Abstract
An orbital energy-based reaction analysis theory is presented as an extension of the orbital-based conceptual density functional theory. In the orbital energy-based theory, the orbitals contributing to reactions are interpreted to be valence orbitals giving the largest orbital energy variation from reactants to products. Reactions are taken to be electron transfer-driven when they provide small variations for the gaps between the contributing occupied and unoccupied orbital energies on the intrinsic reaction coordinates in the initial processes. The orbital energy-based theory is then applied to the calculations of several 2 reactions. Using a reaction path search method, the Cl− + CH3I → ClCH3 + I− reaction, for which another reaction path called “roundabout path” is proposed, is found to have a precursor process similar to the roundabout path just before this SN2 reaction process. The orbital energy-based theory indicates that this precursor process is obviously driven by structural change, while the successor SN2 reaction proceeds through electron transfer between the contributing orbitals. Comparing the calculated results of the SN2 reactions in gas phase and in aqueous solution shows that the contributing orbitals significantly depend on solvent effects and these orbitals can be correctly determined by this theory.
1. Introduction
Reaction analyses based on molecular orbitals take on increasing importance. This is because the experimental imaging of molecular orbitals has recently been made possible [1], and the recent progress in attosecond science enables us to perform the time-resolved analysis of electron dynamics. Even time-resolved chemical reaction tracking using orbitals is going to be realized by combining the molecular orbital imaging and the time-resolved electron dynamics simulation. In this context, reaction analyses based on orbitals raises the stakes again. Major orbital-based reaction analysis theories are the frontier orbital theory [2] and conceptual density functional theory (DFT) [3]. Electronic state variations from the initial to terminal states of reactions are explicitly taken into consideration in these theories, while it is usually neglected in the mainstream reaction analyses based on reaction energy diagrams using potential energy surfaces. Since these theories reveal the highly reactive sites of molecules, they have been used as a guiding principle of reaction designs and molecular syntheses. Note, however, that these theories are essentially unavailable in quantitative reaction analyses because molecular orbitals are too insensible to be quantitative. Actually, molecular orbitals hardly depend on electron correlation that is usually required to discuss reactions quantitatively.
Orbital energies are one of the most prospective alternatives to solve this problem [4]. Different from molecular orbitals, corresponding orbital energies strongly depend on electron correlation [5]. However, there had been no theory to quantitatively reproduce orbital energies until very recently. It was recently revealed that long-range corrected (LC) DFT [6,7] simultaneously provide accurate occupied and unoccupied valence orbital energies [8]. Combining LC-DFT with the pseudospectral regional self-interaction correction [9,10], accurate core orbital energies are also given with keeping or improving the accuracy of valence orbital energies [11]. By using the accurate highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies, it is found that global hardnesses hardly change in the initial processes of two Diels–Alder reactions on their intrinsic reaction coordinates (IRCs) [12]. This indicates that these Diels–Alder reactions initially proceed through only electron transfer because outermost orbital energies hardly change by the donation and acceptance of fractional number of electrons [13], despite molecular structures change. Based on this observation, Tsuneda and Singh have developed a reaction analysis method based on orbital energies [4]. As detailed in Section 2, the normalized orbital energy gap, which is the normalized difference between the orbital energies of the occupied and unoccupied orbitals contributing to reactions, is plotted on the normalized IRCs to obtain the orbital energy gap gradient at the reactant structure. The orbital energy gap gradient is taken as the reactivity index to confirm if the reaction initially proceeds through electron transfer. Applying this reactivity index to many fundamental reactions shows that more than 60% of the forward processes make progress through electron transfer, while more than 60% of the backward processes run through structural change. Moreover, anti-activation-energy reactions, which have higher barrier heights in the forward processes than those in the reverse processes, are found to mostly proceed through electron transfer for the forward processes and run through structural change for the backward processes. These results assure the high availability of the orbital energy-based reaction analysis method.
SN2 reactions are noteworthy in this orbital energy-based reaction analysis because the initial reaction processes of several SN2 reactions are figured out to be structural change-driven on the minimum energy paths despite these reactions being exothermic [4]. For SN2 reactions, Hase and coworkers have experimentally questioned the optimum reaction path [14,15,16,17]. Based on a time-resolved ion-molecule cross-beam imaging spectroscopy analysis, they suggested the roundabout reaction path for the Cl− + CH3I → CH3Cl + I− reaction [15], in which the CH3 group of CH3I spins around I− to move to Cl−. They also studied the CH3I + F− → CH3F + I− reaction mechanism using ion imaging experiments and direct chemical dynamics simulations. Consequently, they proposed three pathways: direct rebound, direct stripping, and indirect hydrogen bonding paths. They also indicated that the results of the experimental rapid intramoleclar vibrational energy distributions and reaction energies are inconsistent with the optimum reaction pathway [16]. Szabó, Czakó, and coworkers performed the dynamics simulation of the F− + CH3Cl → CH3F + Cl− reaction by determining its full-dimensional potential energy surfaces [18,19,20]. As a result, they found that the direct rebound mechanism dominates at high collision energies, while the indirect mechanism mainly proceeds at low collision energies [18]. Moreover, they suggested the significance of a double-inversion mechanism, in which the hydrogen atom of the methyl group first moves to F− and the methyl group then transfers to F− [19]. Comparing the results of a cross-beam imaging experiment with the dynamics simulation calculation, they, however, found that the mechanism significantly depends on the leaving group, which is X of the F− + CH3X → CHF + X− reaction (X = Cl or I) [20]. Based on the above-mentioned orbital energy-based reaction analysis, it is recently found that two SN2 reactions, F− + CH3Cl → Cl− + CH3F and OHCH3 + F− → OH− + CH3F reactions, are also expected to avoid the minimum energy paths because these reactions give large reactivity indices indicating structural change-driven processes for the initial processes. This suggests that these SN2 reactions also take other reaction paths than the minimum energy paths to avoid initial structural change-driven processes.
In this study, we lay the foundation of the orbital energy-based reaction analysis theory [4] and then apply this theory to the SN2 reaction calculations. First, the orbital energy-based reaction analysis theory is formed on the basis of the conceptual DFT [3] in Section 2. After detailing the computational methods in Section 3, the orbital energy-based theory is applied to the calculations of the Cl− + CHI → CHCl + I reaction and two S2 reactions, the Menschutkin reaction (NH + CHCl → NHCH + Cl) and Cl...CHCl → ClCH...Cl reaction, to evaluate the applicability of this orbital energy-based theory in Section 4.
2. Orbital Energy-Based Reaction Analysis Theory
In this section, this orbital energy-based reaction analysis theory [4] is proven to be the expansion of the conceptual DFT [3], which forms the foundation of the frontier orbital theory [2]. In the field of chemistry, the reactivity indices in the conceptual DFT have been used to explain the reactivities and highly reactive regions of molecules. These reactivity indices are proposed in the concept of Legendre variable transformation [3]. The orbital energy-based reaction analysis theory is also formed in this concept.
The conceptual DFT interprets the first Hohenberg–Kohn Theorem [21] as the Legendre transformation of energy functional E from intensive external potential V-dependent to extensive density ρ-dependent ones: . Here, systems are defined to have four independent variables: chemical potential μ, occupation number n, V and ρ. According to this theorem, the energy functional becomes independent of V variation in reaction pathways. Moreover, the Levy constrained search formulation [22] corresponds to finding the optimum μ in this concept. For independent variables and , the Maxwell cross relation is established in classical thermodynamics as:
where F is arbitrary thermodynamics potential. Using and , we obtain
which indicates that chemical potential variation with respect to structural change with a constant occupation number equals density variation with respect to occupation number change with no structural change. Function f in Equation (2) is called “Fukui function” because it is identical to the reactivity index of the frontier orbital theory that Fukui and coworkers suggested [2]. For electrophilic reactions,
for nucleophilic reactions,
and for radical reactions, and
where , and are the electron densities of the highest occupied, lowest unoccupied, and singly occupied molecular orbitals.
These orbital-based reactivity indices are transformed to the orbital energy-based ones [23] using the Janak theorem [24]:
where is the outermost orbital energy. Using this chemical potential, the Fukui function in Equation (2) is expressed as:
Equation (6) has been used to derive various reactivity indices in the conceptual DFT such as global hardness:
However, these orbital energy-based reactivity indices have been infrequently used because it has been hard to quantitatively reproduce orbital energies until recently. For the global hardness in Equation (8), the approximated form on the right-hand side is usually used in reaction analyses. LC-DFT has made all the difference. Recently, LC-DFT is found to quantitatively reproduce occupied and unoccupied valence orbital energies simultaneously for the first time as mentioned in Section 1 [8].
The quantitative LC-DFT orbital energies make it possible to carry out orbital energy-based reaction analyses. As mentioned in Section 1, the orbital energy gaps of occupied and unoccupied orbitals, which mainly contribute to reactions, are found to be kept almost constant in the initial processes of many reactions [4]. This orbital energy gap behavior is interpreted as follows: For the orbital energy gap,
where and are acceptor and donor orbital energies mainly contributing to the reaction, respectively, and the orbital energy gap gradient on the IRCs is expressed as:
In Equation (10), the partial derivatives of are represented as:
where and are the electron densities of the acceptor and donor orbitals, respectively, and is the nuclear-electron electrostatic potential, and
When the orbital energy gap gradient hardly changes by the external potential variation i.e., , and reactions proceed only through electron transfer, the orbital energy gap gradient is supposed to be almost zero, , on the basis of the Sham–Schlüter relation for orbital energies [13]:
where is the -th orbital energy of the system containing n electrons. Therefore, the near-zero orbital energy gap gradient indicates that reactions mainly proceed through electron transfer.
