1. Introduction
Partial atomic charges are useful descriptors for interpreting the results of quantum chemical calculations in a chemically intuitive fashion. It comes down to the description of the electron charge distribution within molecules through assigning a partial charge to each atom of the molecules. Because partial atomic charges are not physical observables and there is no strict quantum mechanical definition of them, many different models have been proposed to extract partial atomic charges from the molecular charge distribution. In principle, the models of partial atomic charges may be classified into three groups [
1]. The first one covers models based on partitioning the molecular wave function into atomic contributions in terms of basis functions used to construct this wave function. The Mulliken population analysis [
2] and the natural population analysis (NPA) [
3,
4] are typical examples of such models. The Mulliken population analysis is probably the best known of all models of partial atomic charge. Due to its simplicity, this model is computationally very attractive. However, its results tend to vary with the basis set employed and they are unrealistic in some cases [
5]. These drawbacks partially arise from the fact that the Mulliken population analysis utilizes a nonorthogonal basis set. This problem is overcome by the NPA in which orthonormal natural atomic functions are constructed. The distributed multipole analysis of Gaussian wave functions (GDMA) [
6] is also ranked among the models of the first group. In this analysis, the distribution of the molecular electron density is represented by multipoles located on the individual atoms of a molecule. The partitioning of the molecular electron density between the atoms is carried out in the basis-function space because the multipoles are obtained by using the products of Gaussian basis functions. The second group of partial atomic charge models comprises models that partition the molecular electron density into atomic domains in the physical space. These atomic domains are defined by means of Bader’s atoms-in-molecules (AIM) topological analysis of the molecular electron density [
7] or using the so-called “promolecular” density proposed originally by Hirshfeld [
8]. To be more specific, the AIM division of a molecule into atoms is based on the zero-flux surfaces of the molecular electron density, and then this density is integrated over the resulting atomic domains to obtain their partial charges. In the Hirshfeld model the partial charge of each atom is calculated by assuming that the electron density at each point is shared between the surrounding atoms in direct proportion to their free-atom electron densities at the corresponding distances from the nuclei. The original partial atomic charges obtained from the Hirshfeld population analysis can be improved through parametrization to reproduce accurately a particular molecular property. For instance, the recently developed charge model 5 (CM5) utilizes a set of parameters derived by fitting to reference values of gas-phase dipole moments [
9]. As for the models belonging to the third group, their partial atomic charges are based on the reproduction of the molecular electrostatic potential. Various schemes that fit partial atomic charges to the molecular electrostatic potential are reported, e.g., the Merz-Kollman-Singh (MKS) scheme [
10,
11], charges from electrostatic potentials (CHELP) [
12], charges from electrostatic potentials using a grid (CHELPG) [
13] and the Hu-Lu-Yang (HLY) scheme [
14]. The MKS, CHELP and CHELPG schemes differ in the selection of points surrounding a molecule. Such points are used for the calculation of the molecular electrostatic potential in the fitting procedure. The MKS scheme employs points located at four shells at a distance of 1.4, 1.6, 1.8 and 2.0 times the van der Waals radii of the atoms constituting a molecule. The CHELP scheme samples the molecular electrostatic potential at points at a distance of 2.5, 3.5, 4.5, 5.5 and 6.5 Å from the van der Waals surface. In the CHELPG scheme the points are selected from a regularly spaced grid, between 0 and 2.8 Å from the van der Waals surface. The HLY scheme introduces the so-called object function in the entire molecular volume space instead of the points around the molecule. The application of the object function improves the numerical stability of fitting results.
Partial atomic charges find application in the molecular modeling of several chemically relevant areas, such as explaining structural and reactivity differences between various molecules and their conformers [
15,
16,
17], investigating charge transfers within a single molecule and between several molecules [
18,
19,
20], and predicting p
Ka variations in series of molecules [
21,
22]. Since the definition of partial atomic charges is not strict, various models of partial atomic charges can, however, differ significantly in the reliability of their predictions. Therefore, an evaluation of the performance of a partial atomic charge model for a given problem is necessary before using it for making reliable predictions [
23,
24,
25,
26,
27,
28].
