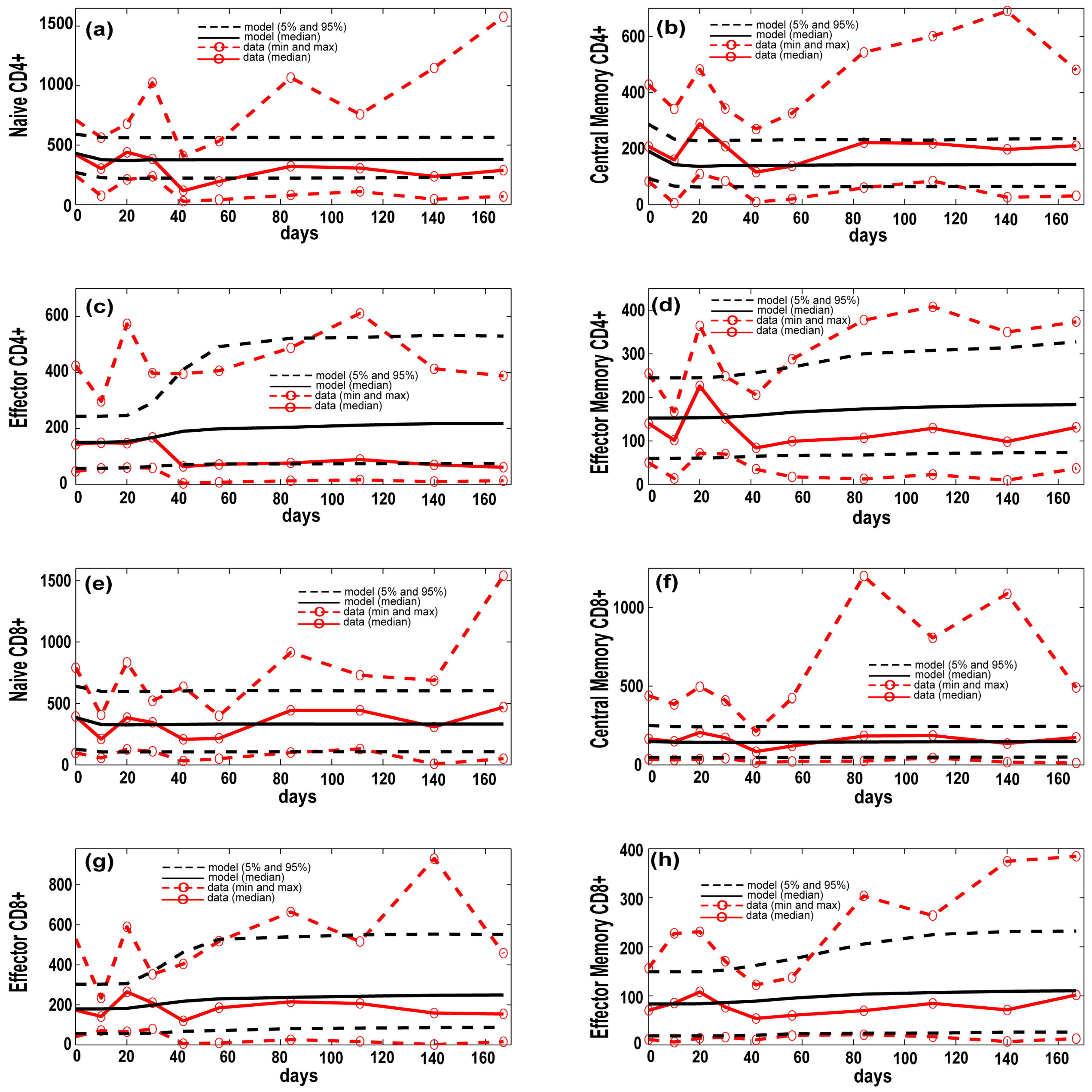
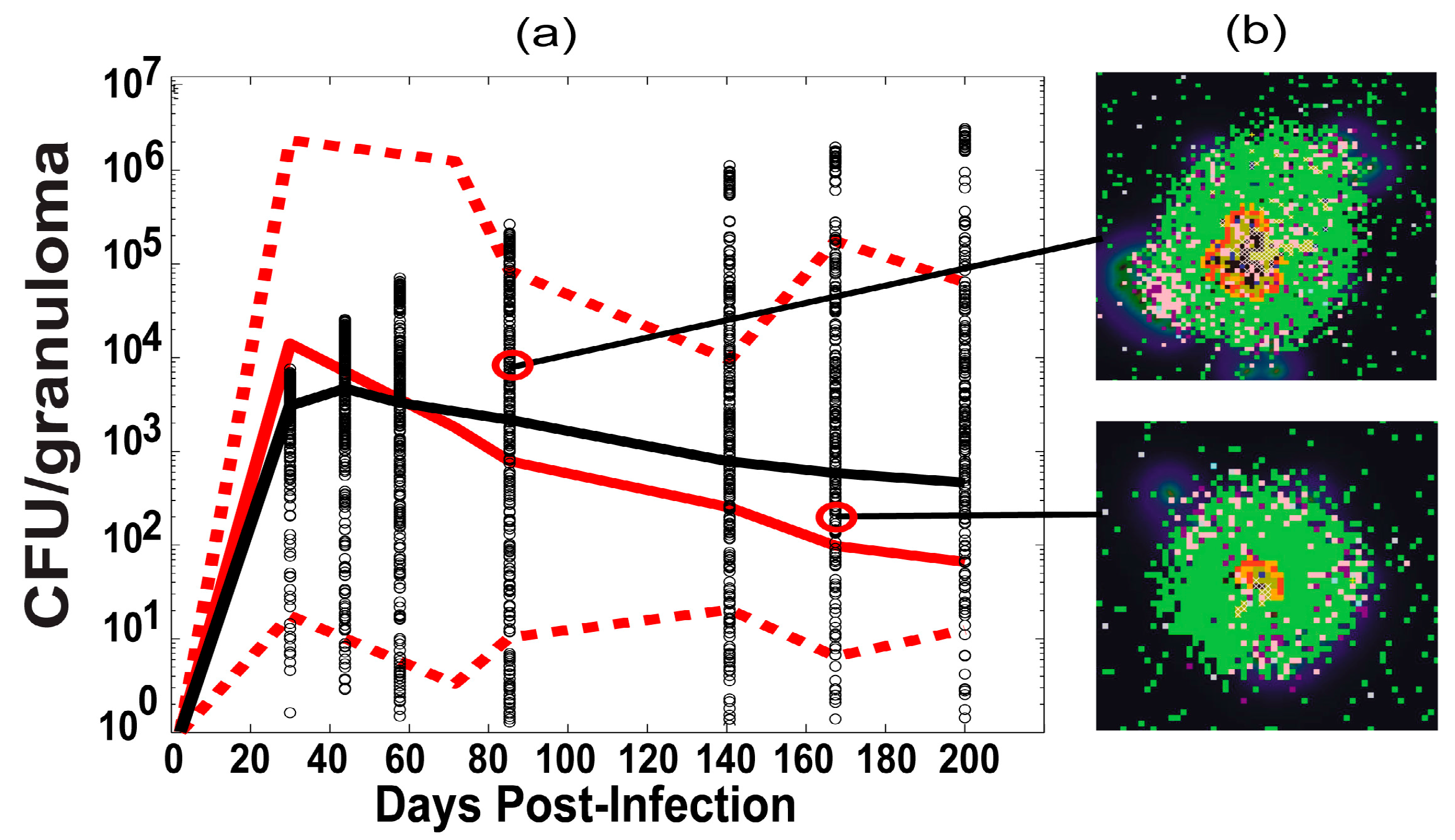
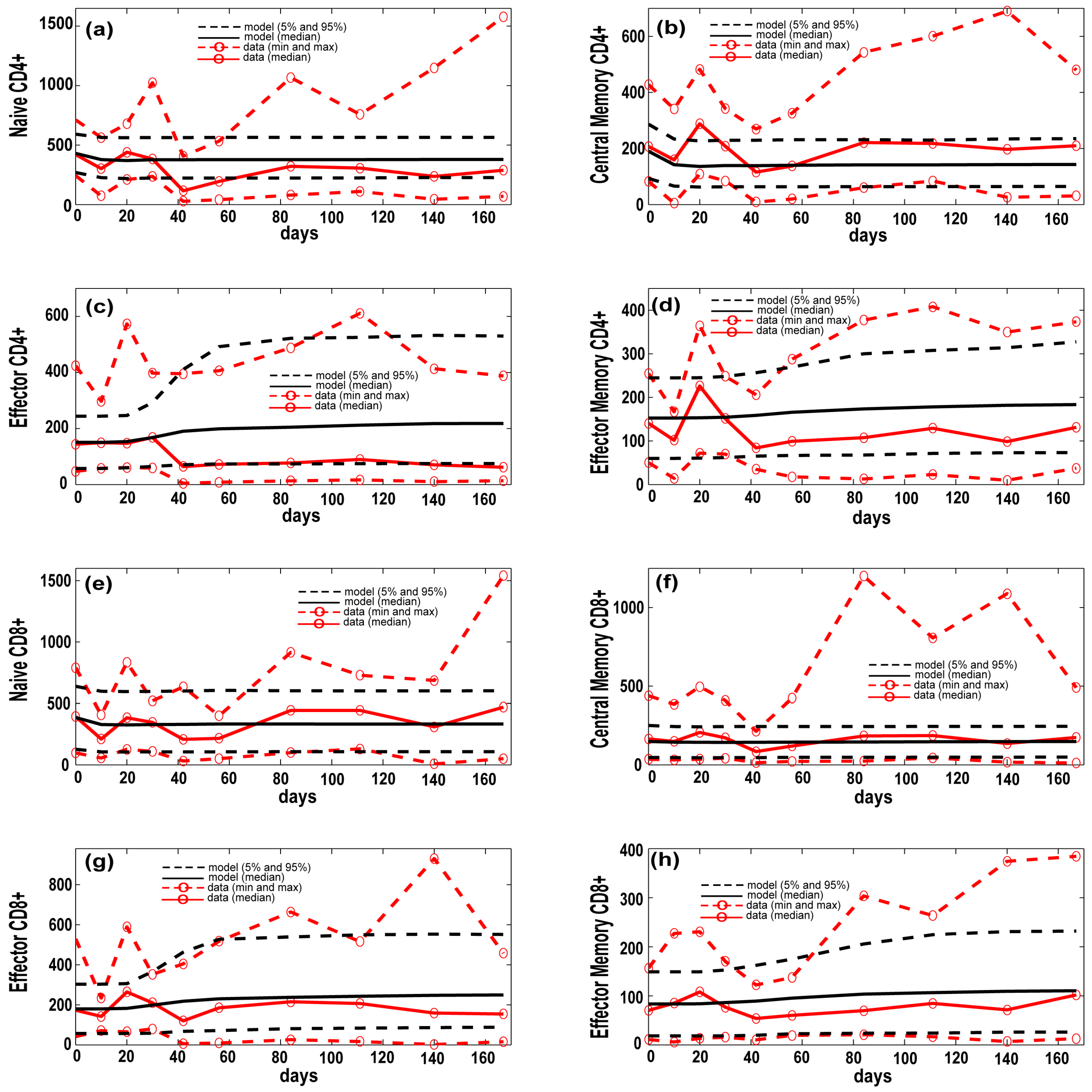
3.1. Model Calibration—Lung and Blood Compartments
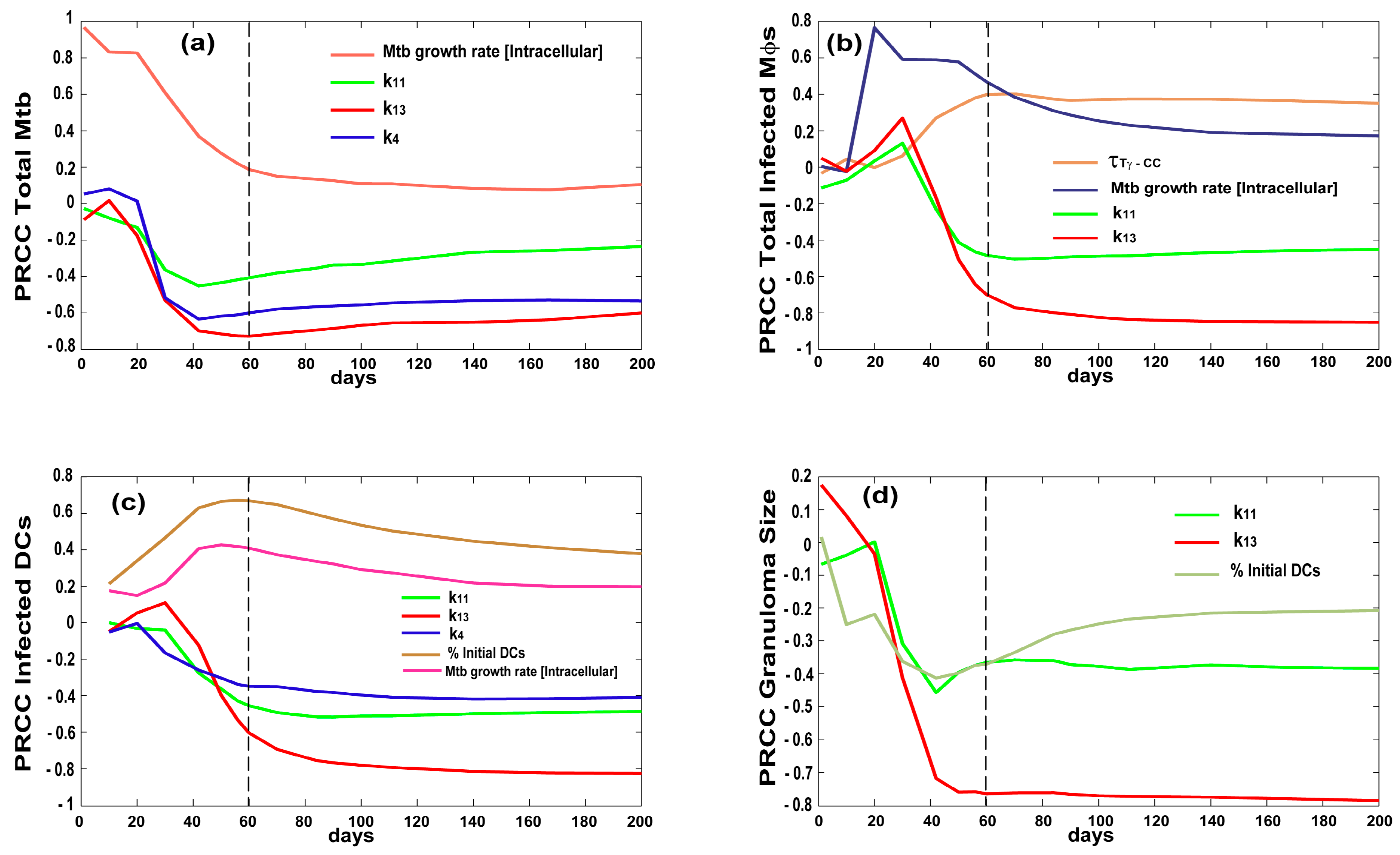
3.2. Bacterial, CD4 and CD8 Proliferation Impact Infection Burden at the Granuloma Site
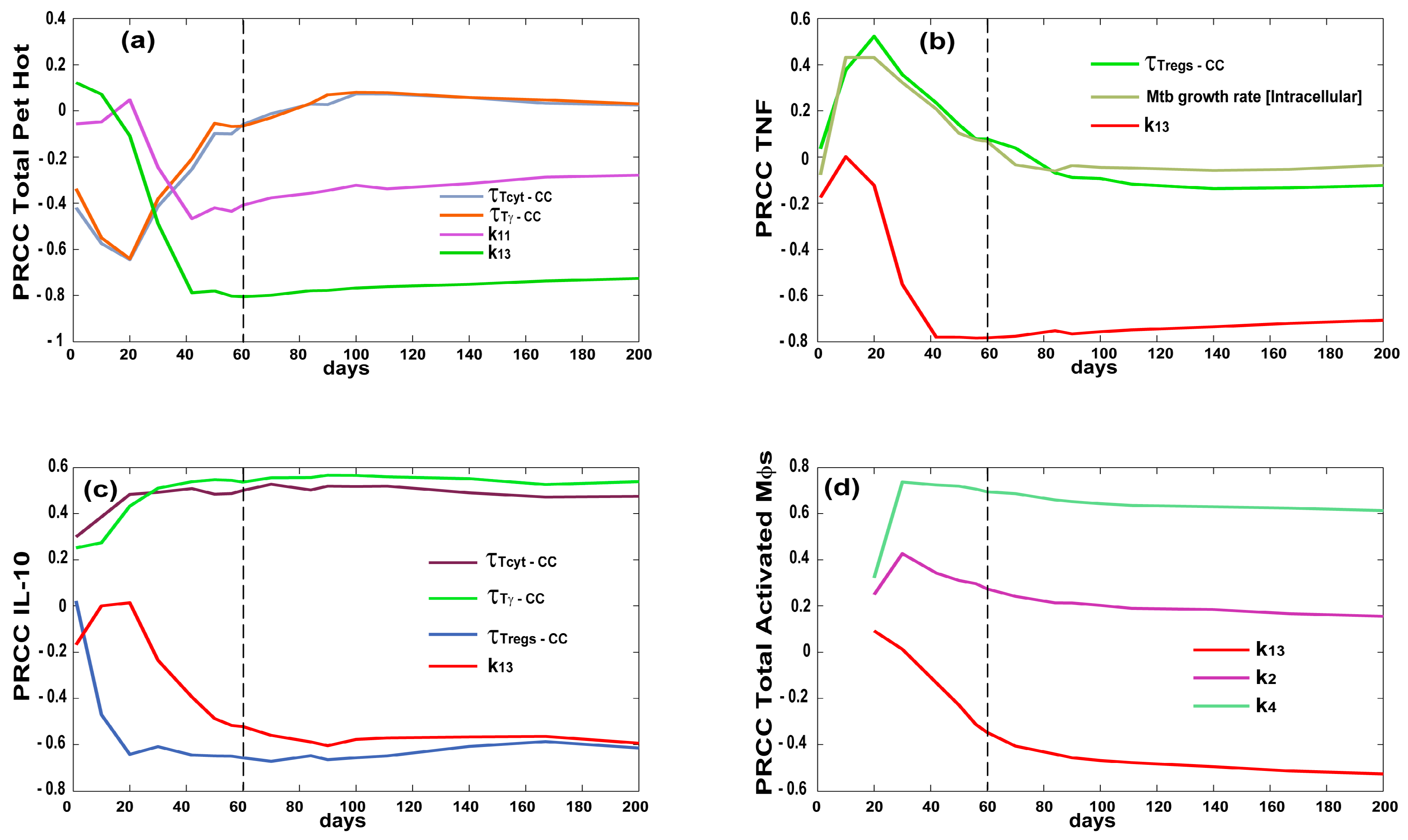
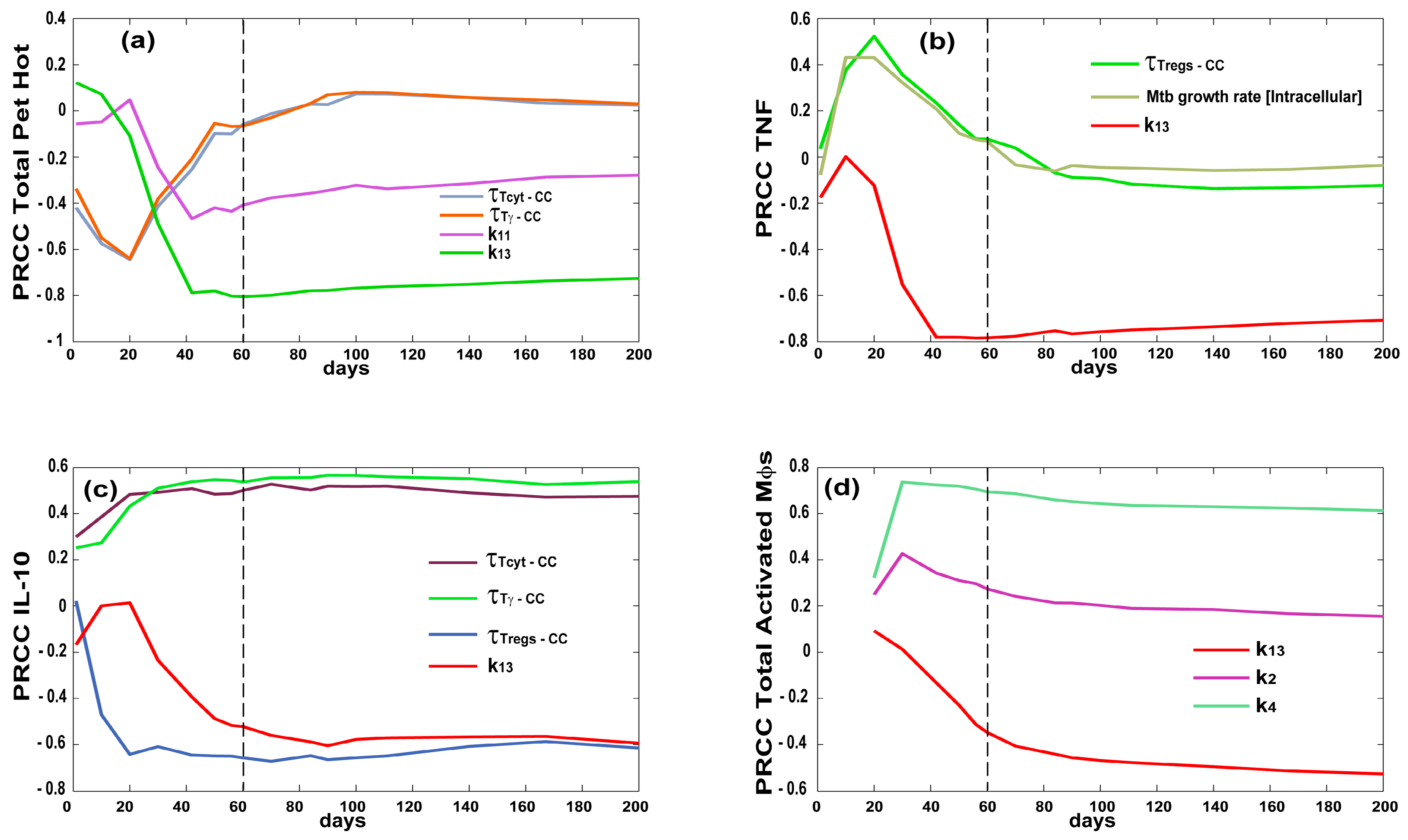
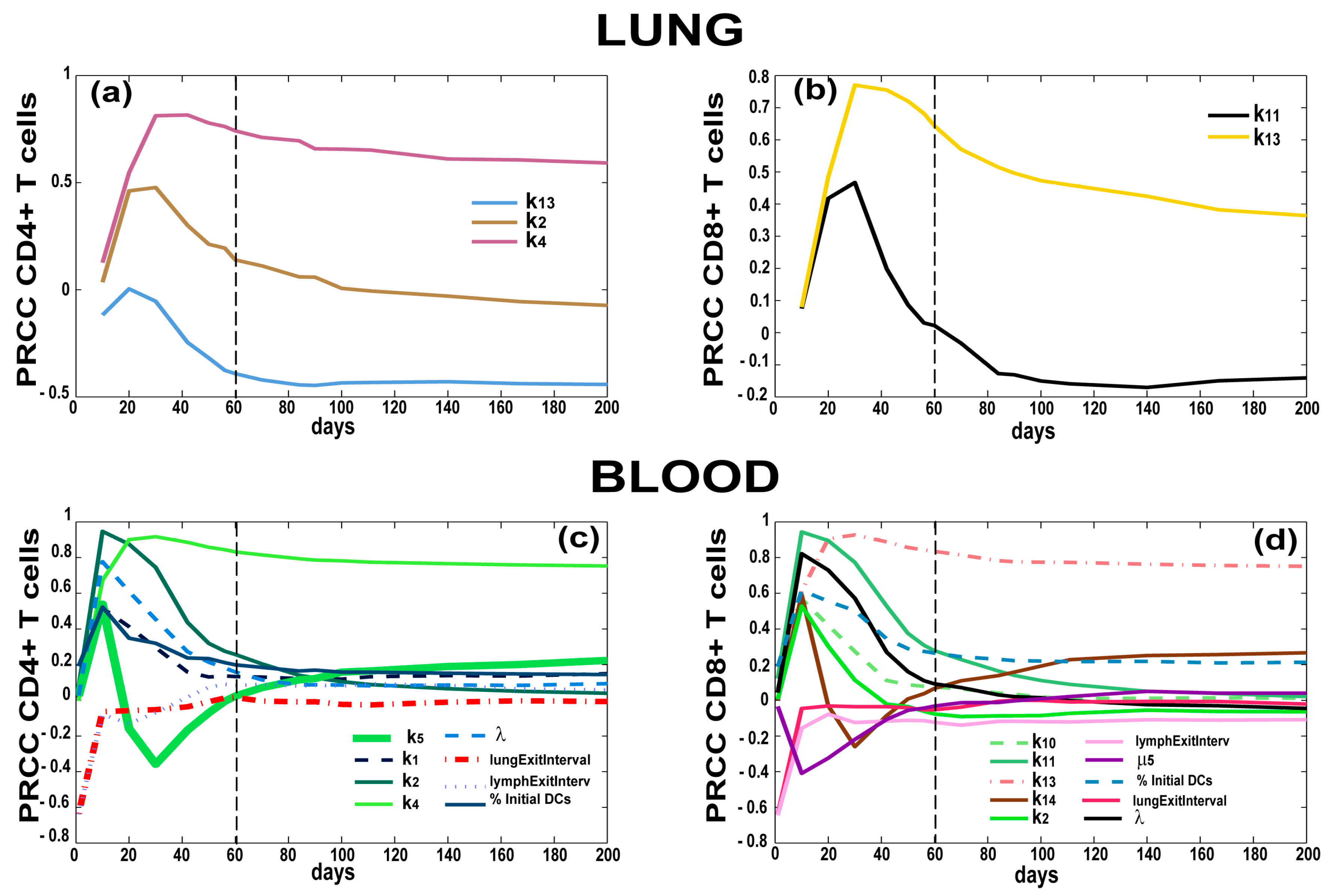
3.3. Priming and Proliferation in the LN Drives Inflammation at the Site of Infection
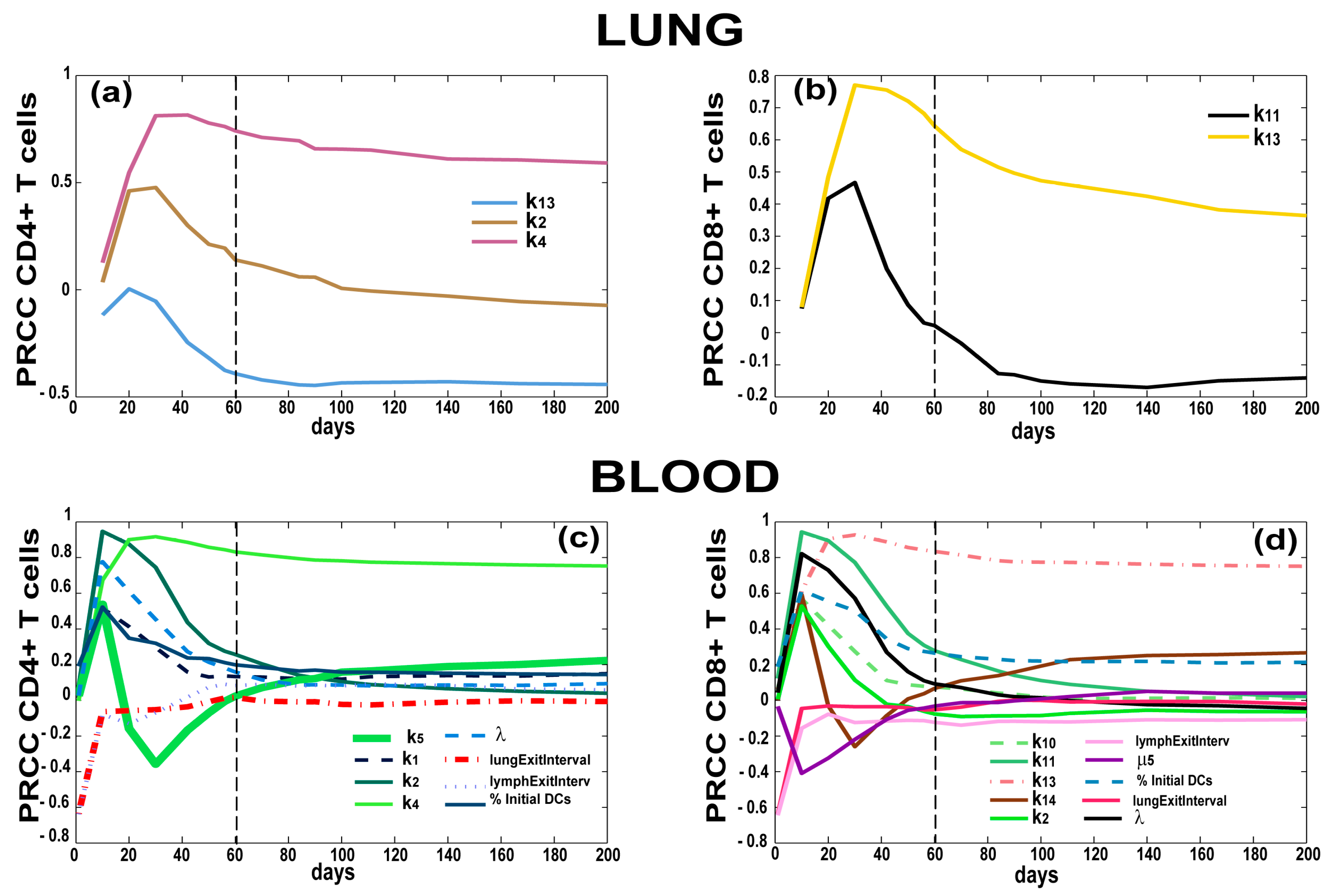
3.4. T Cell Priming, Proliferation and Trafficking Determine the Timing and Magnitude of the Immune Response at the Site of Infection and in the Blood
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| TB | tuberculosis |
| Mtb | Mycobacterium tuberculosis |
| LN | lymph node |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value | Units | Description | Reference |
|---|---|---|---|---|
| α | 5.6 × 105 | µL | Conversion factor from Blood to Ln (max. blood volume) | Estimated and [36] |
| host_Ln | [1, 50] | count | Number of involved lymph nodes in the host | Estimated |
| λ | [10−5, 10−3] | “” | Frequency of Mycobacterium tuberculosis (Mtb)-specific Naïve T cells in the blood/LN | [75,76,77] |
| scalingMDC | [5, 15] | Count | Scaling to host factor representing the number of granulomas developing in the whole lung at time of infection | [52] |
| Sn4 * | NLN,4 × (α/host_Ln) | Cell/µL * day | Thymic output of Naïve CD4+ T cells | Estimated from Uncertainty Analysis |
| Sn8 * | NLN,8 × (α/host_Ln) | Cell/µL * day | Thymic output of Naïve CD8+ T cells | Estimated from Uncertainty Analysis |
| hs1 | 25 | Cell count | Naïve CD4+ T cell recruitment half saturation | Estimated from Uncertainty Analysis |
| hs4 | 10 | Cell count | Precursor CD4+ T cell proliferation half saturation | Estimated from Uncertainty Analysis |
| hs5 | 10 | Cell count | Precursor CD4+ T cell differentiation half saturation | Estimated from Uncertainty Analysis |
| hs8 | 40 | Cell count | Central Memory CD4+ T cell recruitment half saturation | Estimated from Uncertainty Analysis |
| hs10 | 25 | Cell count | Naïve CD8+ T cell recruitment half saturation | Estimated from Uncertainty Analysis |
| hs11 | 10 | Cell count | Naïve CD8+ T cell priming half saturation | Estimated from Uncertainty Analysis |
| hs13 | 10 | Cell count | Precursor CD8+ T cell proliferation half saturation | Estimated from Uncertainty Analysis |
| hs14 | 10 | Cell count | Precursor CD8+ T cell differentiation half saturation | Estimated from Uncertainty Analysis |
| hs17 | 157 | Cell count | Central Memory CD8+ T cell recruitment half saturation | Estimated from Uncertainty Analysis |
| k1 | [5 × 10−3, 1] | day−1 | Naïve CD4+ T cell recruitment rate | Estimated from Uncertainty Analysis |
| k2 | [10−6, 10−1] | day−1 | Naïve CD4+ T cell Priming rate | Estimated from Uncertainty Analysis |
| k3 | [10−7, 10−2] | day−1 | Central Memory CD4+ T cell reactivation rate | Estimated from Uncertainty Analysis |
| k4 | [10−2, 1.2] | day−1 | Precursor CD4+ T cell proliferation rate | Estimated from Uncertainty Analysis |
| k5 | [0.01, 0.75] | day−1 | Precursor CD4+ T cell differentiation to Effector rate | Estimated from Uncertainty Analysis |
| k6 | 0.001 | day−1 | Precursor CD4+ T cell differentiation to Central Memory | Estimated from Uncertainty Analysis |
| k7 | [0.05, 0.75] | day−1 | Effector CD4+ T cell differentiation to Effector Memory | Estimated from Uncertainty Analysis |
| k8 | [0.1, 0.5] | day−1 | Central Memory CD4+ T cell recruitment rate | Estimated from Uncertainty Analysis |
| k10 | [5 × 10−3, 1] | day−1 | Naïve CD8+ T recruitment cell rate | Estimated from Uncertainty Analysis |
| k11 | [10−6, 10−1] | day−1 | Naïve CD8+ T cell priming rate | Estimated from Uncertainty Analysis |
| k12 | [10−7, 10−2] | day−1 | Central Memory CD8+ T cell reactivation rate | Estimated from Uncertainty Analysis |
| k13 | [10−2, 1.2] | day−1 | Precursor CD8+ T cell proliferation rate | Estimated from Uncertainty Analysis |
| k14 | [0.01, 0.75] | day−1 | Precursor CD8+ T cell differentiation to Effector rate | Estimated from Uncertainty Analysis |
| k15 | 0.001 | day−1 | Precursor CD8+ T cell differentiation to Central Memory | Estimated from Uncertainty Analysis |
| k16 | [0.05, 0.75] | day−1 | Effector CD8+ T cell differentiation to Effector Memory | Estimated from Uncertainty Analysis |
| k17 | [0.05, 0.75] | day−1 | Central Memory CD8+ T cell recruitment rate | Estimated from Uncertainty Analysis |