Based on the above concepts, the orbital energy-base reaction analysis is performed as follows [4]:
- First, LC-DFT valence orbital energies are plotted on IRC, which is constructed by, e.g., the predictor-corrector integrator method [25,26]. In this method, step sizes are placed at the same intervals by using the Hessian-based predictor-corrector algorithm [25].
- Then, the target orbitals are selected to be the occupied and unoccupied orbitals giving the most varied valence orbital energies. This selection is based on the concept of Equation (7) that reactions proceed to maximize the variation of chemical potentials, i.e., outermost orbital energies.
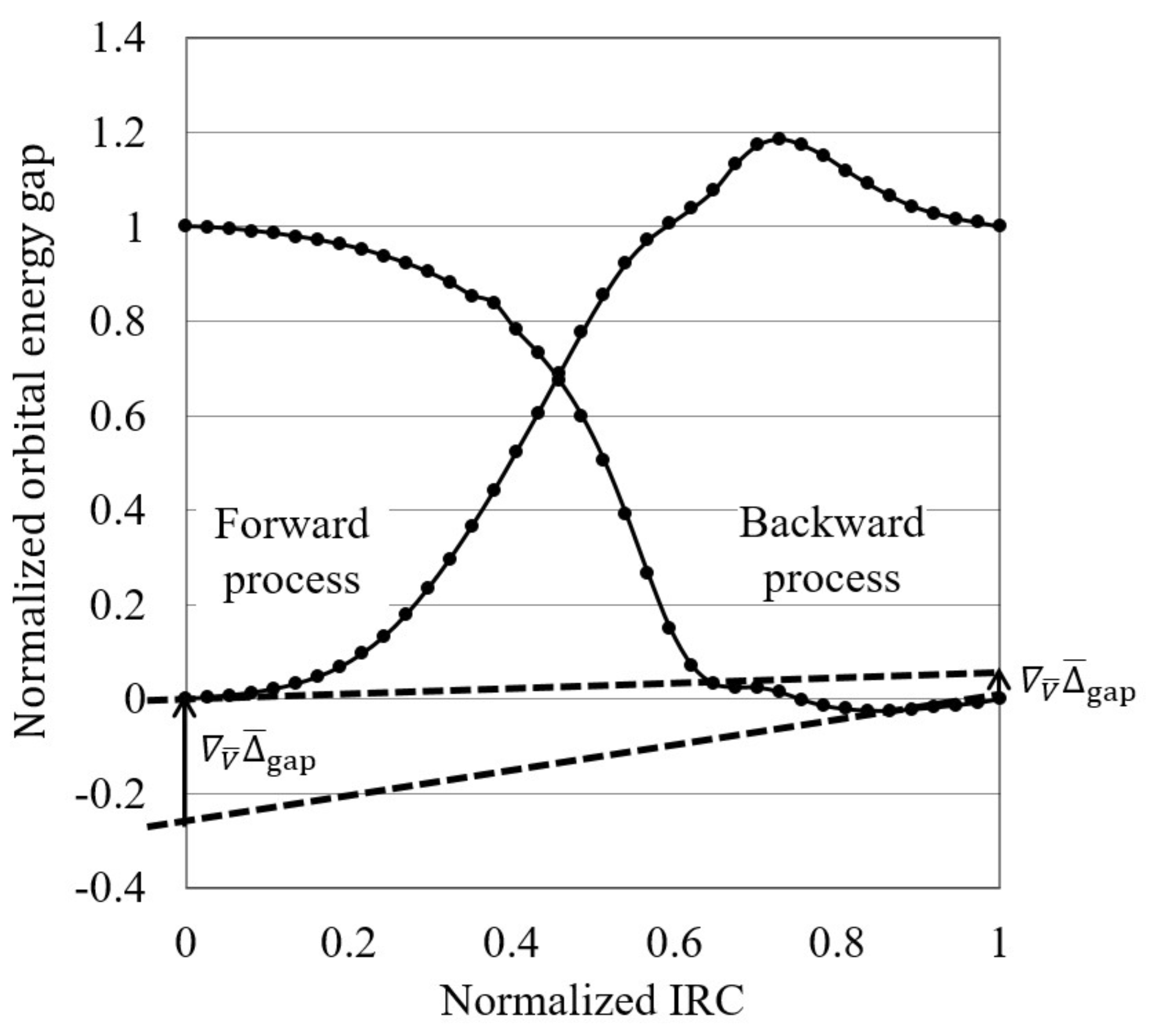
- The normalized reaction diagram is illustrated by plotting the normalized orbital energy gap:where and are the target orbital energy gaps at the initial and terminal steps of each process, on the normalized IRCs, , as shown in Figure 1.
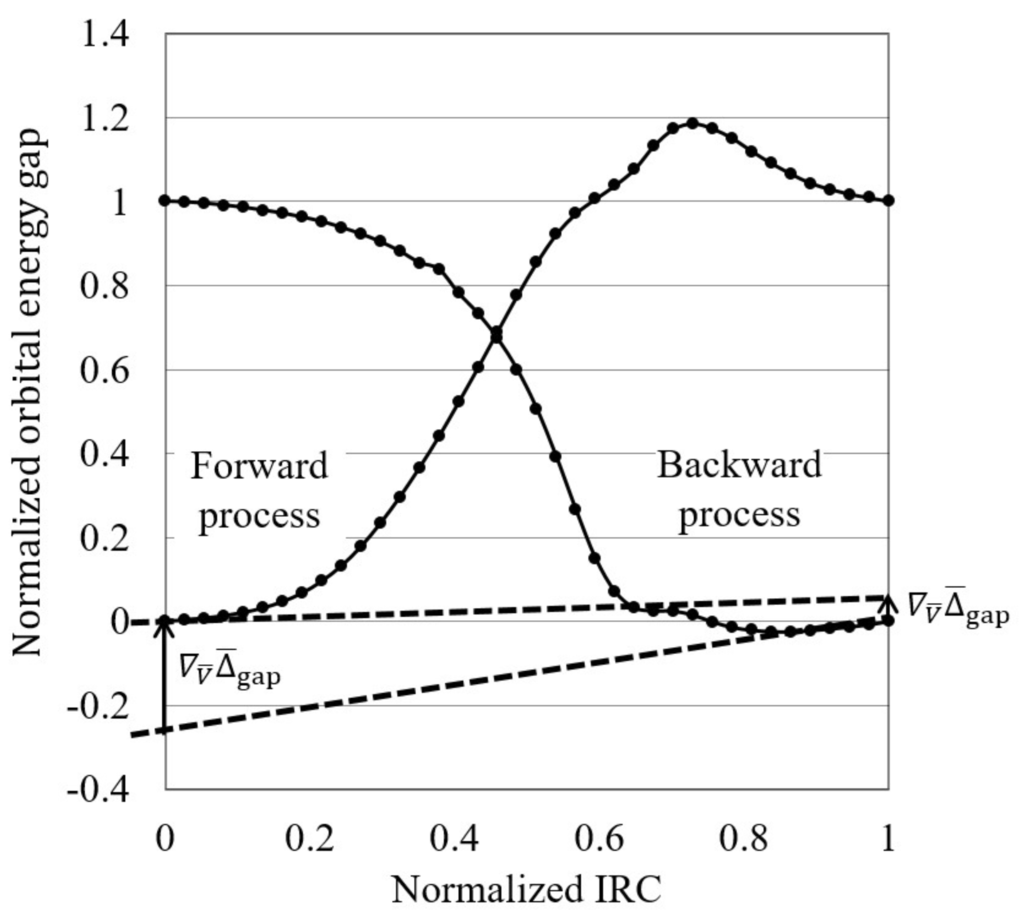 Figure 1. A normalized reaction diagram, in which both the gaps and intrinsic reaction coordinates are normalized by setting the values of the reactant and product to be zero and one, respectively. For both forward and backward reactions, the orbital energy gap gradients are estimated by fit to polynomial functions.
Figure 1. A normalized reaction diagram, in which both the gaps and intrinsic reaction coordinates are normalized by setting the values of the reactant and product to be zero and one, respectively. For both forward and backward reactions, the orbital energy gap gradients are estimated by fit to polynomial functions. - Finally, the orbital energy gap gradient in terms of the normalized IRCs, , is calculated at the initial reaction stage as a “reactivity index”. Reactions are interpreted to be electron transfer-driven when the orbital energy gap gradient is less than a threshold value, which is temporarily 0.250 in this study.
3. Computational Details
In this study, we perform LC-DFT calculations [6,7] on the basis of the unrestricted Kohn–Sham method [27]. In the IRC calculations, we use the predictor-corrector integrator method [25,26], which resets reference points at regular intervals after evaluating IRCs. The Def2-TZVPD basis functions [28,29] are used as the basis set. To explore the S2 reaction of Cl + CHI → CHCl + I in further detail, we also carry out the reaction path search calculations using the artificial force induced reaction method in the global reaction route mapping (GRRM) program [30,31] by taking van der Waals (vdW) correlations into consideration. The van der Waals calculation uses the LC+vdW method [32,33,34], which combines LC-DFT with the van der Waals correlation of, e.g., a van der Waals functional. As the van der Waals correlation, we adopt the local response dispersion (LRD) vdW functional [35]. The long-range correction is done for the Becke 1988 (B88) exchange [36] + one-parameter progressive (OP) correlation [37] functional (LC-BOP), to which the LRD functional is optimized. For S2 reactions in aqueous solutions, we employ the conductor-like polarizable continuum model (CPCM) [38] self-consistent reaction field (SCRF) method to evaluate the solvent effects. In the calculations of the solution reactions, we use the long-range correction for the B88 exchange + the Lee–Yang–Parr correlation [39] (BLYP) functional (LC-BLYP) with the aug-cc-pVTZ basis set. All calculations are performed on the development version of the GAMESS program [40] (Iowa State University, Ames, IA, USA).
