
Computational Studies of the Intestinal Host-Microbiota Interactome

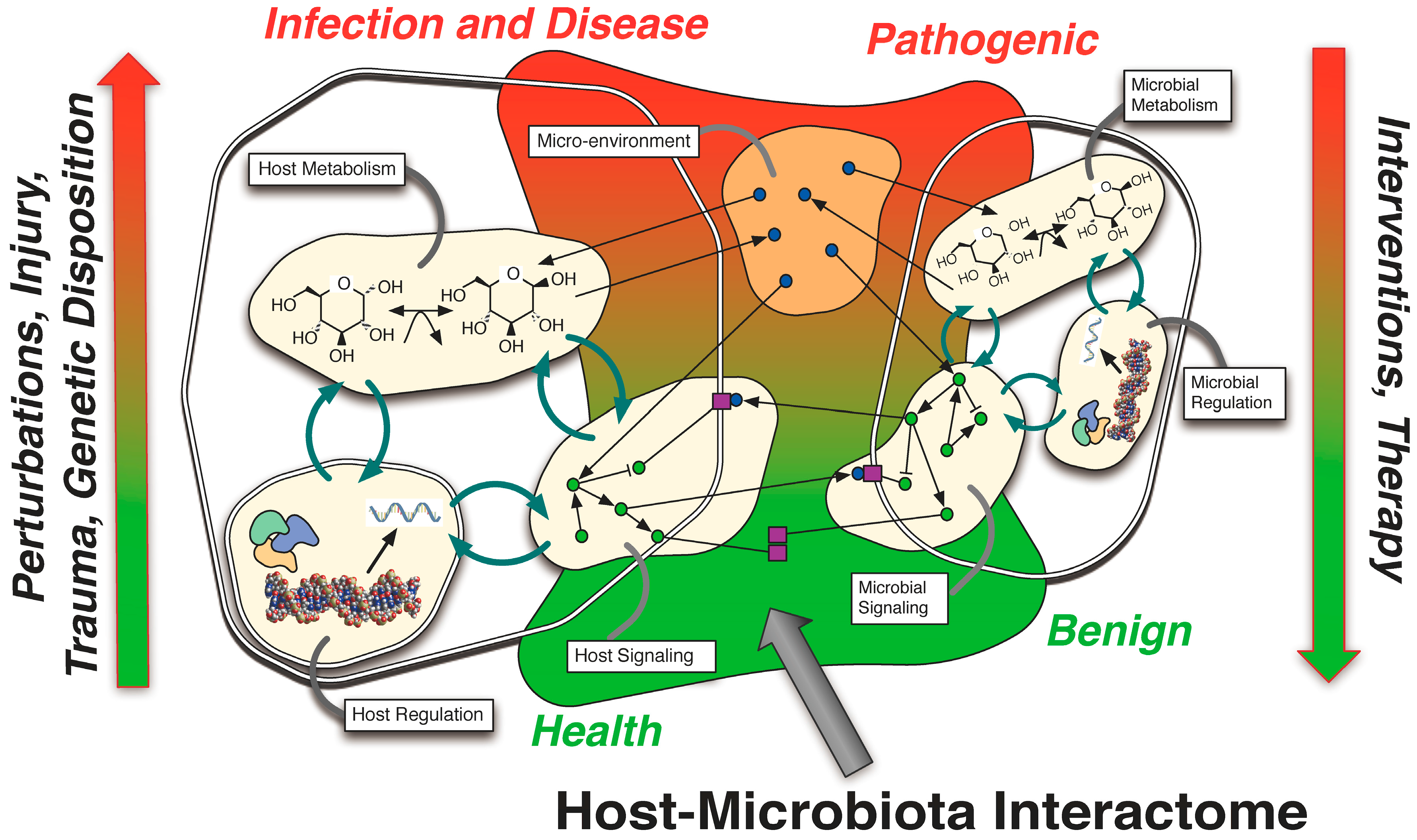{kind=link}
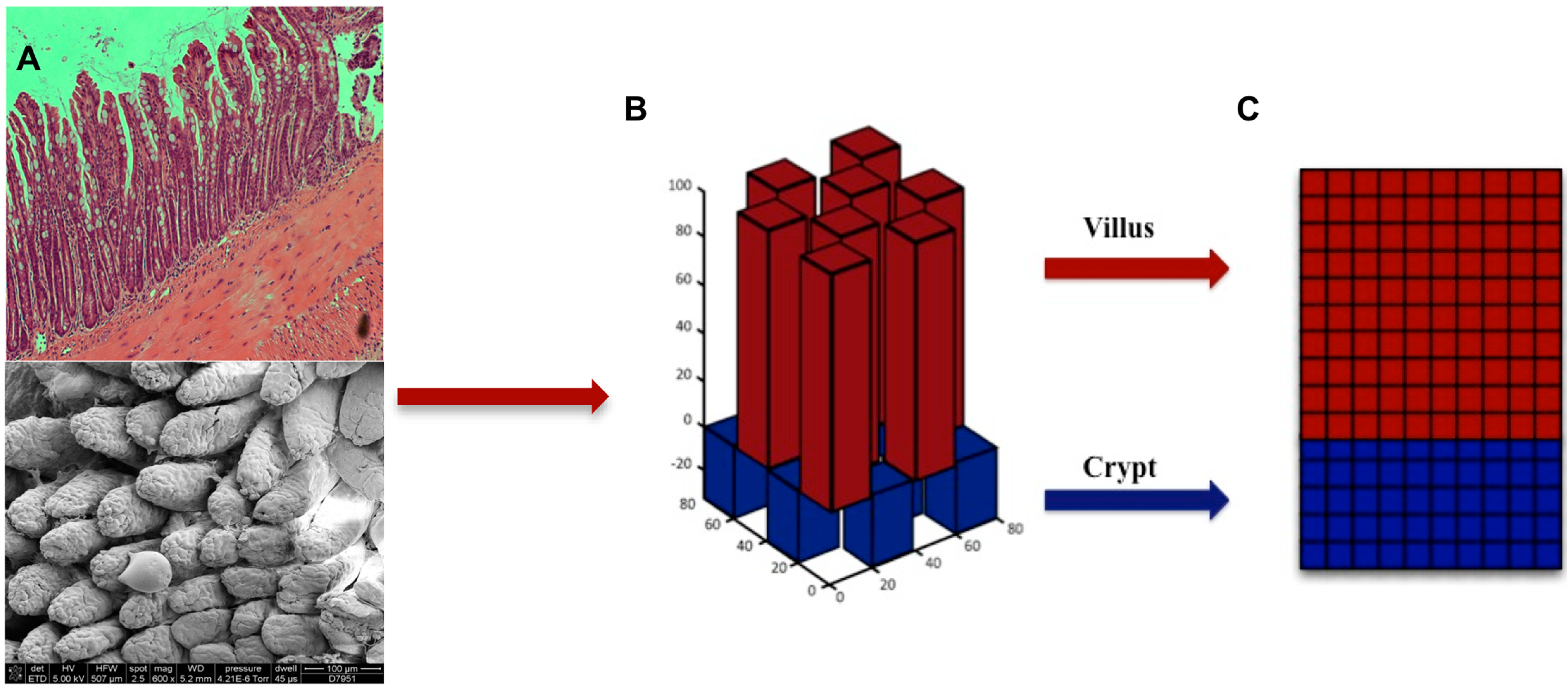
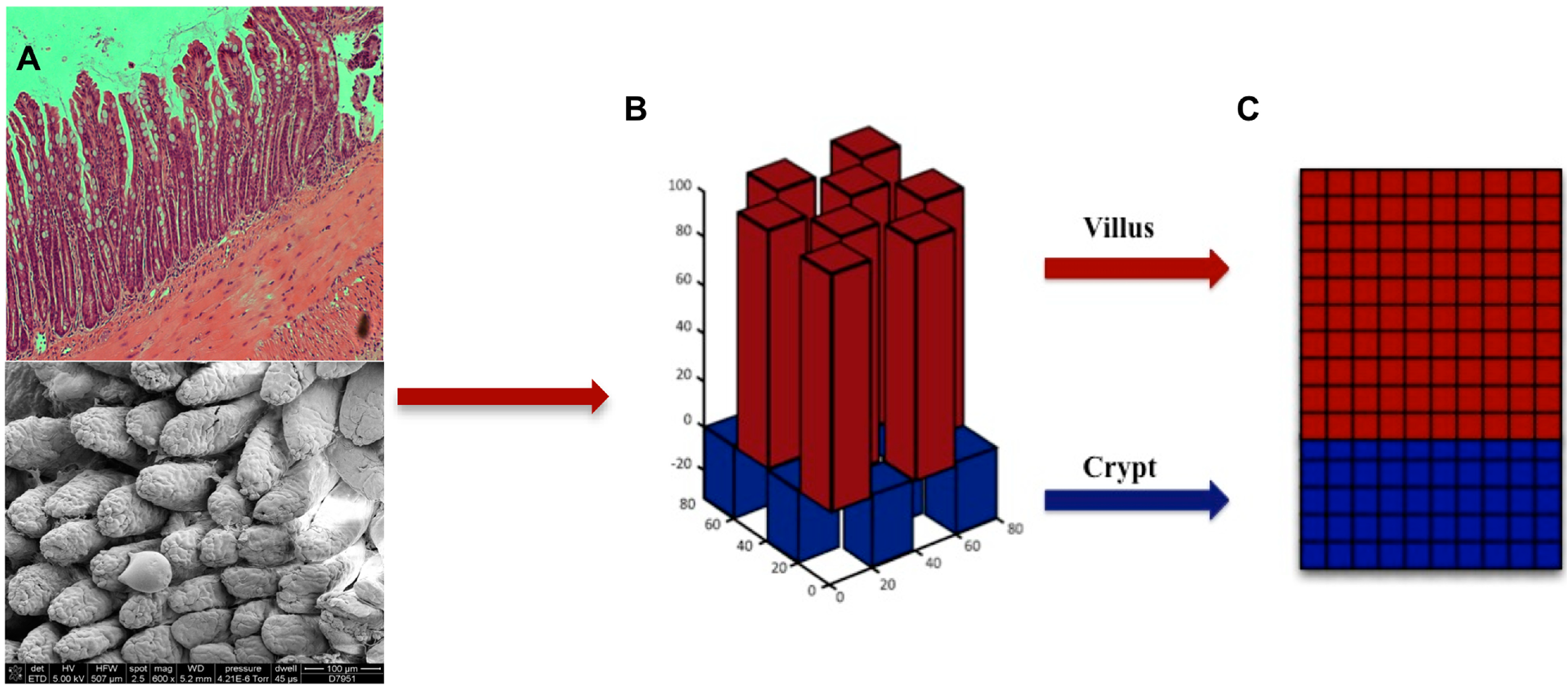{kind=link}
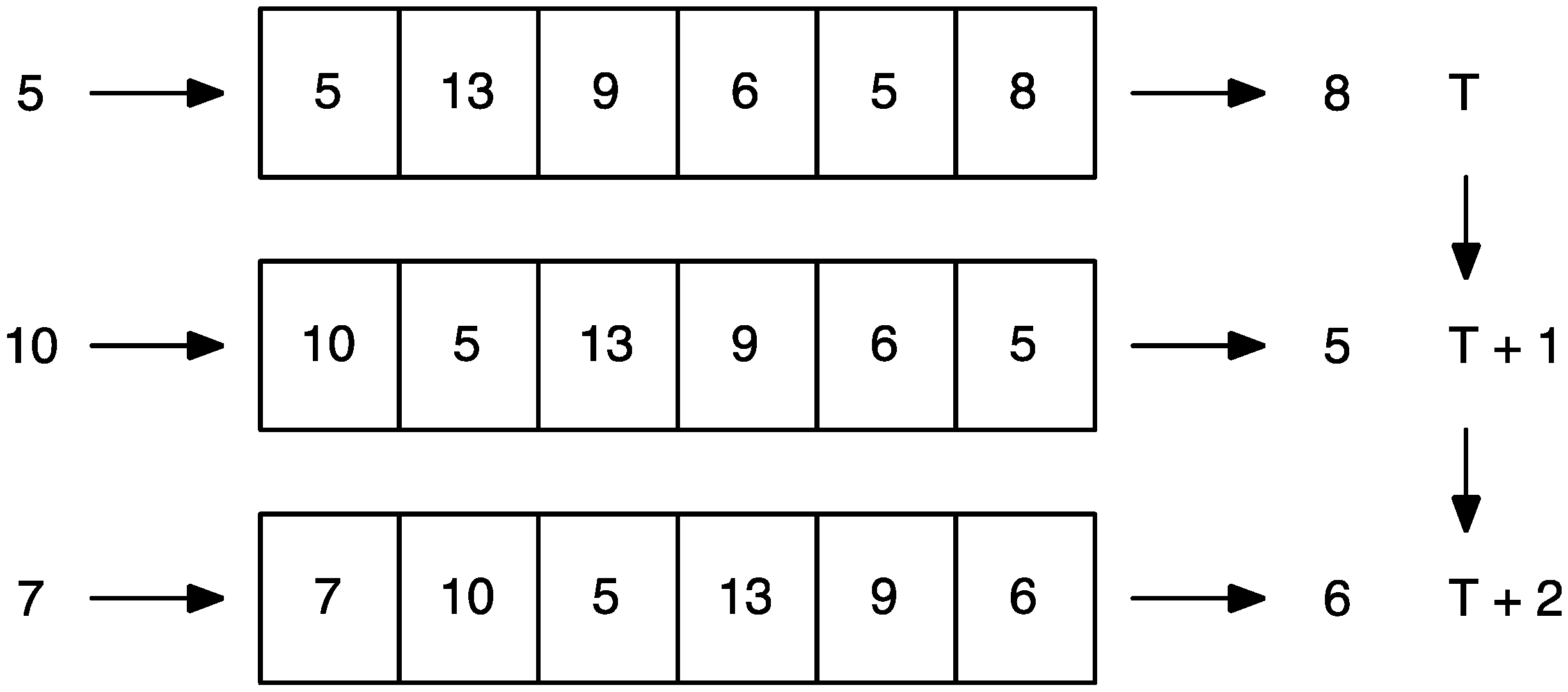{kind=link}
Abstract
:1. Introduction

2. Microbiota Studies
2.1. Marker Gene Profiling
2.2. Metagenomics
2.3. Metatranscriptomics
2.4. Computational Modeling and Simulation
3. Intestinal Host-Microbiota Interactome Studies
3.1. Computational Modeling of Host Immune System
3.2. Inflammatory Diseases and the Intestinal Host-Microbiota Interactome
4. Conclusions
Acknowledgements
Conflicts of Interest
References
- Consortium, H.M.P. A framework for human microbiome research. Nature 2012, 486, 215–221. [Google Scholar] [CrossRef]
- Consortium, H.M.P. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Costello, E.K.; Berg-Lyons, D.; Gonzalez, A.; Stombaugh, J.; Knights, D.; Gajer, P.; Ravel, J.; Fierer, N.; et al. Moving pictures of the human microbiome. Genome Biol. 2011, 12, R50. [Google Scholar] [CrossRef] [PubMed]
- Nava, G.M.; Friedrichsen, H.J.; Stappenbeck, T.S. Spatial organization of intestinal microbiota in the mouse ascending colon. ISME J. 2011, 5, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Geng, J.; Tang, X.; Fan, H.; Xu, J.; Wen, X.; Ma, Z.S.; Shi, P. Spatial heterogeneity and co-occurrence patterns of human mucosal-associated intestinal microbiota. ISME J. 2013, 8, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Koenig, J.E.; Spor, A.; Scalfone, N.; Fricker, A.D.; Stombaugh, J.; Knight, R.; Angenent, L.T.; Ley, R.E. Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. USA 2011, 108, 4578–4585. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Gordon, J.I. Commensal host-bacterial relationships in the gut. Science 2001, 292, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Xavier, R.J.; Gevers, D. The microbiome in inflammatory bowel diseases: Current status and the future ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Carlisle, E.M.; Morowitz, M.J. The intestinal microbiome and necrotizing enterocolitis. Curr. Opin. Pediatr. 2013, 25, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Mittal, R.; Coopersmith, C.M. Redefining the gut as the motor of critical illness. Trends Mol. Med. 2013, 20, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Serban, D.E. Gastrointestinal cancers: Influence of gut microbiota, probiotics and prebiotics. Cancer Lett. 2014, 345, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Rogler, G. Chronic ulcerative colitis and colorectal cancer. Cancer. Lett. 2014, 345, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Vayssier-Taussat, M.; Albina, E.; Citti, C.; Cosson, J.