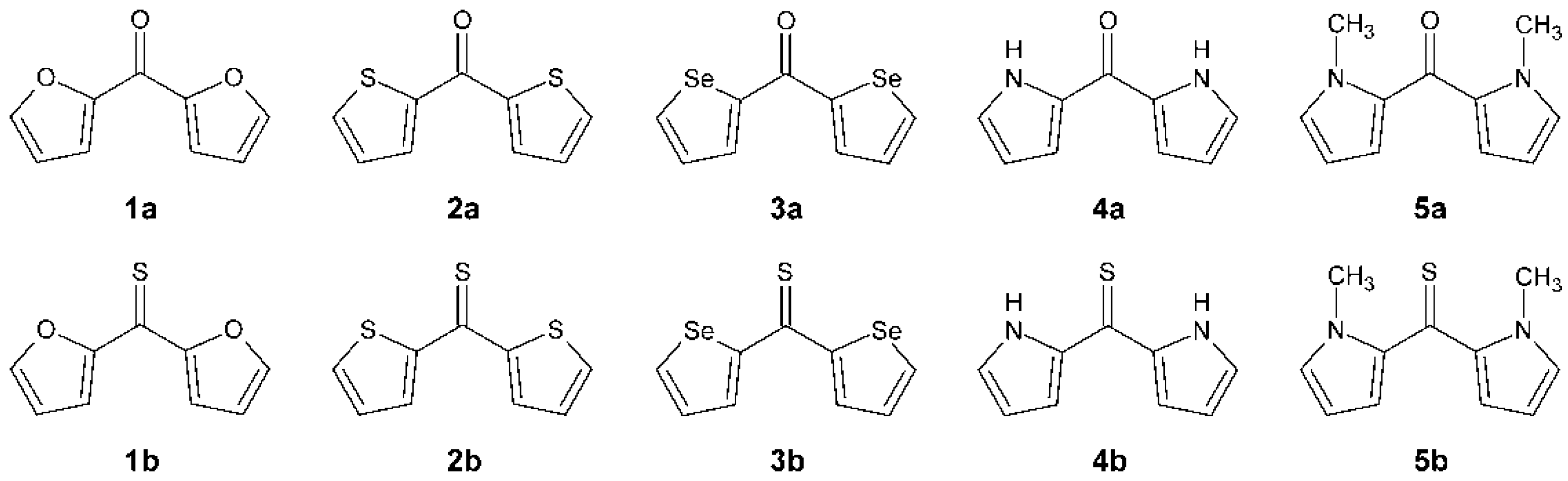
In this work, various models of partial atomic charge are tested to establish their efficiency for computations on diheteroaryl ketones and thioketones. A series of five diheteroaryl ketones and their thiocarbonyl analogs is considered. These ketones and thioketones are formed by the disubstitution of formaldehyde and thioformaldehyde with a five-membered heterocyclic group. The series includes di(furan-2-yl)methanone (
1a), di(thiophen-2-yl)methanone (
2a), di(selenophen-2-yl)methanone (
3a), di(1
H-pyrrol-2-yl)methanone (
4a), di(1-methylpyrrol-2-yl)methanone (
5a) and their thiocarbonyl analogs (
1b–
5b). The structural skeletal formulas of
1a–
5a and
1b–
5b are shown in
Figure 1. The thioketones
1b–
5b have recently been synthesized by the O/S exchange in the corresponding ketones by treatment with Lawesson’s reagent [
29]. In the first stage of the present work, various models of partial atomic charge are tested in terms of their ability to reproduce the molecular dipole moments of
1a–
5a and
1b–
5b. Next, the efficiency of the models for predicting the conformational behavior of
1a–
5a and
1b–
5b is studied. The prediction of the conformational behavior will be restricted here to the determination of a preferred conformer and the sequence of higher-energy conformers.
Figure 1.
The structural skeletal formulas of 1a–5a and 1b–5b.
Figure 1.
The structural skeletal formulas of 1a–5a and 1b–5b.
2. Computational Details
The molecular structures of
1a–
5a and
1b–
5b are taken from our previous works [
30,
31,
32] in which Becke’s three-parameter hybrid exchange functional combined with the correlation functional of Lee, Yang and Parr (B3LYP) [
33,
34,
35] and the def2-QZVPP basis set [
36] were used to optimize the geometries of the isolated molecules of
1a–
5a and
1b–
5b. It was also established there that for each of the compounds its three conformations could be formed by the rotation of heteroaryl substituents about the single C-C bonds linking these substituents with the C atom of carbonyl/thiocarbonyl group. The resulting conformers of
1a–
5a and
1b–
5b are denoted by the prefixes
cc,
ct and
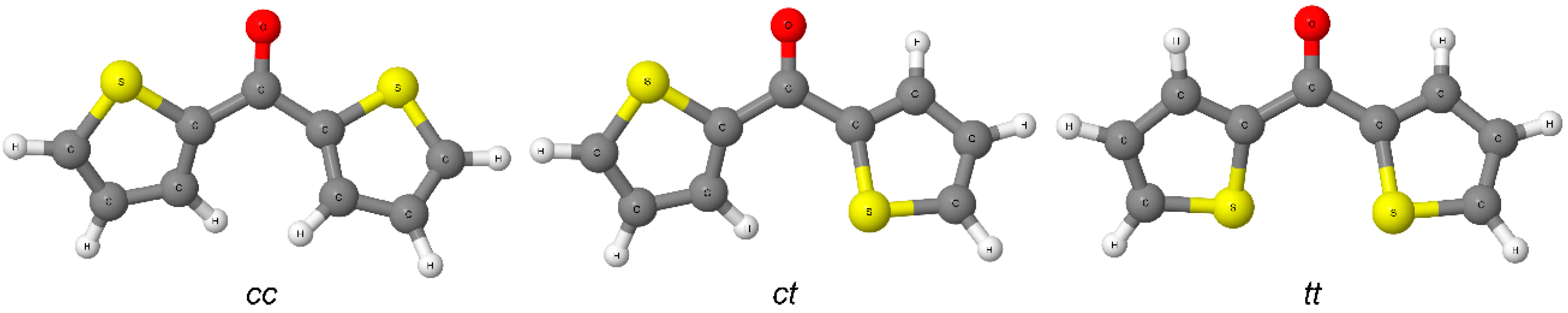
tt, indicating the spatial arrangement of ring heteroatoms with respect to the O/S atom of carbonyl/thiocarbonyl group (see
Figure 2).
Figure 2.
Three conformations of the investigated diheteroaryl ketones and thioketones. The B3LYP/def2-QZVPP-optimized conformers of 2a are shown as an example.
Figure 2.