4. Results and Discussion
4.1. Orbital Energy-Based Reaction Analysis of Cl + CHI → CHCl + I Reaction
Let us first consider the Cl + CHI → CHCl + I S2 reaction, which has been questioned for the reaction path [15], using the orbital energy-based reaction analysis theory in Section 2. We have found that some S2 reactions provide exceptionally large values for the reactivity index, which is the orbital energy gap gradient, , in the normalized reaction diagram, for the forward processes [4]. This indicates that these S2 reactions initially proceed not through electron transfer but structural change. In particular, large reactivity indices are given for all the examined S2 reactions involving the substitution of different ions: X + CHY → CHX + Y. We, therefore, applied the orbital energy-based reaction analysis theory to the Cl + CHI → CHCl + I reaction, which also involves the substitution of ions, to determine if this reaction is included in these exceptional S2 reactions.
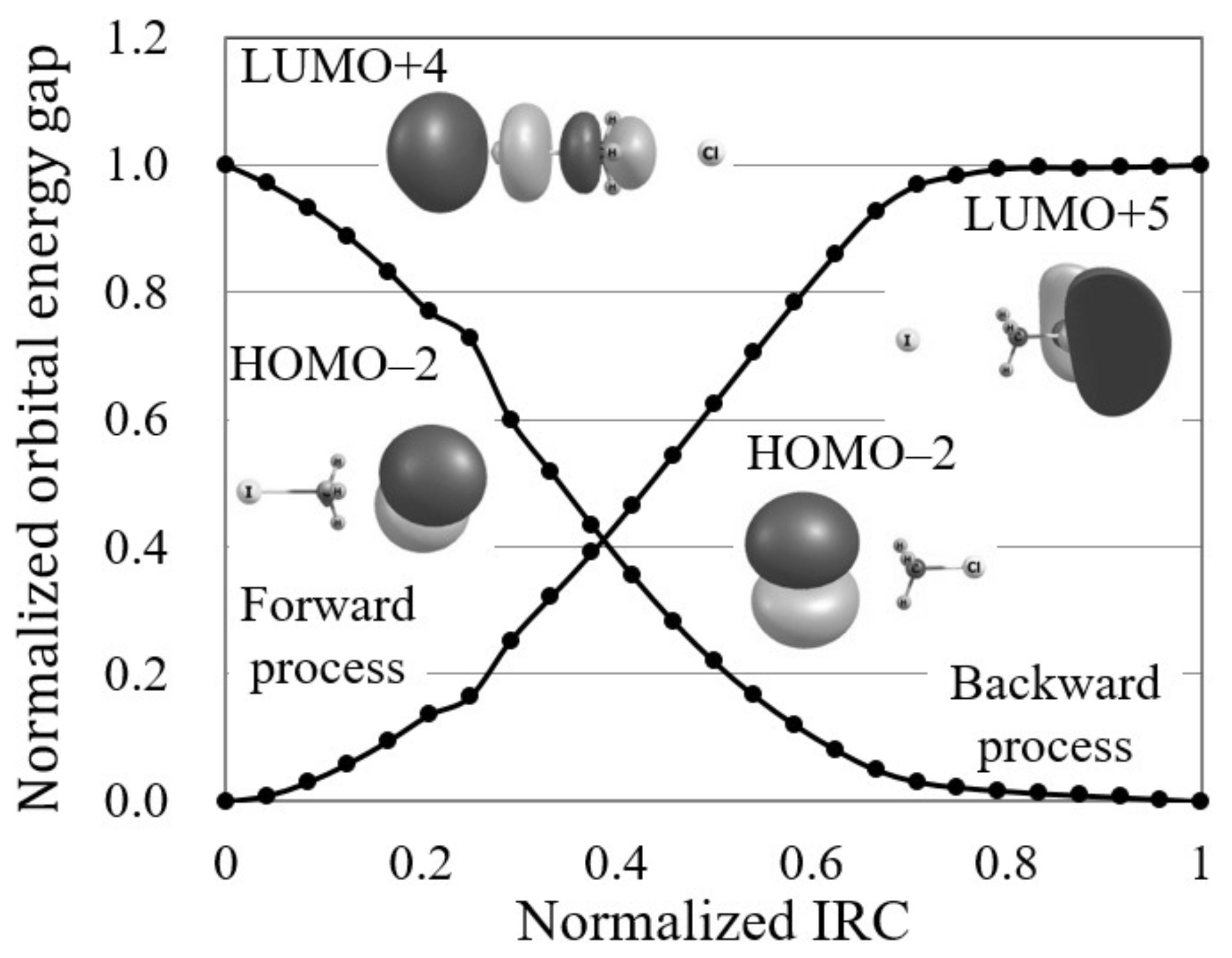
Figure 2 illustrates the normalized reaction diagram of the Cl + CHI → CHCl + I reaction. As shown in the figure, the forward process of this S2 reaction gives a larger reactivity index, , than that of the backward process, . Though both these index values are so small that these reactions are interpreted as electron transfer-driven for the threshold (0.250) in this study, the relatively-large index of the forward process could indicate the possibility of avoiding the minimum energy path as Hase and coworkers have suggested [15].
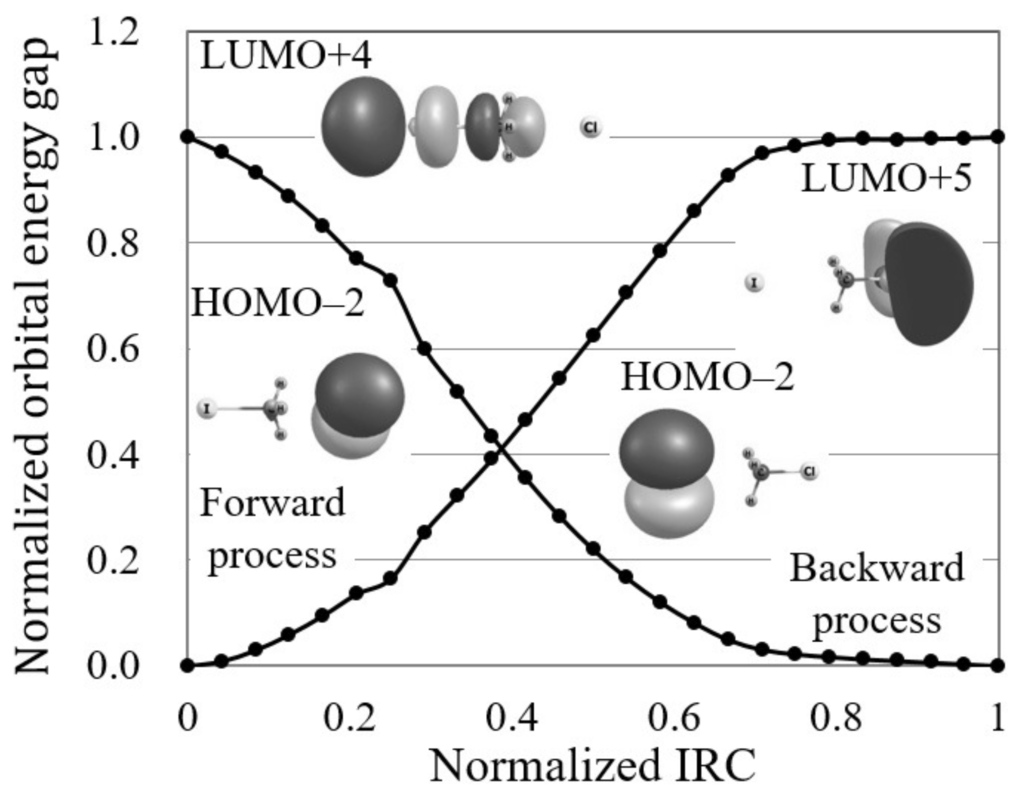
Figure 2.
The normalized reaction diagram of the Cl + CHI → CHCl + I reaction by LC-BOP/Def2-TZVPD calculation. The images of the molecular orbitals mainly contributing to each reaction process are also shown.
4.2. Reaction Path Search for Cl + CHI → ClCH + I Reaction
To investigate the Cl + CHI → CHCl + I reaction in further detail, it is meaningful to explore the overall process of this S2 reaction by the arficial force induced reaction method. As mentioned in Section 1, a different reaction path called a “roundabout path” has been proposed besides the minimum energy path for this reaction. In the roundabout path, a methyl (CH) group turns around an iodine ion (I) to move to chloride ion (Cl) [15]. Though this roundabout path has been proposed using molecular dynamics (MD) simulations, it has not yet been revealed why this path is tracked.