-F.; Jacques, M.-A.; Lebrun, M.-H.; Le Loir, Y.; Ogliastro, M.; Petit, M.-A.; Roumagnac, P.; et al. Shifting the paradigm from pathogens to pathobiome: New concepts in the light of meta-omics. Front. Cell. Infect. Microbiol. 2014. [Google Scholar] [CrossRef]
- Brown, E.M.; Sadarangani, M.; Finlay, B.B. The role of the immune system in governing host-microbe interactions in the intestine. Nat. Immunol. 2013, 14, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Kamada, N.; Seo, S.-U.; Chen, G.Y.; Núñez, G. Role of the gut microbiota in immunity and inflammatory disease. Nat. Rev. Immunol. 2013, 13, 321–355. [Google Scholar] [CrossRef] [PubMed]
- Jarchum, I.; Pamer, E.G. Regulation of innate and adaptive immunity by the commensal microbiota. Curr. Opin. Immunol. 2011, 23, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Knights, D.; Lassen, K.G.; Xavier, R.J. Advances in inflammatory bowel disease pathogenesis: Linking host genetics and the microbiome. Gut 2013, 62, 1505–1510. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.H.; Russell, J.A.; Fjell, C.D. The meta-genome of sepsis: Host genetics, pathogens and the acute immune response. J Innate Immun. 2014, 272–283. [Google Scholar]
- Kamada, N.; Chen, G.Y.; Inohara, N.; Núñez, G. Control of pathogens and pathobionts by the gut microbiota. Nat. Immunol. 2013, 14, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Faust, K.; Raes, J. Microbial interactions: From networks to models. Nat. Rev. Microbiol. 2012, 10, 538. [Google Scholar] [CrossRef]
- Stecher, B.; Maier, L.; Hardt, W.-D. “Blooming” in the gut: How dysbiosis might contribute to pathogen evolution. Nat. Rev. Microbiol. 2013, 11, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Boon, E.; Meehan, C.J.; Whidden, C.; Wong, D.H.-J.; Langille, M.G.I.; Beiko, R.G. Interactions in the microbiome: Communities of organisms and communities of genes. FEMS Microbiol. Rev. 2013. [Google Scholar] [CrossRef]