| µ1 | 0.2 | day−1 | Effector CD4+ T cell death rate | [19,20,22,23,36] |
| µ2 | 0.04 | day−1 | Effector Memory CD4+ T cell death rate | [19,20,22,23,36] |
| µ3 | 0.2 | day−1 | Effector CD8+ T cell death rate | [19,20,22,23,36] |
| µ4 | 0.04 | day−1 | Effector Memory CD8+ T cell death rate | [19,20,22,23,36] |
| µ5 | [0.1, 1] | day−1 | APC death rate | [19,20,22,23,36] |
| µ6 | 0.1 | day−1 | Precursor CD4+ T cell death rate | [19,20,22,23,36] |
| µ7 | 0.1 | day−1 | Precursor CD8+ T cell death rate | [19,20,22,23,36] |
| µ8 * | 3.93 × 10−4 | day−1 | Naïve CD4+ T cell death rate | |
| µ9 * | 2.27 × 10−4 | day−1 | Naïve CD8+ T cell death rate | |
| ρ1 | 3 × 108 | Cell count | Precursor carrying capacity | [19,20,22,23,36] |
| Wp4 | 0.735 | “” | Weight factor for Precursor CD4+ T in CD8+ T cell priming | Estimated from Uncertainty Analysis |
| ξ1 * | ξ2 × (NLn,nc4/NB,nc4)/α | day−1 | Naïve CD4 Lymph Influx | |
| ξ2 | [0.6, 1] | day−1 | Naïve CD4 Lymph Efflux | Estimated from Uncertainty Analysis |
| ξ3 | [2, 5] | day−1 | Effector CD4 Lymph Efflux | Estimated from Uncertainty Analysis |
| ξ4 * | ξ5 × (CMLn,nc4/CMB,nc4)/α | day−1 | Central Memory CD4 Lymph Influx | |
| ξ5 | 0.489 | day−1 | Central Memory CD4 Lymph Efflux | Estimated from Uncertainty Analysis |
| ξ6 | [2, 5] | day−1 | Effector Memory CD4 Lymph Efflux | Estimated from Uncertainty Analysis |
| ξ7 * | ξ8 × (NLn,nc8/NB,nc8)/α | day−1 | Naïve CD8 Lymph Influx | |
| ξ8 | [0.6, 1] | day−1 | Naïve CD8 Lymph Efflux | Estimated from Uncertainty Analysis |
| ξ9 | [2, 5] | day−1 | Effector CD8 Lymph Efflux | Estimated from Uncertainty Analysis |
| ξ10 * | ξ11 × (CMLn,nc8/CMB,nc8)/α | day−1 | Effector CD8 Lymph Influx | |
| ξ11 | [2, 5] | day−1 | Central Memory CD8 Lymph Efflux | Estimated from Uncertainty Analysis |
| ξ12 | [2, 5] | day−1 | Effector Memory CD8 Lymph Efflux | Estimated from Uncertainty Analysis |
| proliferationTime | 8 | h | Doubling time for cognate T cells in the lung | [40,71] |
| maxDivisions | 4 | - - | Max number of divisions for T cells in the lung | [40,71] |
| τTγ−CC | [1, 20] | # molecules | Chemokine threshold for Tγ recruitment | Estimated from Uncertainty Analysis |
| τTγ−TNF | [1, 5] | # molecules | Tumor necrosis factor (TNF) threshold for Tγ recruitment | Estimated from Uncertainty Analysis |
| τTCyt−CC | [1, 20] | # molecules | Chemokine threshold for Tcyt recruitment | Estimated from Uncertainty Analysis |
| τTCyt−TNF | [1, 5] | # molecules | TNF threshold for Tcyt recruitment | Estimated from Uncertainty Analysis |
| τTreg−CC | [1, 10] | # molecules | Chemokine threshold for Treg recruitment | Estimated from Uncertainty Analysis |
| τTreg−TNF | [1, 5] | # molecules | TNF threshold for Treg recruitment | Estimated from Uncertainty Analysis |
| ProbKillMac | [0.05, 0.21] | probability | Probability of Tcyt to kill Macs | [19,20,22,23,36] |
| probKillMacCleanly | [0.15, 0.31] | probability | Probability of Tcyt to kill Macs and all their intracellular Mtb load | [19,20,22,23,36] |
| probApoptosisFasFasL | [0.001, 0.02] | probability | Probability of undergoing apotposis induced by Tγ | [19,20,22,23,36] |
| lungExitInterval | [6, 144] | 10 min | Time it takes a stimulated dendritic cell (DC) to exit the lung through lymphatics | [19,20,22] |
| lymphaticsExitInterval | [6, 40] | 10 min | Time a DC takes to traffic through the lymphatics and reach the lymph node (LN) | [19,20,22] |
| percentOfMacInitNumber | [0.05, 0.25] | %, and used as probability as well | Percentages of DCs that populates the grid initially (calculated as a percentage of initial resident macrophages). It is also used for recruitment on new DC into the grid, at the time a macrophage is recruited | [20,22] |
| growthRateIntMtb | [1.0029, 1.0035] | 10 min | Doubling time of intracellular Mtb | [23] |
| growthRateExtMtb | [1.00124, 1.0014] | 10 min | Doubling time of extracellular Mtb | [23] |
Appendix B
| Outcome of Interest | Compartment | Definition |
| Inflammation | ||
| ‘TotalMr’ | Lung | Total Resting Macrophages |
| ‘TotalDCellMr’ | Lung | Total Unstimulated Dendritic Cells |
| ‘TotalMa’ | Lung | Total Activated Macrophages |
| ‘TotPethot’ | Lung | Total Pet Hot reading from the PET-CT scan |
| ‘NrCaseated’ | Lung | Number of caseated compartments in the granuloma |
| ‘TNF’ | Lung | Tumor Necrosis Factor molecues |
| ‘IL10’ | Lung | Interlukin 10 molecules |
| Infection | ||
| ‘TotMtb’ | Lung | Total Mycobacterium tuberculosis (Mtb) burden |
| ‘IntMtb’ | Lung | Intracellular Mtb burden |
| ‘ExtMtb’ | Lung | Extracellular Mtb burden |