Three conformations of the investigated diheteroaryl ketones and thioketones. The B3LYP/def2-QZVPP-optimized conformers of 2a are shown as an example.
For the
cc-,
ct- and
tt-conformers of
1a–
5a and
1b–
5b, their molecular wave functions/electron densities are calculated at three levels of theory, that is, HF/def2-QZVPP [
36,
37,
38], B3LYP/def2-QZVPP [
33,
34,
35,
36] and MP2/def2-QZVPP [
36,
39]. These molecular wave functions/electron densities are the starting point for deriving partial atomic charges from various models belonging to the three groups mentioned in the introduction. The partial atomic charges on all atoms of each conformer are determined by means of ten models, namely Mulliken, NPA, GDMA, AIM, Hirshfeld, CM5, MKS, CHELP, CHELPG and HLY. Then, the magnitude of the dipole moment μ of the conformer is approximated by the following formula:
where the index
k runs over all atoms of the conformer having
N atoms,
Q denotes the Cartesian coordinates of the corresponding atom and
q is the partial charge of the atom. In order to assess the accuracy of the dipole moments calculated using Equation (1), their values need to be compared with the corresponding reference results, preferably obtained from experimental measurements. However, such reference results are not available for individual gas-phase conformers of
1a–
5a and
1b–
5b. Therefore, the dipole moments calculated by the ten models of partial atomic charge are compared with the corresponding dipole moments obtained from the regular quantum chemical calculations involving the full molecular electron density (to be strict, these dipole moments are calculated as an expectation value of the appropriate quantum operator, which is well defined for the dipole moment).
The calculated partial atomic charges are also used for a rough estimation of the magnitude of electrostatic effects occurring in the conformers of
1a–
5a and
1b–
5b. On this basis, the conformational behavior of these compounds can be predicted. Interaction energies in the pairs of partial atomic charges (
EC(
i,
j)) are calculated using classical Coulomb’s law. These interaction energies are summed up over all pairs of partial atomic charges within each conformer (Σ
i>j EC(
i,
j)) to roughly estimate the energy
Eelst associated with electrostatic effects occurring in the conformer. The conformers of each compound can be ordered with respect to their
Eelst values. In the resulting sequence, the lower (that is, the more negative) the value of
Eelst is obtained for a conformer, the more stable the conformer is. Such a procedure taking only
Eelst into consideration assumes that the electrostatic effects play an important role in governing the conformational behavior of investigated compounds. These effects, indeed, contribute significantly to the ordering of conformers for diheteroaryl ketones and thioketones [
30,
32].
The GDMA
2.2.09 [
6] and AIMAll 14.06.21 [
40] programs have been used to calculate partial atomic charges derived from the GDMA and AIM models, respectively. All the remaining calculations have been carried out with the Gaussian 09 D.01 program [
41]. In this program, the fitting of MKS, CHELP, CHELPG and HLY partial atomic charges to reproduce the molecular electrostatic potential has involved no additional constraint of reproducing the molecular dipole moment.
3. Results and Discussion
Since the molecular dipole moment is a primary quantity providing an essential insight into the distribution of electron charge within a molecule, a reasonable test for any model of partial atomic charge is to inspect the performance of the model in predicting such a quantity. Therefore, we start testing ten models of partial atomic charge by establishing their ability to reproduce the magnitude of the dipole moments for the conformers of
1a–
5a and
1b–
5b.
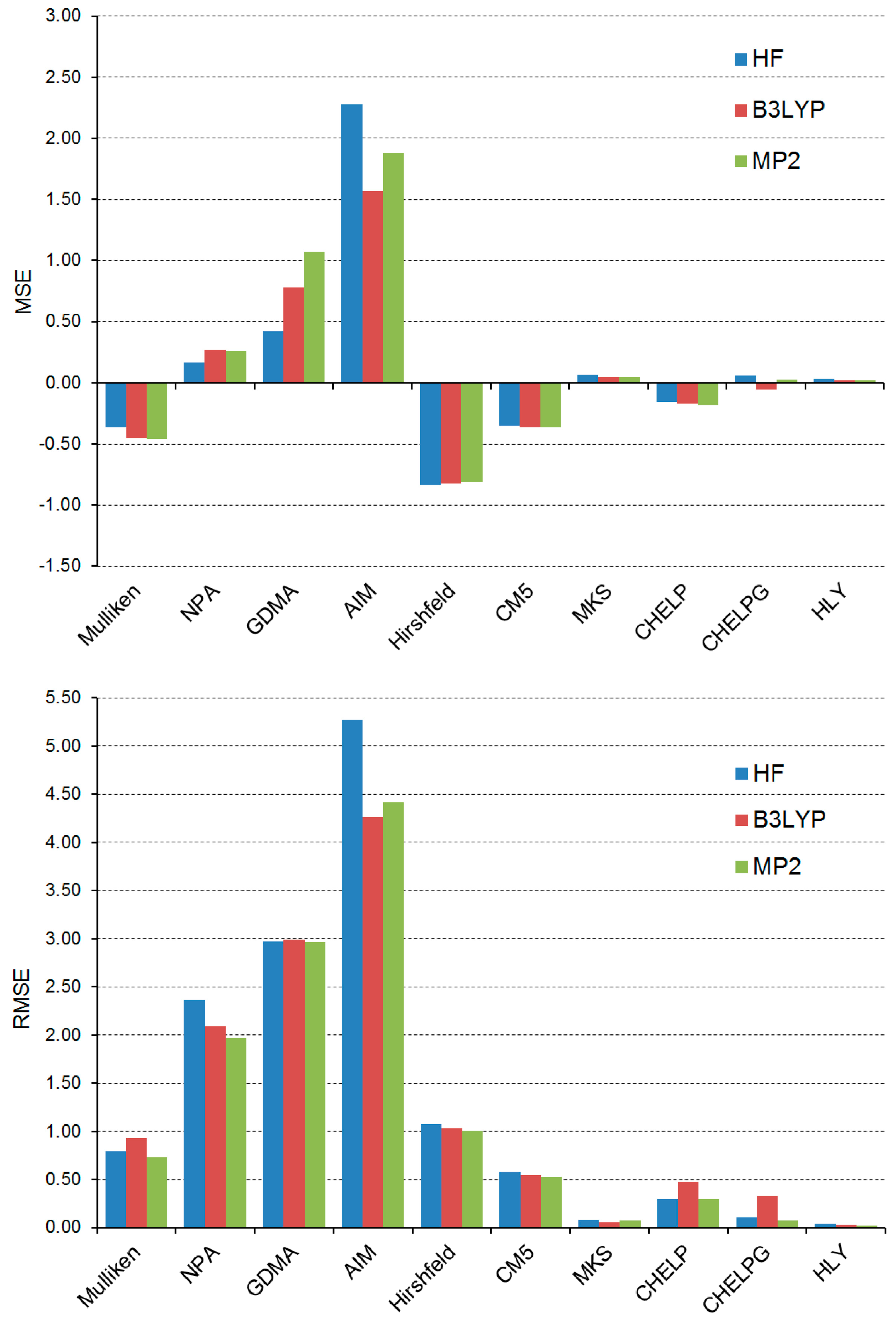
Figure 3 shows the mean signed error (MSE) and root mean square error (RMSE) in the μ values approximated by the partial atomic charges derived from each model with respect to the reference results obtained from the full-density calculations. Additionally, the values of MSE and RMSE are determined for three levels of theory. When comparing here the approximated values of μ with the corresponding reference values, both the former and the latter are obtained from the molecular wave functions/electron densities generated at the same level of theory. The approximated and reference μ values used for the calculations of the MSE and RMSE presented in
Figure 3 can be found in