- Sommer, F.; Bäckhed, F. The gut microbiota—Masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Darveau, R.P.; Curtis, M.A. The keystone-pathogen hypothesis. Nat. Rev. Microbiol. 2012, 10, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L. The gut microbiota and obesity: From correlation to causality. Nat. Rev. Microbiol. 2013, 11, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, V.M.; Chen, I.-M.A.; Chu, K.; Szeto, E.; Palaniappan, K.; Pillay, M.; Ratner, A.; Huang, J.; Pagani, I.; Tringe, S.; et al. IMG/M 4 version of the integrated metagenome comparative analysis system. Nucleic Acids Res. 2014, 42, D568–D573. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Begley, T.; Butler, R.M.; Choudhuri, J.V.; Chuang, H.-Y.; Cohoon, M.; de Crécy-Lagard, V.; Diaz, N.; Disz, T.; Edwards, R.; et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005, 33, 5691–5702. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16s rRNA gene database and workbench compatible with ARB. App. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The rast server: Rapid annotations using subsystems technology. BMC Genom. 2008. [Google Scholar] [CrossRef]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics rast server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef]
- . The Department of Energy Systems Biology Knowledgebase. Available online: http://www.kbase.us (accessed on 5 January 2015).
- Hucka, M.; Finney, A.; Sauro, H.M.; Bolouri, H.; Doyle, J.C.; Kitano, H.; Arkin, A.P.; Bornstein, B.J.; Bray, D.; Cornish-Bowden, A.; et al. The systems biology markup language (SBML): A medium for representation and exchange of biochemical network models. Bioinformatics 2003, 19, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Le Novère, N.; Bornstein, B.; Broicher, A.; Courtot, M.; Donizelli, M.; Dharuri, H.; Li, L.; Sauro, H.; Schilstra, M.; Shapiro, B.; et al. Biomodels database: A free, centralized database of curated, published, quantitative kinetic models of biochemical and cellular systems. Nucleic Acids Res. 2006, 34, D689–D691. [Google Scholar] [CrossRef] [PubMed]
- Kuczynski, J.; Lauber, C.L.; Walters, W.A.; Parfrey, L.W.; Clemente, J.C.; Gevers, D.; Knight, R. Experimental and analytical tools for studying the human microbiome. Nat. Rev. Genet. 2012, 13, 47–58. [Google Scholar] [CrossRef]