| ‘repExtMtb’ | Lung | Extracellular replicating Mtb burden |
| ‘NonReplExtMtb’ | Lung | Extracellular non-replicating Mtb burden |
| ‘TotalMi’ | Lung | Total Infected Macrophages |
| ‘TotalMci’ | Lung | Total Chronically Infected Macrophages |
| ‘TotalDCellMi’ | Lung | Total Infected Dendritic Cells |
| ‘TotalDCellMci’ | Lung | Total Chronically Infected Dendritic Cells |
| ‘LesionSize’ | Lung | Diameter of the granuloma lesion |
| Adaptive Immune Response | Compartment | Definition |
| ‘TγCognate’ | Lung | Number of Mtb-specific Tγ cells present in the lung |
| ‘TcytCognate’ | Lung | Number of Mtb-specific Tcyt cells present in the lung |
| ‘TgamRecruitedCognate’ | Lung | Number of Mtb-specific Tγ cells recruited to the lung |
| ‘TcytRecruitedCognate’ | Lung | Number of Mtb-specific Tcyt cells recruited to the lung |
| ‘DCellStimulated’ | Lung | Number of Dendritic Cells that have been stimulated |
| ‘DCellExitedLung’ | Lung→lymphatics | Number of Dendritic Cells that have left the lung upon stimulation |
| ‘DCellExitedLymphatics’ | Lymphatics→LN | Number of Dendritic Cells that have left the lymphatics to enter the lymph node |
| ‘BlN4C’ | Blood | Concentration of Mtb-Specific Naïve CD4+ T cells |
| ‘BlE4C’ | Blood | Concentration of Mtb-Specific Effector CD4+ T cells |
| ‘BlCM4C’ | Blood | Concentration of Mtb-Specific Central Memory CD4+ T cells |
| ‘BlEM4C’ | Blood | Concentration of Mtb-Specific Effector Memory CD4+ T cells |
| ‘BlN8C’ | Blood | Concentration of Mtb-Specific Naïve CD8+ T cells |
| ‘BlE8C’ | Blood | Concentration of Mtb-Specific Effector CD8+ T cells |
| ‘BlCM8C’ | Blood | Concentration of Mtb-Specific Central Memory CD8+ T cells |
| ‘BlEM8C’ | Blood | Concentration of Mtb-Specific Effector Memory CD8+ T cells |
| ‘APC’ | Lymph node | Number of Dendritic Cells in the Lymph Node [LN] |
| ‘LnN4C’ | Lymph node | Number of Mtb-Specific Naïve CD4+ T cells |
| ‘LnP4C’ | Lymph node | Number of Mtb-Specific Precursor CD4+ T cells |
| ‘LnE4C’ | Lymph node | Number of Mtb-Specific Effector CD4+ T cells |
| ‘LnCM4C’ | Lymph node | Number of Mtb-Specific Central Memory CD4+ T cells |
| ‘LnEM4C’ | Lymph node | Number of Mtb-Specific Effector Memory CD4+ T cells |
| ‘LnN8C’ | Lymph node | Number of Mtb-Specific Naïve CD8+ T cells |
| ‘LnP8C’ | Lymph node | Number of Mtb-Specific Precursor CD8+ T cells |
| ‘LnE8C’ | Lymph node | Number of Mtb-Specific Effector CD8+ T cells |
| ‘LnCM8C’ | Lymph node | Number of Mtb-Specific Central Memory CD8+ T cells |
| ‘LnEM8C’ | Lymph node | Number of Mtb-Specific Effector Memory CD8+ T cells |
References
- World Health Organization. Global Tuberculosis Report 2014; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Mattila, J.T.; Maiello, P.; Sun, T.; Via, L.E.; Flynn, J.L. Granzyme B-expressing neutrophils correlate with bacteria load in granulomas from Mycobacterium tuberculosis-infected cynomolgus macaques. Cell. Microbiol. 2015, 17, 1085–1097. [Google Scholar] [CrossRef] [PubMed]
- Barry, C.E., 3rd; Boshoff, H.I.; Dartois, V.; Dick, T.; Ehrt, S.; Flynn, J.; Schnappinger, D.; Wilkinson, R.J.; Young, D. The spectrum of latent tuberculosis: Rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 2009, 7, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Redford, P.S.; Murray, P.J.; O’Garra, A. The role of IL-10 in immune regulation during M. tuberculosis infection. Mucosal Immunol. 2011, 4, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of monocytes, macrophages, and dendritic cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Saunders, B.M.; Frank, A.A.; Orme, I.M.; Cooper, A.M. CD4 is required for the development of a protective granulomatous response to pulmonary tuberculosis. Cell. Immunol. 2002, 216, 65–72. [Google Scholar] [CrossRef]
- Hao, S.; Baltimore, D. The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat. Immunol. 2009, 10, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Scanga, C.A.; Mohan, V.P.; Yu, K.; Joseph, H.; Tanaka, K.; Chan, J.; Flynn, J.L. Depletion of CD4(+) T cells causes reactivation of murine persistent tuberculosis despite continued expression of interferon gamma and nitric oxide synthase 2. J. Exp. Med. 2000, 192, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Green, A.M.; Difazio, R.; Flynn, J.L. IFN-gamma from CD4 T cells is essential for host survival and enhances CD8 T cell function during Mycobacterium tuberculosis infection. J. Immunol. 2013, 190, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Benoit, M.; Desnues, B.; Mege, J.L. Macrophage polarization in bacterial infections. J. Immunol. 