Tables S1–S6 in Supplementary Materials. The MSE provides information about systematic errors occurring in the approximated values of μ, whereas the RMSE allows us to rank the accuracy of individual models for reproducing the reference values of μ.
It is evident from what the lower plot in
Figure 3 shows that the μ values obtained from the MKS and HLY partial atomic charges reproduce best the μ values calculated from the full density. Among the models that are not based on electrostatic potential fitting, the CM5 model offers the highest accuracy of the approximated μ values. The μ values approximated by the AIM model lead to the largest RMSE values. The accuracy of four models that are based on the molecular electrostatic potential is not very differentiated: the RMSE values of these models fall in the range from 0.03 D (for the HLY model) to 0.48 D (for the CHELP model). As indicated by the values of the MSE in the upper plot in
Figure 3, the AIM and GDMA models overestimate the magnitude of μ significantly, whereas the Mulliken, Hirshfeld, CM5 and CHELP models tend to underestimate the values of μ. The differences in the MSE and RMSE values obtained from various levels of theory are usually relatively small, and thus all the above-mentioned findings are common to the HF, B3LYP and MP2 levels.
Figure 3.
MSE and RMSE (in Debyes) in the μ values approximated by 10 models of partial atomic charge for 30 conformers of 1a–5a and 1b–5b. The errors are calculated relative to the reference results obtained from the full density at the HF/def2-QZVPP, B3LYP/def2-QZVPP and MP2/def2-QZVPP levels of theory.
Figure 3.
MSE and RMSE (in Debyes) in the μ values approximated by 10 models of partial atomic charge for 30 conformers of 1a–5a and 1b–5b. The errors are calculated relative to the reference results obtained from the full density at the HF/def2-QZVPP, B3LYP/def2-QZVPP and MP2/def2-QZVPP levels of theory.
Our findings should now be collated with previous observations on the performance of various models of partial atomic charge. It is known that the NPA partial atomic charges overestimate the dipole moments of polypeptides [
42] and in the present work this observation is validated for smaller molecular systems (see the positive values of the MSE for the NPA model in
Figure 3). The overestimation of the μ values predicted by the NPA model seems to be associated with the fact that this model generally overestimates the amplitude of the electrostatic potential, as it was reported for a large set of 500 simple organic molecules [
43]. In another work taking a large set of simple molecules into account [
9], it was found that the molecular dipole moment is usually underestimated when it is approximated by the Hirshfeld model. It is valid in our case, as evidenced by the negative MSE values of the Hirshfeld model in
Figure 3. Both the MSE and RMSE values in
Figure 3 indicate that the CM5 model outperforms the Hirshfeld one, which is also in accord with previous reports [
9,
44]. Furthermore, the CM5 model turns out to be slightly more accurate than the Mulliken one, and this observation is shared by the results of dipole moment calculations using the MG3S basis set [
9]. It should, however, be stressed here that the CM5 model is essentially independent of a basis set [
9], while the basis set dependence is the well-known weakness of the Mulliken model [
1,
4]. The poor performance of the AIM model results from its excessive partial atomic charges. The severe overestimation of dipole moment magnitude is typical of this model and it is observed even for the water molecule [
45]. As noted in the previous paragraph, the MKS and HLY models provide excellent results for the μ values of the conformers of
1a–
5a and
1b–
5b. The CHELP and CHELPG models demonstrate larger RMSE values but both are still fairly successful in reproducing the μ values from the full density. This seems to be in line with what was previously reported for the MKS and CHELPG models [
46]. The former turned out to be superior to the latter for the representation of dipole moments in ionic liquids. One of the reasons for the good performance of the MKS, CHELP, CHELPG and HLY models in reproducing μ for the conformers of
1a–
5a and
1b–
5b is that the molecular shape of these conformers is not very complex (the heteroaryl substituents are planar although they are most often not coplanar with one another) and almost all their atoms are near their molecular van der Waals surfaces. This practically eliminates the occurrence of the so-called “buried atoms” for which the electrostatic potential fitting is inaccurate, and thus, the resulting partial atomic charges are poorly determined [
47].