- Weinstock, G.M. Genomic approaches to studying the human microbiota. Nature 2012, 489, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Pepke, S.; Wold, B.; Mortazavi, A. Computation for chip-seq and rna-seq studies. Nat. Methods 2009, 6, S22–S32. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. Rna-seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Carvalhais, L.C.; Dennis, P.G.; Tyson, G.W.; Schenk, P.M. Application of metatranscriptomics to soil environments. J. Microbiol. Methods 2012, 91, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Frank, D.N.; Robertson, C.E.; Hung, S.S.; Markle, J.; Canty, A.J.; McCoy, K.D.; Macpherson, A.J.; Poussier, P.; Danska, J.S.; et al. Generation and analysis of a mouse intestinal metatranscriptome through illumina based RNA-sequencing. PLoS ONE 2012. [Google Scholar] [CrossRef]
- Thiele, I.; Palsson, B.Ø. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.S.; DeJongh, M.; Best, A.A.; Frybarger, P.M.; Linsay, B.; Stevens, R.L. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat. Biotech. 2010, 28, 977–982. [Google Scholar] [CrossRef]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What is flux balance analysis? NatBiotech. 2010, 28, 245–248. [Google Scholar]
- Oberhardt, M.A.; Palsson, B.Ø.; Papin, J.A. Applications of genome-scale metabolic reconstructions. Mol. Syst. Biol. 2009. [Google Scholar] [CrossRef]
- Bucci, V.; Bradde, S.; Biroli, G.; Xavier, J.B. Social interaction, noise and antibiotic-mediated switches in the intestinal microbiota. PLoS Comput. Biol. 2012. [Google Scholar] [CrossRef]
- Stein, R.R.; Bucci, V.; Toussaint, N.C.; Buffie, C.G.; Rätsch, G.; Pamer, E.G.; Sander, C.; Xavier, J.B. Ecological modeling from time-series inference: Insight into dynamics and stability of intestinal microbiota. PLoS Comput. Biol. 2013. [Google Scholar] [CrossRef]
- Sanghvi, J.C.; Regot, S.; Carrasco, S.; Karr, J.R.; Gutschow, M.V.; Bolival, B.; Covert, M.W. Accelerated discovery via a whole-cell model. Nat. Methods 2013, 10, 1192–1195. [Google Scholar] [CrossRef] [PubMed]
- Earth Microbiome Project. Available online: http://www.earthmicrobiome.org/emp-standard-protocols/dna-extraction-protocol/ (accessed on 19 March 2014).
- Luo, C.; Tsementzi, D.; Kyrpides, N.; Read, T.; Konstantinidis, K.T. Direct comparisons of illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample. PLoS One 2012. [Google Scholar] [CrossRef]
- Navas-Molina, J.A.; Peralta-Sánchez, J.M.; González, A.; McMurdie, P.J.; Vázquez-Baeza, Y.; Xu, Z.; Ursell, L.K.; Lauber, C.; Zhou, H.; Song, S.J.; et al. Advancing our understanding of the human microbiome using QIIME. Methods Enzymol. 2013, 531, 371–444. [Google Scholar] [PubMed]