2008, 181, 3733–3739. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Huang, D.; Wang, R.C.; Shen, L.; Zeng, G.; Yao, S.; Shen, Y.; Halliday, L.; Fortman, J.; McAllister, M.; et al. A critical role for CD8 T cells in a nonhuman primate model of tuberculosis. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Benoit, M.; Barbarat, B.; Bernard, A.; Olive, D.; Mege, J.L. Coxiella burnetii, the agent of Q fever, stimulates an atypical M2 activation program in human macrophages. Eur. J. Immunol. 2008, 38, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Diedrich, C.R.; Mattila, J.T.; Klein, E.; Janssen, C.; Phuah, J.; Sturgeon, T.J.; Montelaro, R.C.; Lin, P.L.; Flynn, J.L. Reactivation of latent tuberculosis in cynomolgus macaques infected with SIV is associated with early peripheral T cell depletion and not virus load. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiszewski, A.E.; Becerril, E.; Aguilar, L.D.; Kader, I.T.; Myers, W.; Portaels, F.; Hernandez Pando, R. The local immune response in ulcerative lesions of Buruli disease. Clin. Exp. Immunol. 2006, 143, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.L.; Beauchemin, C.A.; Perelson, A.S. Agent-based modeling of host-pathogen systems: The successes and challenges. Inf. Sci. (Ny) 2009, 179, 1379–1389. [Google Scholar] [CrossRef] [PubMed]
- Chavali, A.K.; Gianchandani, E.P.; Tung, K.S.; Lawrence, M.B.; Peirce, S.M.; Papin, J.A. Characterizing emergent properties of immunological systems with multi-cellular rule-based computational modeling. Trends Immunol. 2008, 29, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Greve, L.; Dyson, S. Saddle fit and management: An investigation of the association with equine thoracolumbar asymmetries, horse and rider health. Equine Vet. J. 2015, 47, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Hellweger, F.L.; Clegg, R.J.; Clark, J.R.; Plugge, C.M.; Kreft, J.U. Advancing microbial sciences by individual-based modelling. Nat. Rev. Microbiol. 2016, 14, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.; El-Kebir, M.; Kirschner, D. A hybrid multi-compartment model of granuloma formation and T cell priming in Tuberculosis. J. Theor. Biol. 2011, 280, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.; Kirschner, D.E. The human immune response to Mycobacterium tuberculosis in lung and lymph node. J. Theor. Biol. 2004, 227, 463–486. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.; Linderman, J.J.; Kirschner, D.E. A multifaceted approach to modeling the immune response in tuberculosis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.; Pawar, S.; Fuller, C.L.; Reinhart, T.A.; Flynn, J.L.; Kirschner, D.E. Dendritic cell trafficking and antigen presentation in the human immune response to Mycobacterium tuberculosis. J. Immunol. 2004, 173, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Wigginton, J.E.; Kirschner, D. A model to predict cell-mediated immune regulatory mechanisms during human infection with Mycobacterium tuberculosis. J. Immunol. 2001, 166, 1951–1967. [Google Scholar] [CrossRef] [PubMed]
- Segovia-Juarez, J.L.; Ganguli, S.; Kirschner, D. Identifying control mechanisms of granuloma formation during M. tuberculosis infection using an agent-based model. J. Theor. Biol. 2004, 231, 357–376. [Google Scholar] [CrossRef] [PubMed]
- Ray, J.C.; Flynn, J.L.; Kirschner, D.E. Synergy between individual TNF-dependent functions determines granuloma performance for controlling Mycobacterium tuberculosis infection. J. Immunol. 2009, 182, 3706–3717. [Google Scholar] [CrossRef] [PubMed]
- Gammack, D.; Ganguli, S.; Marino, S.; Segovia-Juarez, J.; Kirschner, D.E. Understanding the immune response in tuberculosis using different mathematical models and biological scales. Multiscale Model. Simul. 2005, 3, 312–345. [Google Scholar] [CrossRef]
- Cardona, P.J.; Amat, I.; Gordillo, S.; Arcos, V.; Guirado, E.; Diaz, J.; Vilaplana, C.; Tapia, G.; Ausina, V. Immunotherapy with fragmented Mycobacterium tuberculosis cells increases the effectiveness of chemotherapy against a chronical infection in a murine model of tuberculosis. Vaccine 2005, 23, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
- Cardona, P.J.; Llatjos, R.; Gordillo, S.; Diaz, J.; Vinado, B.; Ariza, A.; Ausina, V. Towards a ‘human-like’ model of tuberculosis: Intranasal inoculation of LPS induces intragranulomatous lung necrosis in mice infected aerogenically with Mycobacterium tuberculosis. Scand. J. Immunol. 2001, 53, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Files for GranSim. Available online: http://malthus.micro.med.umich.edu/GranSim (accessed on 18 October 2016).