Aside from the statistical comparison of the approximated μ values with the reference results from the full densities computed at the HF/def2-QZVPP, B3LYP/def2-QZVPP or MP2/def2-QZVPP level of theory, another comparison of the calculated μ values is meaningful. Such a comparison utilizes only a single set of reference μ values, irrespective of the level of theory which produces the molecular wave functions/electron densities for the models of partial atomic charge. This comparison allows us to directly examine whether the level of theory affects the performance of the ten models in predicting the values of μ. Because the range of the quantum chemical methods used in this work includes both the simplest
ab initio method (that is, HF) and two more advanced methods (that is, B3LYP and MP2), the effect of electron correlation on the performance of the models can be studied. The B3LYP/def2-QZVPP level of theory is selected to provide the reference full-density values of μ for the conformers of
1a–
5a and
1b–
5b. Of three levels considered in this work, the μ values obtained from the B3LYP/def2-QZVPP full density turned out to be closest to experiment for a test set of simple molecules being the building blocks of
1a–
5a and
1b–
5b (see
section S2 in Supplementary Materials). On this basis, the μ values calculated using the B3LYP/def2-QZVPP full density [
32] are also assumed to be the most realistic for the conformers of
1a–
5a and
1b–
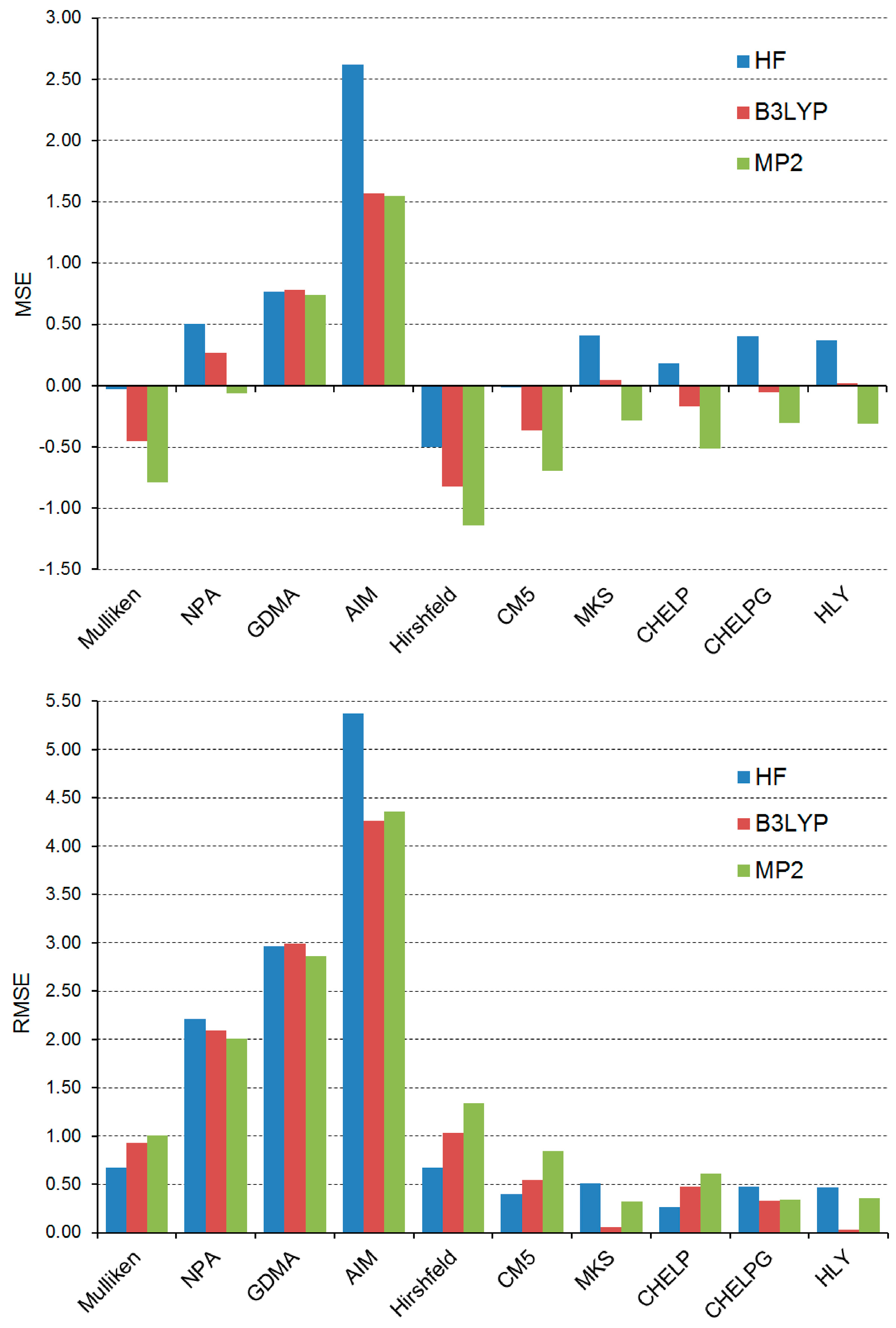
5b. These values are now used as the only reference results for the calculations of the MSE and RMSE in the μ values approximated by the ten models that, in turn, operate on the HF/def2-QZVPP, B3LYP/def2-QZVPP and MP2/def2-QZVPP wave functions/electron densities. The calculated MSE and RMSE values are presented graphically in
Figure 4. For the B3LYP method, its results shown in this figure are obviously identical to those depicted in
Figure 3.