- Huson, D.H.; Mitra, S.; Ruscheweyh, H.-J.; Weber, N.; Schuster, S.C. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011, 21, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, S.; Mende, D.R.; Zeller, G.; Izquierdo-Carrasco, F.; Berger, S.A.; Kultima, J.R.; Coelho, L.P.; Arumugam, M.; Tap, J.; Nielsen, H.B.; et al. Metagenomic species profiling using universal phylogenetic marker genes. Nat. Methods 2013, 10, 1196–1199. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; Mcdonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16s rRNA marker gene sequences. Nat. Biotech. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Sharon, I.; Bercovici, S.; Pinter, R.Y.; Shlomi, T. Pathway-based functional analysis of metagenomes. J. Compt. Biol. 2011, 18, 495–505. [Google Scholar] [CrossRef]
- Abubucker, S.; Segata, N.; Goll, J.; Schubert, A.M.; Izard, J.; Cantarel, B.L.; Rodriguez-Mueller, B.; Zucker, J.; Thiagarajan, M.; Henrissat, B.; et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput. Bol. 2012. [Google Scholar] [CrossRef]
- Nakano, Y.; Takeshita, T.; Kamio, N.; Shiota, S.; Shibata, Y.; Suzuki, N.; Yoneda, M.; Hirofuji, T.; Yamashita, Y. Supervised machine learning-based classification of oral malodor based on the microbiota in saliva samples. Artif. Intell. Med. 2014, 60, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Statnikov, A.; Henaff, M.; Narendra, V.; Konganti, K.; Li, Z.; Yang, L.; Pei, Z.; Blaser, M.J.; Aliferis, C.F.; Alekseyenko, A.V. A comprehensive evaluation of multicategory classification methods for microbiomic data. Microbiome 2013. [Google Scholar] [CrossRef]
- Knights, D.; Costello, E.K.; Knight, R. Supervised classification of human microbiota. FEMS Microbiol. Rev. 2011, 35, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Christley, S.; Nie, Q.; Xie, X. Incorporating existing network information into gene network inference. PLoS One 2009. [Google Scholar] [CrossRef] [Green Version]
- De Smet, R.; Marchal, K. Advantages and limitations of current network inference methods. Nat. Rev. Microbiol. 2010, 8, 717–729. [Google Scholar] [PubMed]
- Bonneau, R. Learning biological networks: From modules to dynamics. Nat. Chem. Biol. 2008, 4, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Friedman, N. Inferring cellular networks using probabilistic graphical models. Science 2004, 303, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Faust, K.; Sathirapongsasuti, J.F.; Izard, J.; Segata, N.; Gevers, D.; Raes, J.; Huttenhower, C. Microbial co-occurrence relationships in the human microbiome. PLoS Comput. Biol. 2012. [Google Scholar] [CrossRef]