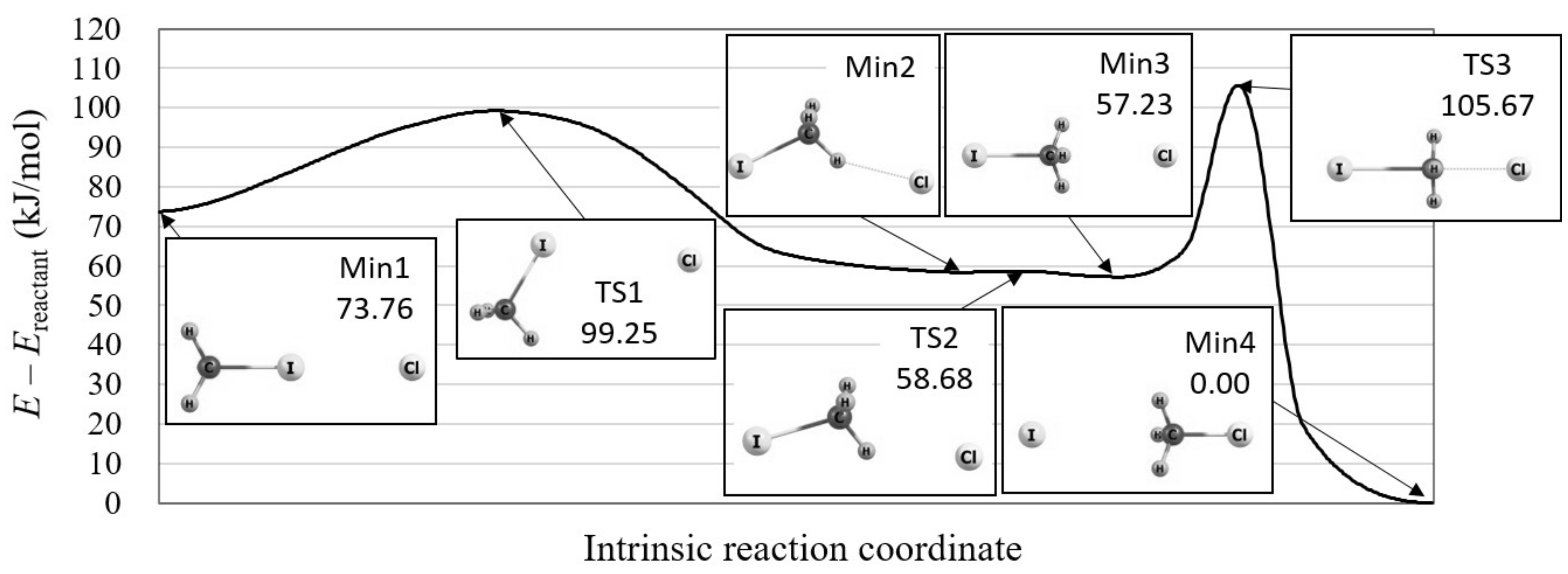
We, therefore, explored the reaction path in the vicinity of this S2 reaction using the reaction path search method in the GRRM program. As a result, we found that there is a precursor process just before this S2 reaction, in which the IRC is very similar to that of the roundabout path. Figure 3 illustrates the calculated overall reaction path and the molecular structures on this path. As shown in the figure, the methyl group rotates around the iodine ion before moving to the chloride ion. Note that the significance of the “Min1” complex is suggested in a recent study using a dynamics simulation [20]. The calculated potential energy curve shows that the barrier height of this precursor process is only 25.49 kJ/mol, which is much lower than that of the successive S2 reaction, 48.44 kJ/mol. This indicates that this precursor process naturally proceeds under the condition that the S2 reaction goes on.
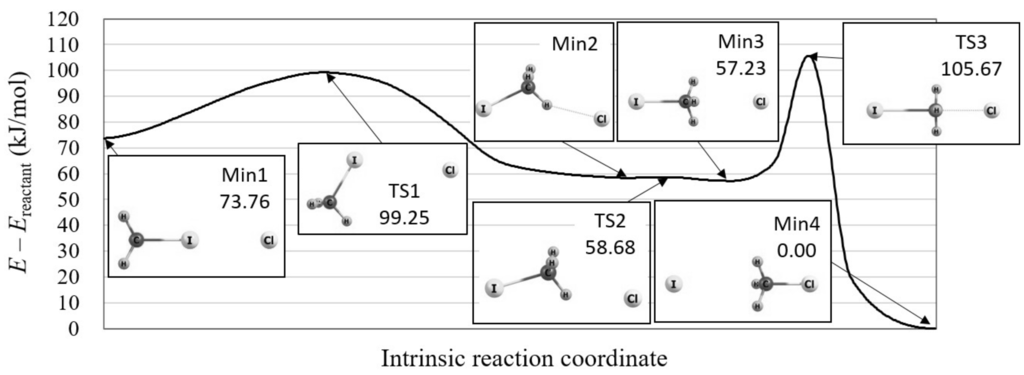
Figure 3.
The potential energy curve of the overall Cl + CHI → CHCl + I reaction including its precursor process, which is calculated by the reaction path search method in the GRRM program with LC-BOP+LRD/Def2-TZVPD. The molecular structures are also shown for the local minima (Min) and transition states (TS) on the potential energy curve.
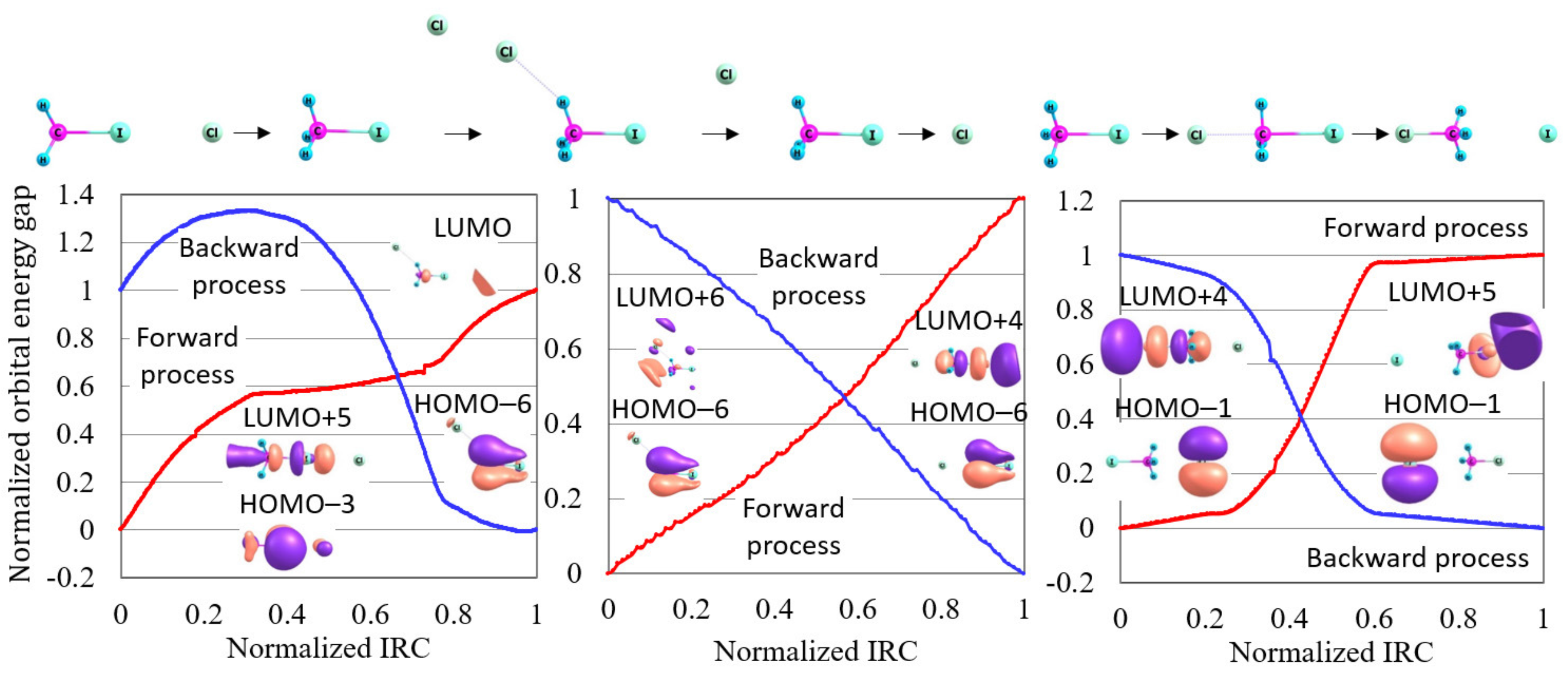
How is this precise reaction pathway interpreted in the orbital energy-based reaction analysis theory? Figure 4 shows the normalized reaction diagram for the three steps of this S2 reaction with the images of molecular orbitals contributing to each reaction step. In this figure, the most remarkable is that the inclusion of the van der Waals correlation decreases the reactivity index for the forward process of the S2 reaction (the third step). This result indicates that van der Waals correlation plays a significant role in the electronic states of the forward process of this S2 reaction. Applying the orbital energy-based reaction analysis shows that the contributing molecular orbitals of this reaction are HOMO−1 and LUMO+4 for the forward process, and HOMO−1 and LUMO+5 for the backward process. As shown in Figure 3, the contributing molecular orbitals correctly correspond to the electron transfer from I to the CH-Cl bond for the forward process and from Cl to CH-I for the backward process. It is, therefore, concluded that this S2 reaction process runs through the electron transfer similarly to many other reactions.
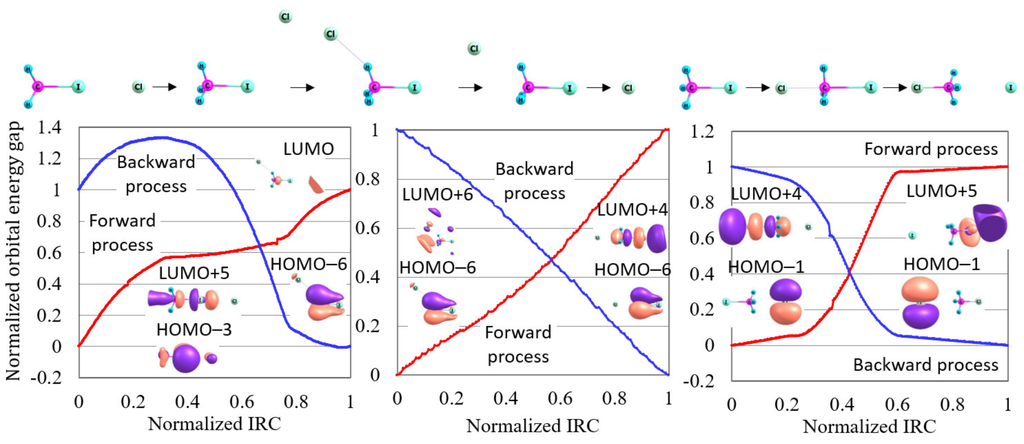
Figure 4.