Results shown in
Figure 4 support our previous findings that such electrostatic potential-based models as MKS, CHELP, CHELPG and HLY are able to predict the values of μ with great accuracy. Out of these four models, it is hard to select a single model that performs best at all three levels of theory. For the molecular wave functions calculated by HF/def2-QZVPP, the CHELP model leads to the lowest RMSE value. For the molecular wave functions computed at the B3LYP/def2-QZVPP and MP2/def2-QZVPP levels, the most accurate values of μ are predicted by the HLY and MKS models, respectively. The positive values of the MSE yielded by the MKS, CHELP, CHELPG and HLY models operating on the HF/def2-QZVPP wave functions illustrate the systematic overestimation of the approximated μ values. This is in line with the previous finding made for the MKS model [
47], and now it is additionally extended to the CHELP, CHELPG and HLY models. All four models consistently change their behavior while employing the MP2/def2-QZVPP wave functions (MSE < 0). Differences in the three RMSE values of each model are usually relatively small and in such cases they do not exceed 0.5 D. This indicates that, in general, the effect of the quantum chemical method applied for obtaining the molecular wave functions/electron densities is rather minor. However, at least one exception to this general conclusion can be seen in
Figure 4. The AIM model combined with the HF/def2-QZVPP electron densities performs particularly badly when compared with its RMSE values obtained from both B3LYP/def2-QZVPP and MP2/def2-QZVPP electron densities. From this perspective, the AIM model seems to be particularly sensitive to the inclusion of electron correlation effects into the molecular electron density. On the other hand, it should be stressed that the performance of the AIM model still remains poor for the B3LYP/def2-QZVPP and MP2/def2-QZVPP electron densities.
Figure 4.
MSE and RMSE (in Debyes) in the μ values approximated by 10 models of partial atomic charge for 30 conformers of 1a–5a and 1b–5b. The errors are calculated relative to the reference results obtained from the full density at the B3LYP/def2-QZVPP level of theory.
Figure 4.
MSE and RMSE (in Debyes) in the μ values approximated by 10 models of partial atomic charge for 30 conformers of 1a–5a and 1b–5b. The errors are calculated relative to the reference results obtained from the full density at the B3LYP/def2-QZVPP level of theory.
A brief search of the literature reveals several previous mentions of the impact of electron correlation on partial atomic charges and resulting μ values. It was reported that this impact is small [
48] or even minimal [
46]. Our results are in agreement with these findings. The usual effect of electron correlation on the distribution of molecular electron density is to deplete electron density from the centers of bonds and increase it in shells around the atomic nuclei and at periphery of molecules [
49]. However, the influence of such a depletion on molecular dipole moments is often smaller than might be expected because the charge reorganization happens around each atom and can result in only a small net change in polarization of whole molecules [
48]. In the case of the AIM model, a significant insensitivity of its many parameters (also those obtained through the integration of electron density) to the addition of electron correlation and the change in basis set was observed [
50,
51]. On the other hand, it was detected for the water molecule that, within the framework of the AIM model, the electron density produced by the HF method leads to a slightly enhanced dipole moment compared to that obtained from the MP2 electron density [
45]. This observation is in agreement with our results that, indeed, show an increase in the RMSE values for the AIM model while going from the correlation-corrected electron densities to the HF/def2-QZVPP densities.
The next stage of the present work is to establish the usability of various partial atomic charge models to predict qualitatively the conformational behavior of
1a–
5a and
1b–
5b. The
Eelst energy calculated using the partial atomic charges determined for each conformer is considered here to be a simple measure of electrostatic effects stabilizing the conformer. It is convenient to express the
Eelst values obtained for the conformers of each compound with respect to the
Eelst energy of the conformer that is most favorable (in other words, with respect to the conformer possessing the lowest
Eelst energy). The resulting relative electrostatic energy Δ
Eelst is equal to zero for the preferred conformer while it is positive for less stable conformers. A part of the Δ
Eelst values characterizing the
cc-,
ct-, and
tt-conformers of
1a–
5a and
1b–
5b is given in
Table 1 and
Table 2. The tabulated values have been calculated using selected models that have operated on the molecular wave functions/electron densities computed at the B3LYP/def2-QZVPP level of theory (a complete set of results obtained from all ten models of partial atomic charge, as well as at the HF/def2-QZVPP, B3LYP/def2-QZVPP and MP2/def2-QZVPP levels of theory can be found in
Tables S8–S13 in Supplementary Materials). Additionally, the relative electron energies Δ
E and full-density dipole moments μ obtained from the previous regular calculations at the B3LYP/def2-QZVPP level [
30,
32] are also presented in
Table 1 and
Table 2. The values of Δ
E allow us to establish the reference orderings of conformers for
1a–
5a and
1b–
5b.