- Friedman, J.; Alm, E.J. Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 2012. [Google Scholar] [CrossRef]
- Marino, S.; Baxter, N.T.; Huffnagle, G.B.; Petrosino, J.F.; Schloss, P.D. Mathematical modeling of primary succession of murine intestinal microbiota. Proc. Natl. Acad. Sci. USA 2014, 111, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Emerson, J.B.; Thomas, B.C.; Andrade, K.; Allen, E.E.; Heidelberg, K.B.; Banfield, J.F. Dynamic viral populations in hypersaline systems as revealed by metagenomic assembly. Appl. Environ. Microbiol. 2012, 78, 6309–6320. [Google Scholar] [CrossRef] [PubMed]
- Narasingarao, P.; Podell, S.; Ugalde, J.A.; Brochier-Armanet, C.; Emerson, J.B.; Brocks, J.J.; Heidelberg, K.B.; Banfield, J.F.; Allen, E.E. De novo metagenomic assembly reveals abundant novel major lineage of archaea in hypersaline microbial communities. ISME J. 2012, 6, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Hess, M.; Sczyrba, A.; Egan, R.; Kim, T.-W.; Chokhawala, H.; Schroth, G.; Luo, S.; Clark, D.S.; Chen, F.; Zhang, T.; et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 2011, 331, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Bashir, A.; Klammer, A.A.; Robins, W.P.; Chin, C.-S.; Webster, D.; Paxinos, E.; Hsu, D.; Ashby, M.; Wang, S.; Peluso, P.; et al. A hybrid approach for the automated finishing of bacterial genomes. Nat. Biotech. 2012, 30, 701–707. [Google Scholar] [CrossRef]
- Goldberg, S.M.D.; Johnson, J.; Busam, D.; Feldblyum, T.; Ferriera, S.; Friedman, R.; Halpern, A.; Khouri, H.; Kravitz, S.A.; Lauro, F.M.; et al. A sanger/pyrosequencing hybrid approach for the generation of high-quality draft assemblies of marine microbial genomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11240–11245. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E.; Biezuner, T.; Linnarsson, S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 2013, 14, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Lasken, R.S. Genomic sequencing of uncultured microorganisms from single cells. Nat. Rev. Microbiol. 2012, 10, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Moran, M.A. Assembly-free metagenomic analysis reveals new metabolic capabilities in surface ocean bacterioplankton. Environ. Microbiol. Rep. 2013, 5, 686–696. [Google Scholar] [PubMed]