The normalized reaction diagrams of the three steps of the Cl + CHI → CHCl + I reaction given in the potential energy curve of Figure 3, which is calculated by LC-BOP+LRD/Def2-TZVPD. The images of the molecular orbitals mainly contributing to each reaction process are also shown.
Figure 4 also shows that large reactivity indices are given for the two precursor steps of this reaction. This indicates that these precursor steps proceed through structural change. This result may be related to the fact that the roundabout path has been found in MD simulations [15]. That is, this precursor process similar to the roundabout path is not a usual chemical reaction induced by the electronic state variation from the electron transfer but a dynamic process depending on the kinetic state of the nuclei. Nonetheless, this dynamic process plays a key role in leading to the front of the S2 reaction. We expect that there are many other reactions which are induced by such dynamic processes.
4.3. S2 Reactions in Aqueous Solution
Next, let us compare the orbital energy-based reaction analysis theory with conventional orbital-based theories using frontier orbitals (HOMO and LUMO) and global hardness by applying them to the calculations of S2 reactions in aqueous solution. Actually, it has been reported that conventional orbital-based reaction analysis theories have poor applicabilities to solution reactions, though they have contributed to the interpretation of various gas-phase reactions such as Diels–Alder reactions. If the orbital energy-based reaction analysis theory is well applicable to solution reactions, this theory is established to extend the applicability of the orbital-based reaction analysis theories. In the orbital-based theories, reactions are explained by the electron transfer between frontier orbitals and by the maximum hardness rule [41], which suggests that reactions proceed to maximize global hardnesses. Though many studies have reported the poor applicability of the orbital-based theories, how to improve the applicability has not been suggested.
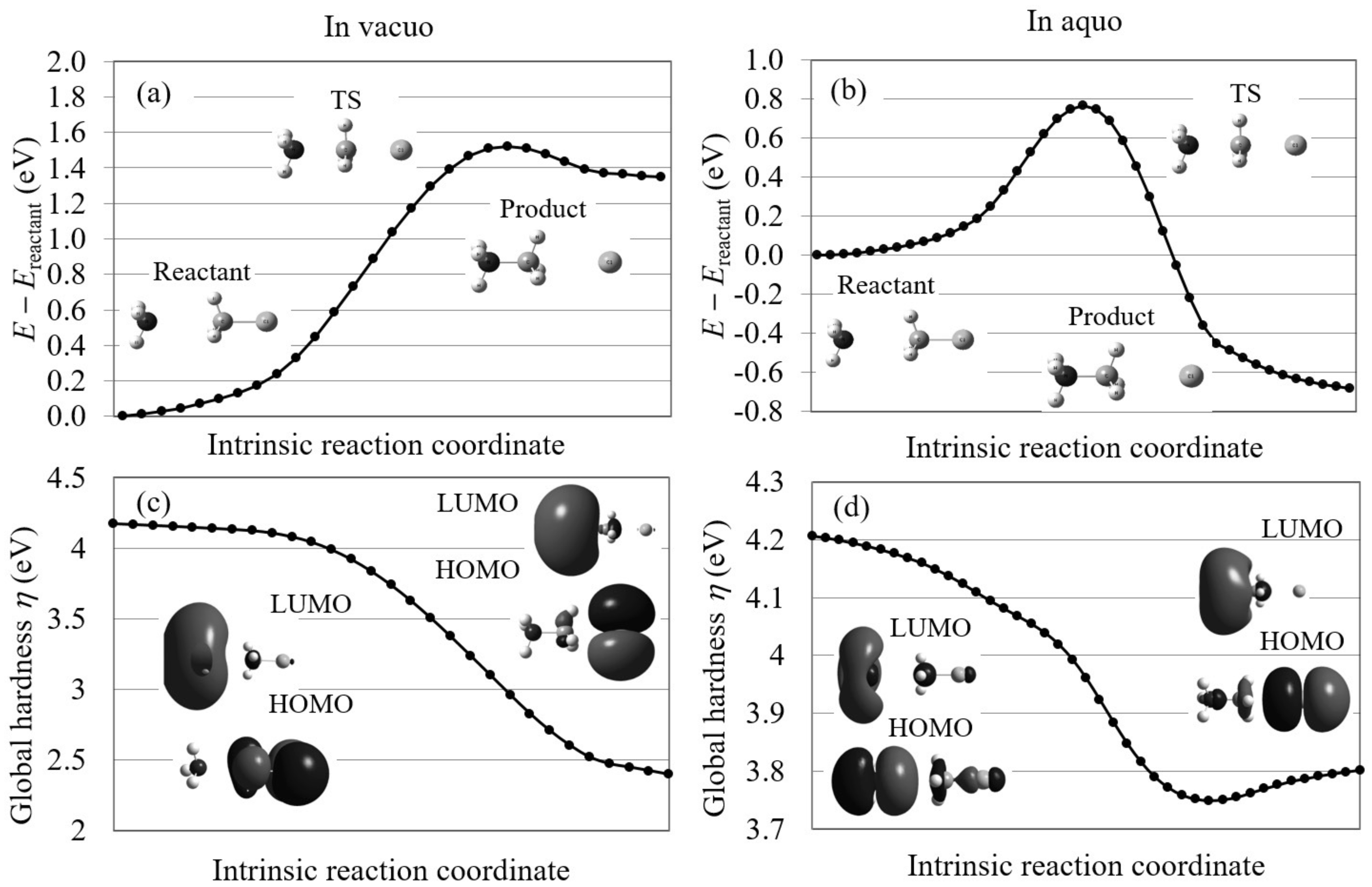
First, we analyzed the Menschutkin reaction (NH + CHCl → NHCH + Cl) using conventional reaction analysis theories. This reaction, which converts neutral reactants into a pair of charged products, is known to proceed only in polar solution. In Figure 5, the calculated potential energy curves on the intrinsic reaction coordinates and the global hardnesses on these coordinates are illustrated for the reaction in gas-phase and in aqueous solution with the molecular structures of the reactants, transition states (TSs), and products and the molecular orbitals contributing to the reactions in the initial processes. The given potential energy curves supports the experimental results that this reaction does not proceed in gas phase because it is endothermic (reaction energy = 1.35 eV) with very large reaction barrier height (1.52 eV) in gas phase, while it is exothermic ( = −0.68 eV) with relatively small barrier height (0.75 eV) in aqueous solution. The figure also shows that the global hardnesses violate the maximum hardness rule both in gas phase and in aqueous solution. In the figure, the HOMO and LUMO images are also inconsistent with the experimental results for the reaction in aqueous solution because the electron distributions of both HOMO and LUMO are biased to the NH group in the forward process. These results indicate that it is hard for conventional orbital-based reaction analysis theories to interpret this S2 reaction.
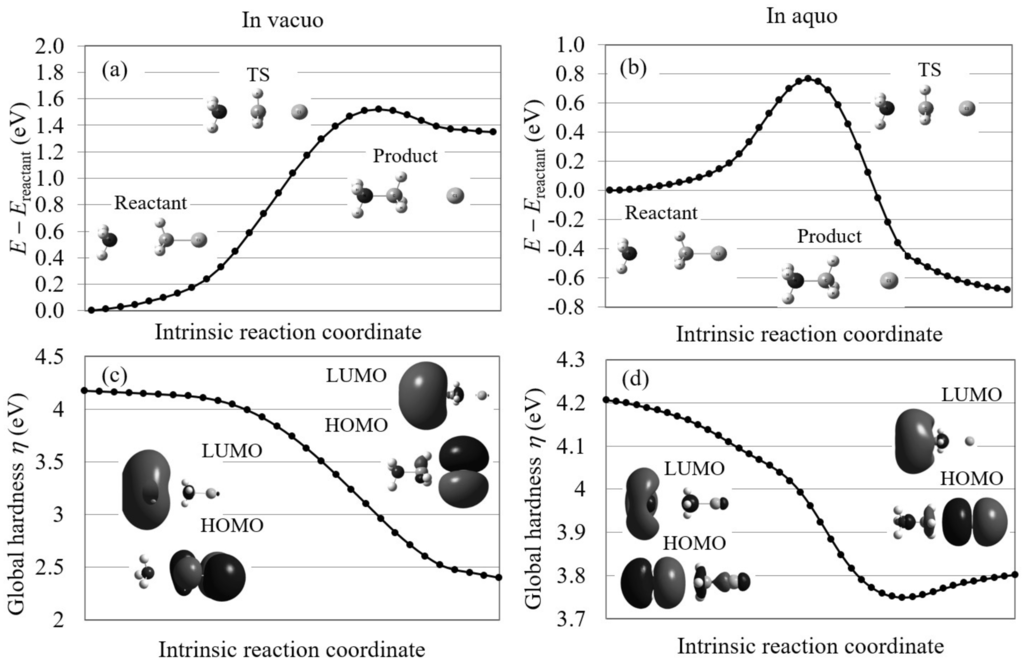
Figure 5.
The potential energy curves and the global hardnesses of the Menschutkin reaction (NH + CHCl → NHCH + Cl) in gas phase and in aqueous solution on their intrinsic reaction coordinates, which are calculated by LC-BLYP/aug-cc-pVTZ. The images of the molecular orbitals mainly contributing to each reaction process are also shown.