Table 1.
Relative electron energies (ΔE in kcal/mol), dipole moments obtained from the full density (μ in Debyes) and relative electrostatic energies (ΔEelst in kcal/mol) for 1a–5a in their three conformations. All the results are calculated at the B3LYP/def2-QZVPP level of theory.
Table 1.
Relative electron energies (ΔE in kcal/mol), dipole moments obtained from the full density (μ in Debyes) and relative electrostatic energies (ΔEelst in kcal/mol) for 1a–5a in their three conformations. All the results are calculated at the B3LYP/def2-QZVPP level of theory.
| Conformer | ΔE a | μ b | ΔEelst |
|---|
| Mulliken | NPA | AIM | Hirshfeld | MKS | HLY |
|---|
| cc-1a | 2.14 | 4.73 | 13.78 | 8.80 | 35.42 | 1.81 | 0.07 | 0.00 |
| ct-1a | 0.00 | 3.91 | 4.13 | 0.00 | 0.95 | 1.10 | 0.47 | 19.65 |
| tt-1a | 0.34 | 2.91 | 0.00 | 0.67 | 0.00 | 0.00 | 0.00 | 25.15 |
| cc-2a | 0.00 | 4.03 | 0.93 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| ct-2a | 0.77 | 3.37 | 0.30 | 5.71 | 5.29 | 0.68 | 44.47 | 64.19 |
| tt-2a | 1.85 | 2.69 | 0.00 | 10.99 | 10.58 | 1.47 | 50.28 | 69.59 |
| cc-3a | 0.00 | 3.65 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| ct-3a | 1.41 | 3.21 | 1.01 | 7.60 | 10.11 | 0.15 | 53.46 | 68.89 |
| tt-3a | 2.93 | 2.81 | 1.42 | 16.44 | 22.96 | 0.05 | 69.25 | 87.14 |
| cc-4a | 0.00 | 0.41 | 0.00 | 0.00 | 5.38 | 0.00 | 0.00 | 0.00 |
| ct-4a | 3.82 | 3.31 | 10.99 | 7.79 | 0.00 | 1.75 | 6.79 | 1.51 |
| tt-4a | 8.99 | 5.31 | 9.77 | 11.96 | 8.28 | 3.34 | 26.92 | 25.70 |
| cc-5a | 0.00 | 0.13 | 0.00 | 0.00 | 3.35 | 0.00 | 0.00 | 0.00 |
| ct-5a | 5.37 | 3.55 | 8.00 | 1.81 | 3.30 | 0.50 | 36.19 | 44.15 |
| tt-5a | 9.63 | 5.29 | 28.21 | 5.54 | 0.00 | 1.36 | 99.21 | 106.26 |
The tabulated values of ΔEelst show that the energy of electrostatic interactions between partial atomic charges is able to give us some indication of the preferred conformations for the investigated compounds, especially for those whose preferred conformation is characterized by either the largest or the smallest values of μ. The most energetically stable conformers of 1a and 1b possess molecular dipole moments whose values lie in the middle between the values obtained for the other two conformers. In such case the Eelst energy usually turns out to be insufficient to identify the preferred conformation. Although the preference of the ct-conformation can be inferred from ΔEelst calculated using the NPA and Mulliken partial atomic charges, none of the two sets of partial atomic charges leads to ct-conformation preference simultaneously for 1a and 1b. Nevertheless, the NPA model is recognized to be most successful in predicting the preferred conformers of 1a–5a and 1b–5b in terms of Eelst. The Mulliken, AIM and Hirshfeld models lead to a slightly greater number of incorrect indications of preferred conformers than the NPA model does. Furthermore, this model always performs best, irrespective of the level of theory used to generate the molecular wave functions. The electrostatic potential-based models are generally less accurate in identifying the preferred conformers of 1a–5a and 1b–5b than the models belonging to the two remaining classes. Of the electrostatic potential-based models, only MKS and HLY are able to correctly indicate the preferred conformers for more than half of the investigated compounds.
Table 2.
Relative electron energies (ΔE in kcal/mol), dipole moments obtained from the full density (μ in Debyes) and relative electrostatic energies (ΔEelst in kcal/mol) for 1b–5b in their three conformations. All the results are calculated at the B3LYP/def2-QZVPP level of theory.
Table 2.