- Carr, R.; Shen-Orr, S.S.; Borenstein, E. Reconstructing the genomic content of microbiome taxa through shotgun metagenomic deconvolution. PLoS Compt. Biol. 2013. [Google Scholar] [CrossRef]
- Wilke, A.; Harrison, T.; Wilkening, J.; Field, D.; Glass, E.M.; Kyrpides, N.; Mavrommatis, K.; Meyer, F. The M5NR: A novel non-redundant database containing protein sequences and annotations from multiple sources and associated tools. BMC Bioinform. 2012, 13, 141. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kristiansson, E.; Hugenholtz, P.; Dalevi, D. Shotgunfunctionalizer: An R-package for functional comparison of metagenomes. Bioinformatics 2009, 25, 2737–2738. [Google Scholar] [PubMed]
- Ye, Y.; Doak, T.G. A parsimony approach to biological pathway reconstruction/inference for genomes and metagenomes. PLoS Comput. Biol. 2009. [Google Scholar] [CrossRef]
- Prakash, T.; Taylor, T.D. Functional assignment of metagenomic data: Challenges and applications. Brief. Bioinform. 2012, 13, 711–727. [Google Scholar] [CrossRef] [PubMed]
- Jiao, D.; Ye, Y.; Tang, H. Probabilistic inference of biochemical reactions in microbial communities from metagenomic sequences. PLoS Comput. Biol. 2013. [Google Scholar] [CrossRef]
- Levy, R.; Borenstein, E. Metabolic modeling of species interaction in the human microbiome elucidates community-level assembly rules. Proc. Natl. Acad. Sci. USA 2013, 110, 12804–12809. [Google Scholar] [CrossRef] [PubMed]
- Borenstein, E. Computational systems biology and in silico modeling of the human microbiome. Brief. Bioinform. 2012, 13, 769–780. [Google Scholar] [CrossRef]
- Nyyssönen, M.; Tran, H.M.; Karaoz, U.; Weihe, C.; Hadi, M.Z.; Martiny, J.B.H.; Martiny, A.C.; Brodie, E.L. Coupled high-throughput functional screening and next generation sequencing for identification of plant polymer decomposing enzymes in metagenomic libraries. Front. Microbiol. 2013. [Google Scholar] [CrossRef]
- Stern, A.; Mick, E.; Tirosh, I.; Sagy, O.; Sorek, R. CRISPR targeting reveals a reservoir of common phages associated with the human gut microbiome. Genome Res. 2012, 22, 1985–1994. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Minot, S.; Bryson, A.; Chehoud, C.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Rapid evolution of the human gut virome. Proc. Natl. Acad. Sci. USA 2013, 110, 12450–12455. [Google Scholar] [CrossRef] [PubMed]
- Greenblum, S.; Turnbaugh, P.J.; Borenstein, E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc. Natl. Acad. Sci. USA 2011. [Google Scholar] [CrossRef]