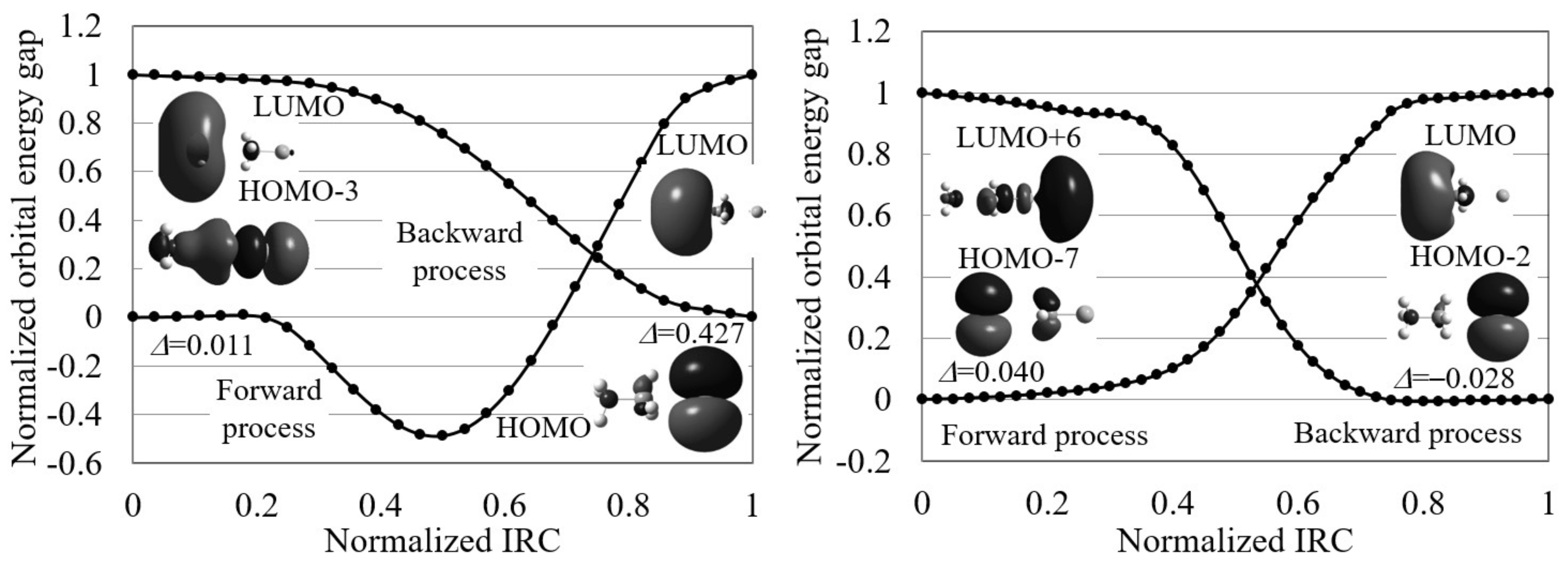
Figure 6 displays the normalized reaction diagram of the Menschutkin reaction. As shown in the figure, this reaction gives small indices indicating electron transfer-driven processes except for the backward process in gas phase. However, the contributing orbitals are not the frontier orbitals except for those of the backward process in gas phase. The orbital images are also very different for the reactions in gas-phase and in aqueous solution: the gas-phase reaction begins with the electron transfer from the CH-Cl bond to NH, while the aqueous solution reaction proceeds through the electron transfer from NHCH to Cl. That is, electrons move to NH in gas phase and to Cl in aqueous solution. This result is consistent with the experimental result that this reaction forms ions from the neutral molecules in aqueous solution. We, therefore, conclude that though electron transfer rapidly take place in the forward process of this reaction, the directions of the transfers are different in gas-phase and in aqueous solution.
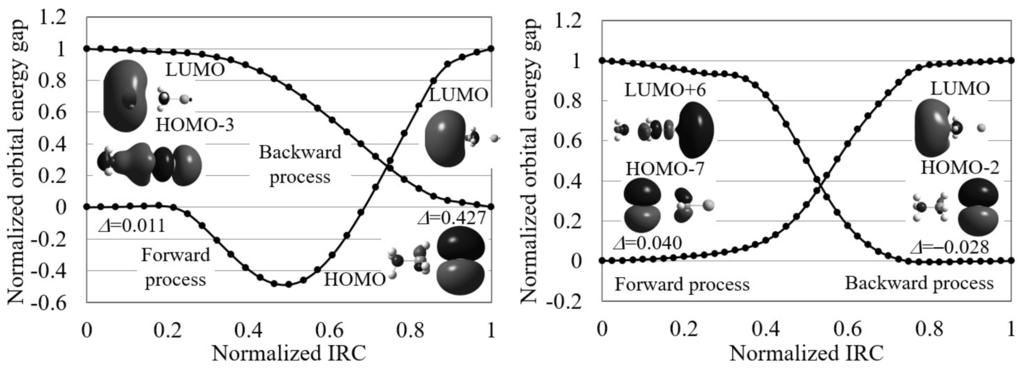
Figure 6.
The normalized reaction diagrams of the three steps of the Menschutkin reaction (NH + CHCl → NHCH + Cl) in gas phase and in aqueous solution, which are calculated by LC-BLYP/aug-cc-pVTZ. The images of the molecular orbitals mainly contributing to each reaction process and the calculated orbital energy gap gradient values are also shown.
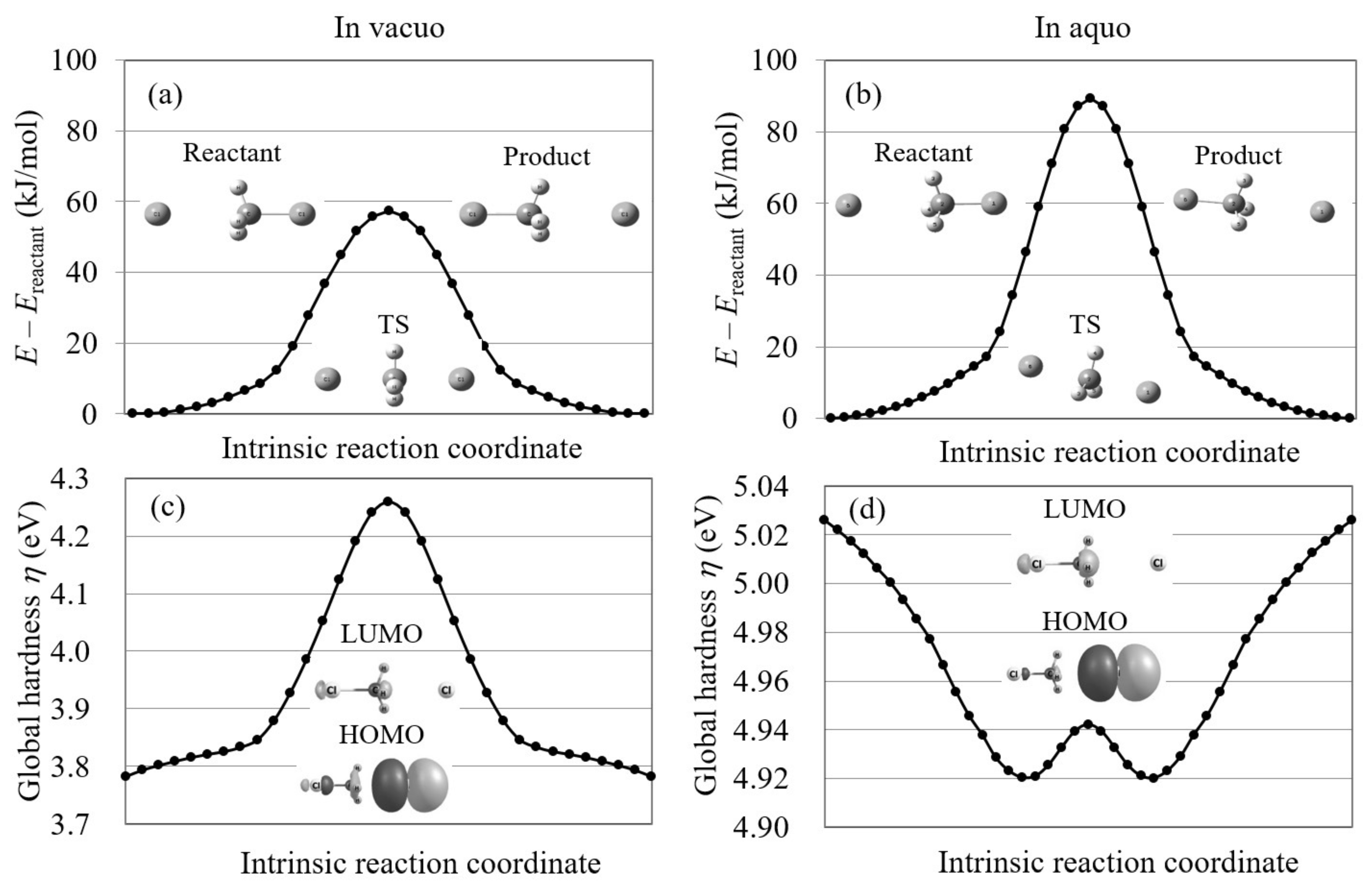
We also explored the Cl...CHCl → ClCH...Cl reaction using conventional reaction analysis theories. For this reaction, the potential energy curve is symmetric because the reactant and product are the same. Figure 7 plots the potential energy curve and global hardness of this symmetric reaction. The figure shows that the global hardness is also symmetric on the IRCs of this reaction and therefore violates the maximum hardness rule. Moreover, the frontier orbitals do not correspond to this reaction as seen in the figure. This indicates that conventional orbital-based reaction analysis theories cannot explain this reaction.