- Rateitschak, K.; Karger, A.; Fitzner, B.; Lange, F.; Wolkenhauer, O.; Jaster, R. Mathematical modelling of interferon-gamma signalling in pancreatic stellate cells reflects and predicts the dynamics of STAT1 pathway activity. Cell Signal. 2010, 22, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.A.; Fribourg, M.; Sealfon, S.C. Cytokine response is determined by duration of receptor and signal transducers and activators of transcription 3 (STAT3) activation. J. Biol. Chem. 2013, 288, 2986–2993. [Google Scholar] [CrossRef] [PubMed]
- Pienaar, E.; Cilfone, N.A.; Lin, P.L.; Dartois, V.; Mattila, J.T.; Butler, J.R.; Flynn, J.L.; Kirschner, D.E.; Linderman, J.J. A computational tool integrating host immunity with antibiotic dynamics to study tuberculosis treatment. J. Theor. Biol. 2015, 367, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.; Sud, D.; Plessner, H.; Lin, P.L.; Chan, J.; Flynn, J.L.; Kirschner, D.E. Differences in reactivation of tuberculosis induced from anti-TNF treatments are based on bioavailability in granulomatous tissue. PLoS Comput. Biol. 2007, 3, 1909–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallahi-Sichani, M.; Flynn, J.L.; Linderman, J.J.; Kirschner, D.E. Differential risk of tuberculosis reactivation among anti-TNF therapies is due to drug binding kinetics and permeability. J. Immunol. 2012, 188, 3169–3178. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.; Fallahi-Sichani, M.; Linderman, J.J.; Kirschner, D.E. Mathematical Models of Anti-TNF Therapies and their Correlation with Tuberculosis. In Antibody-Mediated Drug Delivery Systems; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 83–104. [Google Scholar]
- Marino, S.; Gideon, H.P.; Gong, C.; Mankad, S.; McCrone, J.T.; Lin, P.L.; Linderman, J.J.; Flynn, J.L.; Kirschner, D.E. Computational and Empirical Studies Predict Mycobacterium tuberculosis-Specific T Cells as a Biomarker for Infection Outcome. PLoS Comput. Biol. 2016, 12. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.; Hogue, I.B.; Ray, C.J.; Kirschner, D.E. A methodology for performing global uncertainty and sensitivity analysis in systems biology. J. Theor. Biol. 2008, 254, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Cilfone, N.A.; Perry, C.R.; Kirschner, D.E.; Linderman, J.J. Multi-scale modeling predicts a balance of tumor necrosis factor-alpha and interleukin-10 controls the granuloma environment during Mycobacterium tuberculosis infection. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Fallahi-Sichani, M.; El-Kebir, M.; Marino, S.; Kirschner, D.E.; Linderman, J.J. Multiscale computational modeling reveals a critical role for TNF-alpha receptor 1 dynamics in tuberculosis granuloma formation. J. Immunol. 2011, 186, 3472–3483. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Linderman, J.J.; Kirschner, D. Harnessing the heterogeneity of T cell differentiation fate to fine-tune generation of effector and memory T cells. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Fallahi-Sichani, M.; Kirschner, D.E.; Linderman, J.J. NF-kappaB Signaling Dynamics Play a Key Role in Infection Control in Tuberculosis. Front. Physiol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Cilfone, N.A.; Ford, C.B.; Marino, S.; Mattila, J.T.; Gideon, H.P.; Flynn, J.L.; Kirschner, D.E.; Linderman, J.J. Computational Modeling Predicts IL-10 Control of Lesion Sterilization by Balancing Early Host Immunity-Mediated Antimicrobial Responses with Caseation during Mycobacterium tuberculosis Infection. J. Immunol. 2015, 194, 664–677. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.; Cilfone, N.A.; Mattila, J.T.; Linderman, J.J.; Flynn, J.L.; Kirschner, D.E. Macrophage Polarization Drives Granuloma Outcome during Mycobacterium tuberculosis Infection. Infect. Immun. 2015, 83, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Cilfone, N.A.; Kirschner, D.E.; Linderman, J.J. Strategies for efficient numerical implementation of hybrid multi-scale agent-based models to describe biological systems. Cell. Mol. Bioeng. 2015, 8, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Demangel, C.; Britton, W.J. Interaction of dendritic cells with mycobacteria: Where the action starts. Immunol. Cell Biol. 2000, 78, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Mihret, A. The role of dendritic cells in Mycobacterium tuberculosis infection. Virulence 2012, 3, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, K.A.; Serbina, N.V.; Flynn, J.L. Fate of Mycobacterium tuberculosis within murine dendritic cells. Infect. Immun. 2001, 69, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Mazzini, E.; Massimiliano, L.; Penna, G.; Rescigno, M. Oral tolerance can be established via gap junction transfer of fed antigens from CX3CR1(+) macrophages to CD103(+) dendritic cells. Immunity 2014, 40, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C. Immunology: The Immune System in Health and Disease, 6th ed.; Garland Science Publishing: New York, NY, USA, 2005. [Google Scholar]