Relative electron energies (ΔE in kcal/mol), dipole moments obtained from the full density (μ in Debyes) and relative electrostatic energies (ΔEelst in kcal/mol) for 1b–5b in their three conformations. All the results are calculated at the B3LYP/def2-QZVPP level of theory.
| Conformer | ΔE a | μ b | ΔEelst |
|---|
| Mulliken | NPA | AIM | Hirshfeld | MKS | HLY |
|---|
| cc-1b | 1.46 | 4.69 | 7.79 | 0.00 | 0.00 | 0.80 | 51.02 | 42.63 |
| ct-1b | 0.00 | 4.09 | 0.00 | 1.35 | 2.22 | 0.62 | 10.35 | 11.94 |
| tt-1b | 0.31 | 3.35 | 0.75 | 10.90 | 36.07 | 0.00 | 0.00 | 0.00 |
| cc-2b | 0.00 | 4.13 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| ct-2b | 0.99 | 3.62 | 1.02 | 2.84 | 2.09 | 0.48 | 37.25 | 49.47 |
| tt-2b | 2.22 | 3.08 | 1.23 | 6.27 | 4.48 | 1.15 | 35.82 | 51.96 |
| cc-3b | 0.00 | 3.80 | 0.00 | 0.00 | 0.00 | 0.25 | 0.00 | 0.00 |
| ct-3b | 1.44 | 3.46 | 2.09 | 3.29 | 1.51 | 0.27 | 56.32 | 57.83 |
| tt-3b | 3.00 | 3.17 | 3.05 | 8.54 | 5.66 | 0.00 | 65.31 | 67.72 |
| cc-4b | 0.00 | 1.54 | 0.64 | 0.00 | 0.00 | 0.00 | 27.94 | 31.98 |
| ct-4b | 3.92 | 3.96 | 5.86 | 1.59 | 11.12 | 1.22 | 37.82 | 41.82 |
| tt-4b | 9.41 | 5.66 | 0.00 | 0.01 | 38.50 | 1.95 | 0.00 | 0.00 |
| cc-5b | 0.00 | 1.23 | 0.00 | 0.00 | 0.00 | 0.00 | 180.49 | 180.68 |
| ct-5b | 3.05 | 4.44 | 10.21 | 2.66 | 0.01 | 0.91 | 117.01 | 113.67 |
| tt-5b | 5.04 | 5.62 | 17.64 | 2.61 | 7.42 | 1.60 | 0.00 | 0.00 |
Aside from the application of
Eelst to the identification of the most stable conformation, this quantity allows us to order conformers relative to their
Eelst values and the resulting orderings of conformers for
1a–
5a and
1b–
5b can be compared with the orderings based on Δ
E. The evident relationship between μ and Δ
E for
2a–
5a and their thiocarbonyl counterparts suggests that electrostatic effects are responsible to a certain extent for the ordering of individual conformers relative to their energetic stability. We focus here only on the successful reproduction of conformer orderings and not on the quantitative correlation between individual non-zero Δ
Eelst and Δ
E values. The values of Δ
Eelst should not be compared directly with Δ
E because the former are merely a crude approximation of intramolecular electrostatic effects. The electrostatic effects are undoubtedly important but not the sole factor affecting the stability of the investigated conformers. Therefore, the values of Δ
Eelst are usually far from the corresponding Δ
E values. The values of Δ
Eelst obviously inherit the deficiencies of the applied model of partial atomic charge. The Δ
Eelst values calculated using the AIM partial atomic charges are usually large for
1a–
3a and
1b–
3b, whereas the Hirshfeld partial atomic charges lead to very small Δ
Eelst values for the majority of the investigated compounds. This is due to the fact that AIM partial atomic charges generally tend to adopt large absolute values [
23], while the Hirshfeld model assigns partial atomic charges that are very small in magnitude [
52].
Turning our attention to the predicted conformer orderings, we can see that none of the considered models of partial atomic charge provides the correct orderings of conformers for all ten compounds. Among the ten models, the NPA one appears to be most suitable for illustrating tendencies in electrostatic effects (in terms of
Eelst) in the investigated compounds, although some inconsistencies with the orderings predicted by Δ
E are found. The inconsistencies in the conformer orderings yielded by the NPA model occur mostly for diheteroaryl thioketones. The AIM and Hirshfeld models give less satisfactory results than those of the NPA model. On the other hand, they reproduce the reference orderings of conformers for a greater number of the investigated compounds than the HLY model does. Of the electrostatic potential-based models, HLY produces the conformer orderings that fit best to the corresponding reference orderings indicated by Δ
E. All the aforementioned findings are common to the three levels of theory used to generate the wave functions/electron densities of the conformers. This is additionally supported by the results presented in
Table 3. This table shows the percentage similarity of the conformer sequences deduced from Δ
Eelst to the reference conformer orderings determined in terms of Δ
E. The percentage similarity has been obtained through comparing the conformer orderings of all investigated compounds.
Table 3.
Percentage similarity of the conformer sequences predicted by ΔEelst to the reference sequences determined using ΔE. The values of ΔEelst are calculated by 10 partial atomic charge models operating on the wave functions/electron densities calculated at three levels of theory.
Table 3.
Percentage similarity of the conformer sequences predicted by ΔEelst to the reference sequences determined using ΔE. The values of ΔEelst are calculated by 10 partial atomic charge models operating on the wave functions/electron densities calculated at three levels of theory.
| Model | Level of Theory |
|---|
| HF | B3LYP | MP2 |
|---|
| Mulliken | 50 | 70 | 50 |
| NPA | 83 | 73 | 83 |
| GDMA | 57 | 53 | 57 |
| AIM | 67 | 70 | 80 |
| Hirshfeld | 73 | 70 | 70 |
| CM5 | 37 | 43 | 47 |
| MKS | 60 | 60 | 50 |
| CHELP | 23 | 33 | 20 |
| CHELPG | 57 | 43 | 53 |
| HLY | 60 | 67 | 63 |




{kind=link}
{kind=link}
{kind=link}
{kind=link}