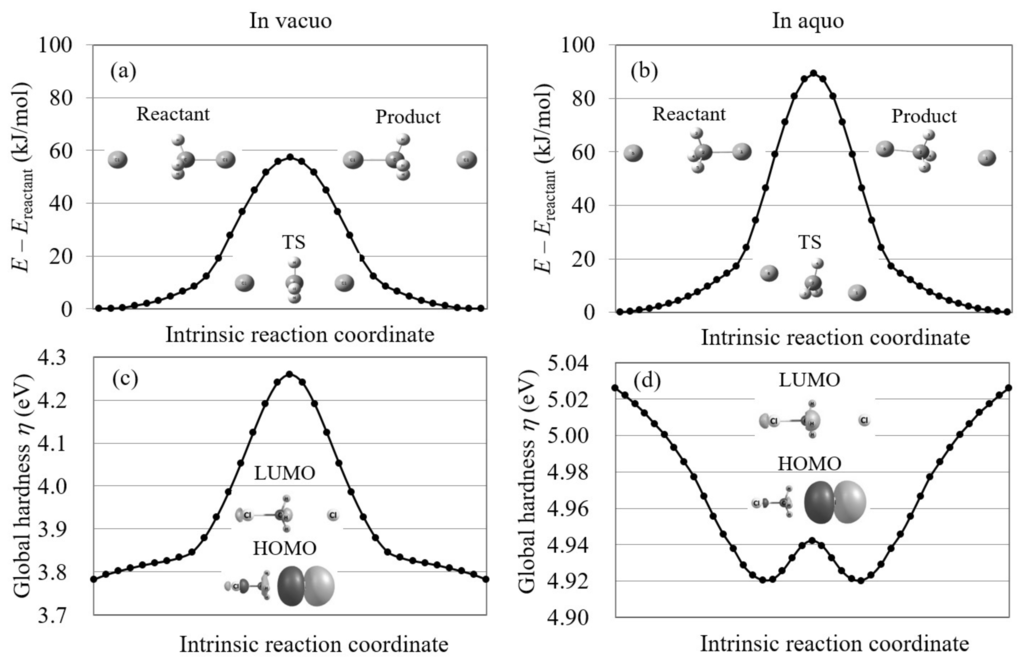
Figure 7.
The potential energy curves and the global hardnesses of the Cl...CHCl → ClCH...Cl reaction in gas phase and in aqueous solution on their intrinsic reaction coordinates, which are calculated by LC-BLYP/aug-cc-pVTZ. The images of the molecular orbitals mainly contributing to each reaction process are also shown.
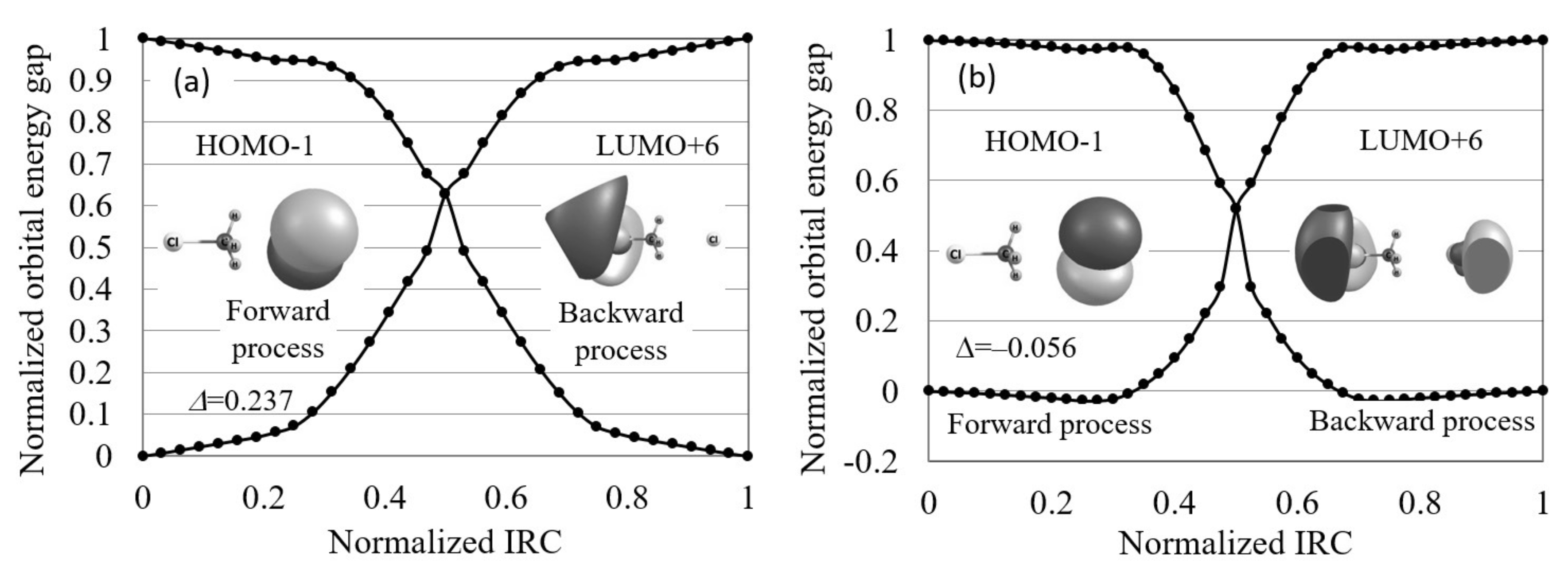
Finally, we applied the orbital energy-based reaction analysis theory to the Cl...CHCl → ClCH...Cl reaction. In Figure 8, the normalized reaction diagram of this symmetric S2 reaction is illustrated. The figure shows that the contributing orbitals are not the frontier orbitals but HOMO−1 and LUMO+6. Using these contributing orbitals, we found that the orbital energy gap increases on the IRCs in agreement with the maximum hardness rule, despite this reaction being symmetric. The orbital images also indicate that this reaction proceeds through the electron transfer from one Cl to another Cl.
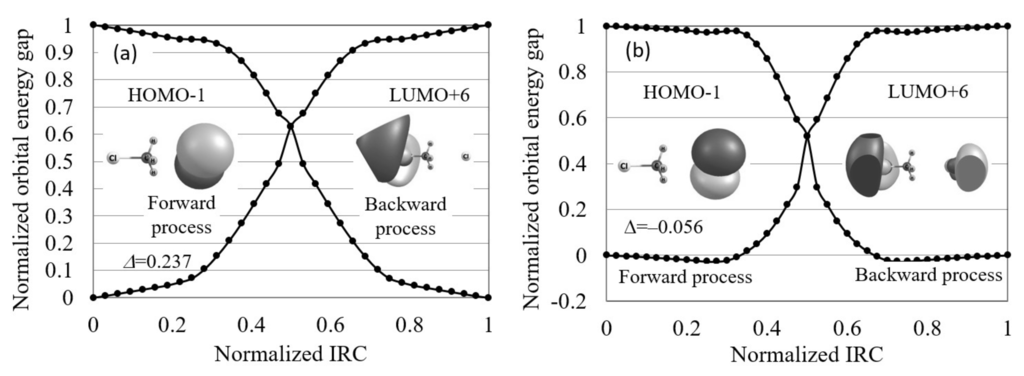
Figure 8.
The normalized reaction diagrams of the Cl...CHCl → ClCH...Cl reaction in gas phase and in aqueous solution, which are calculated by LC-BLYP/aug-cc-pVTZ. The images of the molecular orbitals mainly contributing to each reaction process and the calculated orbital energy gap gradient values are also shown.
As mentioned above, the orbital energy-based reaction analysis theory can reveal the details of the solution reactions that conventional orbital-based reaction analysis theories cannot target. This theory makes it possible to clarify the molecular orbitals contributing to the reaction and to determine if the reaction is electron transfer-driven or structural change-driven. We, therefore, conclude that this orbital energy-based reaction analysis theory can extend the applicability of the orbital-based theories.
5. Conclusions
In this study, we have formed the foundation of the orbital energy-based reaction analysis theory [4] as the extension of the orbital-based reaction analysis theories. Based on this orbital energy-based theory, we have explored the S2 reactions, for which the minimum energy path is questioned, and the difference of the S2 reactions in gas-phase and in aqueous solution.
First, we have extended the conceptual DFT [3], an orbital-based reaction analysis theory, to the orbital energy-based one in order to construct the foundation of the orbital energy-based reaction analysis theory. As a result, we have proven that the Fukui function, which is the density variation in terms of the occupation number and is the main reactivity index of the orbital-based reaction analysis theories, is equal to the outermost orbital energy variation along structural change. In the orbital-based reaction analysis theories, the orbitals contributing to reactions have been interpreted to give large Fukui functions. The contributing orbitals are, therefore, expected to give large orbital energy variations from reactants to products. Note also that small outermost orbital energy variations indicate that the reactions proceed through electron transfer [4]. Consequently, we succeed to lay the foundation of the orbital energy-based reaction analysis theory on the ground of the orbital-based analysis theories and to give physical meanings for determining the contributing molecular orbitals and for specifying the electron transfer-driven reaction processes.
Next, we have applied the orbital energy-based reaction analysis theory to the Cl + CHI → ClCH + I reaction, for which another reaction path than the minimum energy path called the roundabout path has been proposed in MD simulation studies [15]. To investigate this reaction in further detail, we have determined the IRCs in the vicinity of this reaction using a reaction path search method called the artificial force induced reaction method in the GRRM program. As a result, we have found that there is a precursor process just before the usual S2 reaction process. The IRCs of the precursor process are very similar to the roundabout path. The orbital energy-based reaction analysis using these IRCs indicates that the S2 reaction proceeds through electron transfer, while the precursor process runs through structural change. This may cause the precursor process called the roundabout path has been found only in MD simulation studies.