- Lin, P.L.; Coleman, T.; Carney, J.P.; Lopresti, B.J.; Tomko, J.; Fillmore, D.; Dartois, V.; Scanga, C.; Frye, L.J.; Janssen, C.; et al. Radiologic responses in cynomolgous macaques for assessing tuberculosis chemotherapy regimens. Antimicrob. Agents Chemother. 2013. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.T.; Maiello, P.; Tomko, J.; Frye, L.J.; Fillmore, D.; Janssen, C.; Klein, E.; Lin, P.L. Early Changes by (18)Fluorodeoxyglucose positron emission tomography coregistered with computed tomography predict outcome after Mycobacterium tuberculosis infection in cynomolgus macaques. Infect. Immun. 2014, 82, 2400–2404. [Google Scholar] [CrossRef] [PubMed]
- Uncertainty and Sensitivity Analysis. Available online: http://malthus.micro.med.umich.edu/lab/usanalysis.html (accessed on 18 October 2016).
- Blower, S.M.; Dowlatabadi, H. Sensitivity and Uncertainty Analysis of Complex-Models of Disease Transmission—An HIV Model, as an Example. Int. Stat. Rev. 1994, 62, 229–243. [Google Scholar] [CrossRef]
- Greenland, S. Sensitivity analysis, Monte Carlo risk analysis, and Bayesian uncertainty assessment. Risk Anal. 2001, 21, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Kimmick, G.G.; Shelton, B.J.; Case, L.D.; Cooper, M.R.; Muss, H.B. Long-term follow-up of a phase II trial studying a weekly doxorubicin-based multiple drug adjuvant therapy for stage II node-positive carcinoma of the breast. Breast Cancer Res. Treat. 2002, 72, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, M.A.; Blower, S.M. Uncertainty and sensitivity analysis of the basic reproductive rate. Tuberculosis as an example. Am. J. Epidemiol. 1997, 145, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Gammack, D.; Doering, C.R.; Kirschner, D.E. Macrophage response to Mycobacterium tuberculosis infection. J. Math. Biol. 2004, 48, 218–242. [Google Scholar] [CrossRef] [PubMed]
- Ganguli, S.; Gammack, D.; Kirschner, D.E. A metapopulation model of granuloma formation in the lung during infection with mycobacterium tuberculosis. Math. Biosci. Eng. 2005, 2, 535–560. [Google Scholar] [PubMed]
- Marino, S.; Beretta, E.; Kirschner, D.E. The role of delays in innate and adaptive immunity to intracellular bacterial infection. Math. Biosci. Eng. 2007, 4, 261–288. [Google Scholar] [CrossRef] [PubMed]
- Helton, J.C.; Johnson, J.D.; Sallaberry, C.J.; Storlie, C.B. Survey of sampling-based methods for uncertainty and sensitivity analysis. Reliab. Eng. Syst. Saf. 2006, 91, 1175–1209. [Google Scholar] [CrossRef]
- Fallahi-Sichani, M.; Marino, S.; Flynn, J.L.; Linderman, J.J.; Kirschner, D.E. A systems biology approach for understanding granuloma formation and function in tuberculosis. In Systems Biology of Tuberculosis; McFadden, J., Beste, D.J.V., Kierzek, A.M., Eds.; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Fallahi-Sichani, M.; Schaller, M.A.; Kirschner, D.E.; Kunkel, S.L.; Linderman, J.J. Identification of key processes that control tumor necrosis factor availability in a tuberculosis granuloma. PLoS Comput. Biol. 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.Y.; Dodd, L.E.; Lee, M.; Paripati, P.; Hammoud, D.A.; Mountz, J.M.; Jeon, D.; Zia, N.; Zahiri, H.; Coleman, M.T.; et al. PET/CT imaging correlates with treatment outcome in patients with multidrug-resistant tuberculosis. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.L.; Chan, J.; Triebold, K.J.; Dalton, D.K.; Stewart, T.A.; Bloom, B.R. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J. Exp. Med. 1993, 178, 2249–2254. [Google Scholar] [CrossRef] [PubMed]
- Rozot, V.; Vigano, S.; Mazza-Stalder, J.; Idrizi, E.; Day, C.L.; Perreau, M.; Lazor-Blanchet, C.; Petruccioli, E.; Hanekom, W.; Goletti, D.; et al. Mycobacterium tuberculosis-specific CD8+ T cells are functionally and phenotypically different between latent infection and active disease. Eur. J. Immunol. 2013, 43, 1568–1577. [Google Scholar] [CrossRef] [PubMed]
- Shang, S.; Siddiqui, S.; Bian, Y.; Zhao, J.; Wang, C.R. Nonclassical MHC Ib-restricted CD8+ T Cells Recognize Mycobacterium tuberculosis-Derived Protein Antigens and Contribute to Protection against Infection. PLoS Pathog. 2016, 12. [Google Scholar] [CrossRef] [PubMed]
- Sud, D.; Bigbee, C.; Flynn, J.L.; Kirschner, D.E. Contribution of CD8+ T cells to control of Mycobacterium tuberculosis infection. J. Immunol. 2006, 176, 4296–4314. [Google Scholar] [CrossRef] [PubMed]
- Bellone, M.; Calcinotto, A. Ways to enhance lymphocyte trafficking into tumors and fitness of tumor infiltrating lymphocytes. Front. Oncol. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Mattila, J.T.; Miller, M.; Flynn, J.L.; Linderman, J.J.; Kirschner, D. Predicting lymph node output efficiency using systems biology. J. Theor. Biol. 2013, 335, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, H.A.; Schrager, L. TB vaccine development and the End TB Strategy: Importance and current status. Trans. R. Soc. Trop. Med. Hyg. 2016, 110, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M. Dendritic cells and the control of immunity: Enhancing the efficiency of antigen presentation. Mt. Sinai J. Med. 2001, 68, 160–166. [Google Scholar] [PubMed]
- Lindestam Arlehamn, C.S.; Lewinsohn, D.; Sette, A. Antigens for CD4 and CD8 T cells in tuberculosis. Cold Spring Harb. Perspect. Med. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Lindestam Arlehamn, C.S.; Paul, S.; Mele, F.; Huang, C.; Greenbaum, J.A.; Vita, R.; Sidney, J.; Peters, B.; Sallusto, F.; Sette, A. Immunological consequences of intragenus conservation of Mycobacterium tuberculosis T-cell epitopes. Proc. Natl. Acad. Sci. USA 2015, 112, E147–E155. [Google Scholar] [CrossRef] [PubMed]
- Lindestam Arlehamn, C.S.; Sette, A. Definition of CD4 Immunosignatures Associated with MTB. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
| Variable | Value | Units | Description |
|---|---|---|---|
| APC | 0 | Cell count | Antigen presenting cell proxy in the lymph node |
| NLn,4 | NB,4 × (α/host_Ln) | Cell count | Mtb-specific LN Naïve CD4+ T cell |
| PLn,4 | 0 | Cell count | Mtb-specific LN Precursor CD4+ T cell |
| EMLn,4 | 0 | Cell count | Mtb-specific LN Effector Memory CD4+ T cell |
| CMLn,4 | 0 | Cell count | Mtb-specific LN Central Memory CD4+ T cell |
| NB,4 | [255, 610] × λ | Cell/mm3 | Mtb-specific Blood Naïve CD4+ T cell |
| EB,4 | 0 | Cell/mm3 | Mtb-specific Blood Effector CD4+ T cell |
| CMB,4 | 0 | Cell/mm3 | Mtb-specific Blood Central Memory CD4+ T cell |
| EMB,4 | 0 | Cell/mm3 | Mtb-specific Blood Effector Memory CD4+ T cell |
| NLn,8 | NB,8 × (α/host_Ln) | Cell count | Mtb-specific LN Naïve CD8+ T cell |
| PLn,8 | 0 | Cell count | Mtb-specific LN Precursor CD8+ T cell |
| EMLn,8 | 0 | Cell count | Mtb-specific LN Effector Memory CD8+ T cell |
| CMLn,8 | 0 | Cell count | Mtb-specific LN Central Memory CD8+ T cell |
| NB,8 | [255, 610] × λ | Cell/mm3 | Mtb-specific Blood Naïve CD8+ T cell |
| EB,8 | 0 | Cell/mm3 | Blood Effector CD8+ T cell |
| CMB,8 | 0 | Cell/mm3 | Blood Central Memory CD8+ T cell |
| EMB,8 | 0 | Cell/mm3 | Blood Effector Memory CD8+ T cell |
| NLn,nc4 | NB,nc4 × (α/host_Ln) | Cell count | Non-Mtb-specific LN Naïve CD4+ T cell |
| CMLn,nc4 | CMB,nc4 × (α/host_Ln) | Cell count | Non-Mtb-specific LN Central Memory CD4+ T cell |
| NB,nc4 | [255, 610] × (1 − λ) | Cell/mm3 | Non-Mtb-specific Blood Naïve CD4+ T cell |
| EB,nc4 | [47, 254] × (1 − λ) | Cell/mm3 | Non-Mtb-specific Blood Effector CD4+ T cell |
| CMB,nc4 | [83, 300] × (1 − λ) | Cell/mm3 | Non-Mtb-specific Blood Central Memory CD4+ T cell |
| EMB,nc4 | [50, 255] × (1 − λ) | Cell/mm3 | Non-Mtb-specific Blood Effector Memory CD4+ T cell |
| NLn,nc8 | BN,nc8 × (α/host_Ln) | Cell count | Non-Mtb-specific LN Naïve CD8+ T cell |
| CMLn,nc8 | CMN,nc8 × (α/host_Ln) | Cell count | Non-Mtb-specific LN Central Memory CD8+ T cell |
| NB,nc8 | [100, 672] × (1 − λ) | Cell/mm3 | Non-Mtb-specific Blood Naïve CD8+ T cell |
| EB,nc8 | [43, 317] × (1 − λ) | Cell/mm3 | Non-Mtb-specific Blood Effector CD8+ T cell |
| CMB,nc8 | [36, 262] × (1 − λ) | Cell/mm3 | Non-Mtb-specific Blood Central Memory CD8+ T cell |
| EMB,nc8 | [11, 156] × (1 − λ) | Cell/mm3 | Non-Mtb-specific Blood Effector Memory CD8+ T cell |
| INFECTION (LUNG) | |||||
|---|---|---|---|---|---|
| Parameters | Tot Mtb * | Ext Mtb | Total Infected Macs * | Total Infected DCs * | Gran Size * |
| growthRateIntMtb | + + + | + + + | + + + | + 1 | |
| lungExitInterval | + + + | ||||
| τTγ−CC—chemokine threshold for Tγ recruitment | ++ | ||||
| τTreg−TNF—tumor necrosis factor (TNF) threshold for Treg recruitment | |||||
| k4—CD4+ T precursorproliferation | − − − | − − − | − − | ||