To explore the applicability of the orbital energy-based reaction analysis theory, we have also analyzed two S2 reactions in aqueous solution: the Menschutkin reaction (NH + CHCl → NHCH + Cl) and the Cl...CHCl → ClCH...Cl reaction. Consequently, we have found that the Menschutkin reaction, which proceeds only in polar solution, has different contributing orbitals in gas-phase and in aqueous solution: this reaction is driven by the electron transfer from CHCl to NH in gas phase, while it is driven by the electron transfer from NHCH to Cl in aqueous solution. For the Cl...CHCl reaction, we have found that it is driven by the electron transfer from one Cl to another Cl by specifying the pair of the contributing orbitals, in which orbital energy gap increases as the reaction proceeds, in contrast to the HOMO-LUMO gap.
As mentioned above, we have succeeded in developing an orbital energy-based reaction analysis theory as an extension of the orbital-based reaction analysis theories, the frontier orbital theory and the conceptual DFT, in terms of orbital energies. We have also shown that this orbital energy-based theory can interpret various types of S2 reactions from the electronic state viewpoint. We expect that this theory will make it possible to analyze a wide variety of reactions focusing on their electronic state variations.
Supplementary Materials
The following are available online at http://www.mdpi.com/2079-3197/4/3/23/s1. Figures S1–S8: The chosen contributing orbital energies of trial reactions along the IRCs.
Acknowledgments
This research was supported by the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT) (Grants: 23225001 and 24350005) and by the Superlative, Stable, and Scalable Performance Fuel Cell (SPer-FC) projects of the New Energy and Industrial Technology Development Organization (NEDO) of the Japanese Ministry of International Trade and Industry (MITI).
Author Contributions
T.T. developed the orbital energy-based theory; T.T. and S.M. proposed the collaborative study on the application of this theory and the reaction search method to the S2 reaction analyses; S.M. and Y.H. performed the reaction search calculations and determined the roundabout-like reaction path; R.K.S. carried out the S2 reaction calculations based on the orbital energy-based theory; T.T. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Itatani, J.; Levesque, J.; Zeidler, D.; Niikura, H.; Pepin, H.; Kieffer, J.C.; Corkum, P.B.; Vileneuve, D.M. Tomographic imaging of molecular orbitals. Nature 2004, 432, 867–871. [Google Scholar] [CrossRef] [PubMed]
- Fukui, K.; Yonezawa, T.; Shingu, H. A molecular orbital theory of reactivity in aromatic hydrocarbons. J. Chem. Phys. 1952, 20, 722–725. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Parr, R.G. Legendre transforms and Maxwell relations in density functional theory. J. Chem. Phys. 1982, 77, 399–407. [Google Scholar] [CrossRef]
- Tsuneda, T.; Singh, R.K. Reactivity index based on orbital energies. J. Comput. Chem. 2014, 35, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Tsuneda, T. Density Functional Theory in Quantum Chemistry; Springer: Tokyo, Japan, 2014. [Google Scholar]
- Iikura, H.; Tsuneda, T.; Yanai, T.; Hirao, K. A long-range correction scheme for generalized-gradient-approximation exchange functionals. J. Chem. Phys. 2001, 115, 3540–3544. [Google Scholar] [CrossRef]
- Tawada, Y.; Tsuneda, T.; Yanagisawa, S.; Yanai, T.; Hirao, K. A long-range-corrected time-dependent density functional theory. J. Chem. Phys. 2004, 120, 8425–8433. [Google Scholar] [CrossRef] [PubMed]
- Tsuneda, T.; Song, J.W.; Suzuki, S.; Hirao, K. On Koopmans’ theorem in density functional theory. J. Chem. Phys. 2010, 133, 174101. [Google Scholar] [CrossRef] [PubMed]
- Tsuneda, T.; Kamiya, M.; Hirao, K. Regional self-interaction correction of density functional theory. J. Comput. Chem. 2003, 24, 1592–1598. [Google Scholar] [CrossRef] [PubMed]
- Nakata, A.; Tsuneda, T.; Hirao, K. Modified Regional Self-Interaction Correction Method Based on the Pseudospectral Method. J. Phys. Chem. A 2010, 114, 8521–8528. [Google Scholar] [CrossRef] [PubMed]
- Nakata, A.; Tsuneda, T. Density functional theory for comprehensive orbital energy calculations. J. Chem. Phys. 2013, 139, 064102. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Tsuneda, T. Reaction energetics on long-range corrected density functional theory: Diels–Alder reactions. J. Comput. Chem. 2013, 34, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Sham, L.J.; Schlüter, M. Density-functional theory of the band gap. Phys. Rev. B 1985, 32, 3883–3889. [Google Scholar] [CrossRef]
- Hase, W.L. Simulations of Gas-Phase Chemical Reactions: Applications to SN2 Nucleophilic Substitution. Science 1994, 266, 998–1002. [Google Scholar] [CrossRef] [PubMed]
- Mikosch, J.; Trippel, S.; Eichhorn, C.; Otto, R.; Lourderaj, U.; Zhang, J.X.; Hase, W.L.; Weidemuller, M.; Wester, R. Imaging nucleophilic substitution dynamics. Science 2008, 319, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Mikosch, J.; Trippel, S.; Otto, R.; Weidemüller, M.; Wester, R.; Hase, W.L. F− + CH3I → FCH3 + I− reaction dynamics. Nontraditional atomistic mechanisms and formation of a hydrogen-bonded complex. J. Phys. Chem. Lett. 2010, 1, 2747–2752. [Google Scholar] [CrossRef]
- Zhang, J.; Lourderaj, U.; Sun, R.; Mikosch, J.; Wester, R.; Hase, W.L. Simulation studies of the Cl− + CH3I SN2 nucleophilic substitution reaction: Comparison with ion imaging experiments. J. Chem. Phys. 2013, 138, 114309. [Google Scholar] [CrossRef] [PubMed]
- Szabó, I.; Császár, A.G.; Czakó, G. Dynamics of the F− + CH3Cl → Cl− + CH3F SN2 reaction on a chemically accurate potential energy surface. Chem. Sci. 2013, 4, 4362–4370. [Google Scholar] [CrossRef]
- Szabó, I.; Czakó, G. Revealing a double-inversion mechanism for the F−+ CH3Cl SN2 reaction. Nat. Commun. 2015, 6, 5972–5977. [Google Scholar] [CrossRef] [PubMed]
- Stei, M.; Carrascosa, E.; Kainz, M.A.; Kelkar, A.H.; Meyer, J.; Szabó, I.; Czakó, G.; Wester, R. Influence of the leaving group on the dynamics of a gas-phase SN2 reaction. Nat. Chem. 2016, 8, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. B 1964, 136, 864–871. [Google Scholar] [CrossRef]
- Levy, M. Universal variational functionals of electron densities, first-order density matrices, and natural spin-orbitals and solution of the v-representability problem. Proc. Natl. Acad. Sci. USA 1979, 76, 6062–6065. [Google Scholar] [CrossRef] [PubMed]
- Tsuneda, T. Chemical Reaction Analyses Based on Orbitals and Orbital Energies. Int. J. Quantum Chem. 2015, 115, 270–282. [Google Scholar] [CrossRef]
- Janak, J.F. Proof that ∂E/∂ni = ϵ in density-functional theory. Phys. Rev. B 1978, 18, 7165–7168. [Google Scholar] [CrossRef]
- Hratchian, H.P.; Schlegel, H.B. Accurate reaction paths using a Hessian based predictor-corrector integrator. J. Chem. Phys. 2004, 120, 9918–9924. [Google Scholar] [CrossRef] [PubMed]
- Hratchian, H.P.; Schlegel, H.B. Using Hessian updating to increase the efficiency of a Hessian based predictor-corrector reaction path following method. J. Chem. Theory Comput. 2005, 1, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. A 1965, 140, 1133–1138. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Ohno, K.; Morokuma, K. Systematic exploration of the mechanism of chemical reactions: The global reaction route mapping (GRRM) strategy using the ADDF and AFIR methods. Phys. Chem. Chem. Phys. 2013, 15, 3683–3701. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Taketsugu, T.; Morokuma, K. Exploring transition state structures for intramolecular pathways by the artificial force induced reaction method. J. Comput. Chem. 2014, 35, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, M.; Tsuneda, T.; Hirao, K. A density functional study of van der Waals interactions. J. Chem. Phys. 2002, 117, 6010–6015. [Google Scholar] [CrossRef]
- Sato, T.; Tsuneda, T.; Hirao, K. Van der Waals interactions studied by density functional theory. Mol. Phys. 2005, 103, 1151–1164. [Google Scholar] [CrossRef]
- Sato, T.; Tsuneda, T.; Hirao, K. Long-range corrected density functional study on weakly bound systems: Balanced descriptions of various types of molecular interactions. J. Chem. Phys. 2007, 126, 1402–1406. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Nakai, H. Density functional method including weak interactions: Dispersion coefficients based on the local response approximation. J. Chem. Phys. 2009, 131, 224104. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Tsuneda, T.; Suzumura, T.; Hirao, K. A new one-parameter progressive Colle-Salvetti-type correlation functional. J. Chem. Phys. 1999, 110, 10664–10678. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Parr, R.G.; Chattaraj, P.K. Principle of maximum hardness. J. Am. Chem. Soc. 1991, 113, 1854–1855. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).