| k13—CD8+ T precursorproliferation | − − − | − − − | − − − | − − − | early + then − − − |
| scalingMDC—Scaling to host factor representing the number of granulomas developing in the whole lung at time of infection | |||||
| k11—Naïve CD8+ T priming | − − | − − | − − | − − | − − |
| % of Resident DCs | + + | − | |||
| λ | − − | ||||
| INFLAMMATION (LUNG) | |||||
|---|---|---|---|---|---|
| PARAMETERS | Total Activated Macrophages | Tot Pet Hot * | Caseation/Necrosis | TNF * | IL10 * |
| growthRateIntMtb | + early 1 | + + early | |||
| τTγ−CC—chemokine threshold for Tγ recruitment | − − early | − early 1 | + + + | ||
| τTcyt−CC—chemokine threshold for Tcyt recruitment | − − early | − early then + 1 | + + + | ||
| τTreg−CC—chemokine threshold for Treg recruitment | − 1 | + and then 1 | + + early | − − − | |
| k2—Naïve CD4 priming | + + | − early 1 | + 1 | ||
| k4—CD4+ T precursor proliferation | + + + | + 1 | |||
| k13—CD8+ T precursor proliferation | − − − | − − − | + + + early − late | − − − | − − − |
| k14—CD8+ T differentiation—effector | + 1 | + 1 | |||
| k11—Naïve CD8 priming | − | + + early − late | − 1 | − 1 | |
| ADAPTIVE IMMUNE RESPONSE (LUNG) | |||||||
|---|---|---|---|---|---|---|---|
| PARAMETERS | Mtb-Specific Tgam (Pro-Inflammatory) T Cells * | Mtb-Specific Tcyt * (Cytotoxic) T Cells | Recruited Mtb-Specific Treg | Recruited Mtb-Specific Tcyt | DC Stimulated | DC Exited Lung | DC Exited Lymph |
| λ—Frequency of Mtb-specific Naïve T cells in the blood/LN | + then − 1 | ||||||
| k11—CD8 priming | + + then − | ||||||
| k4—CD4 precursor proliferation | + + + | + + | |||||
| k13—CD8 precursor proliferation | − − | + + + | − − − | + + | − − − | − − − | − − − |
| k2—CD4 priming | + + + early | ||||||
| % of Resident DCs | + + | + + | + + | ||||
| τTγ−CC—chemokine threshold for Tγ recruitment | + + 1 | + + | |||||
| τTreg−CC—chemokine threshold for Treg recruitment | − − 1 | ||||||
| BLOOD OUTCOMES—Mtb-Specific T Cells | ||||||||
|---|---|---|---|---|---|---|---|---|
| PARAMETERS | Naïve CD4 | Effector CD4 * | Central Memory CD4 | Effector Memory CD4 | Naïve CD8 | Effector CD8 * | Central Memory CD8 | Effector Memory CD8 |
| lungExitInterval | − − − early | − − − early | − − − early | − − − early | − − − early | − − − early | ||
| lymph_ExitInterval | − − − early | − − − early | − − − early | − − − early | − − − early | − − − early | ||
| % of Resident DCs | + + early | + + early | + + + early | + + + early | ||||
| Initial Conditions for Mtb−specific Naïve CD4+ T cells—BLOOD | + + + | |||||||
| Initial Conditions for Mtb-specific Naïve CD8+ T cells—BLOOD | + + + | + + early | ||||||
| host_LN—Number of involved lymph nodes in the host | − − | + + early | ||||||
| λ—Frequency of Mtb-specific Naïve T cells in the blood/LN | + + + | + + + early | + + early | + + early | + + + | + + + early | + + + early | + + + early |
| k1—Naïve CD4 recruitment rate | − − − | + + + early | + + early | + + + early | ||||
| k10—Naïve CD8 recruitment rate | − − − | + + + early | + + + early | + + + early | ||||
| k2—Naïve CD4 priming | − − − | + + + early | + + early | + + + early | + + + early | + + + early | + + + early | |
| k11—Naïve CD8 priming | − − − | + + + early | + + + early | + + + early | ||||
| k4—CD4 precursor proliferation | + + + | + + + | + + + | |||||
| k13—CD8 precursor proliferation | + + + | + + + | + + + | |||||
| k5—Precursor CD4 differentiation to Effector rate | + + +/−/+ | − − − | + + +/−/+ | |||||
| k14—CD8 differentiation to effector | + + +/−/+ | − − − | + + + early | |||||
| µ5—Mature DC half-life in the LN | − − early | |||||||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marino, S.; Kirschner, D.E. A Multi-Compartment Hybrid Computational Model Predicts Key Roles for Dendritic Cells in Tuberculosis Infection. Computation 2016, 4, 39. https://doi.org/10.3390/computation4040039
Marino S, Kirschner DE. A Multi-Compartment Hybrid Computational Model Predicts Key Roles for Dendritic Cells in Tuberculosis Infection. Computation. 2016; 4(4):39. https://doi.org/10.3390/computation4040039
Chicago/Turabian StyleMarino, Simeone, and Denise E. Kirschner. 2016. "A Multi-Compartment Hybrid Computational Model Predicts Key Roles for Dendritic Cells in Tuberculosis Infection" Computation 4, no. 4: 39. https://doi.org/10.3390/computation4040039
APA StyleMarino, S., & Kirschner, D. E. (2016). A Multi-Compartment Hybrid Computational Model Predicts Key Roles for Dendritic Cells in Tuberculosis Infection. Computation, 4(4), 39. https://doi.org/10.3390/computation4040039