- Gerber, G.K. The dynamic microbiome. FEBS Lett. 2014, 588, 4131–4139. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009. [Google Scholar] [CrossRef]
- Ning, Z.; Cox, A.J.; Mullikin, J.C. Ssaha: A fast search method for large DNA databases. Genome Res. 2001, 11, 1725–1729. [Google Scholar] [CrossRef] [PubMed]
- McNulty, N.P.; Yatsunenko, T.; Hsiao, A.; Faith, J.J.; Muegge, B.D.; Goodman, A.L.; Henrissat, B.; Oozeer, R.; Cools-Portier, S.; Gobert, G.; et al. The impact of a consortium of fermented milk strains on the gut microbiome of gnotobiotic mice and monozygotic twins. Sci. Transl. Med. 2011. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Quince, C.; Faith, J.J.; McHardy, A.C.; Yatsunenko, T.; Niazi, F.; Affourtit, J.; Egholm, M.; Henrissat, B.; Knight, R.; et al. Organismal, genetic, and transcriptional variation in the deeply sequenced gut microbiomes of identical twins. Proc. Natl. Acad. Sci. USA 2010, 107, 7503–7508. [Google Scholar] [CrossRef] [PubMed]
- Zaborin, A.; Smith, D.; Garfield, K.; Quensen, J.; Shakhsheer, B.; Kade, M.; Tirrell, M.; Tiedje, J.; Gilbert, J.A.; Zaborina, O.; et al. Membership and behavior of ultra-low-diversity pathogen communities present in the gut of humans during prolonged critical illness. mBio 2014. [Google Scholar] [CrossRef]
- Gosalbes, M.J.; Durbán, A.; Pignatelli, M.; Abellan, J.J.; Jiménez-Hernández, N.; Pérez-Cobas, A.E.; Latorre, A.; Moya, A. Metatranscriptomic approach to analyze the functional human gut microbiota. PLoS One 2011. [Google Scholar] [CrossRef]
- Xu, G.; Strong, M.J.; Lacey, M.R.; Baribault, C.; Flemington, E.K.; Taylor, C.M. RNA CoMPASS: A dual approach for pathogen and host transcriptome analysis of RNA-Seq datasets. PLoS ONE 2014. [Google Scholar] [CrossRef]
- Leimena, M.M.; Ramiro-Garcia, J.; Davids, M.; van den Bogert, B.; Smidt, H.; Smid, E.J.; Boekhorst, J.; Zoetendal, E.G.; Schaap, P.J.; Kleerebezem, M. A comprehensive metatranscriptome analysis pipeline and its validation using human small intestine microbiota datasets. BMC Genomics 2013. [Google Scholar] [CrossRef]
- Westermann, A.J.; Gorski, S.A.; Vogel, J. Dual RNA-Seq of pathogen and host. Nat. Rev. Microbiol. 2012, 10, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, R.; Edwards, J.S.; Doyle, F.J. Dynamic flux balance analysis of diauxic growth in Escherichia coli. Biophys. J. 2002, 83, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Min Lee, J.; Gianchandani, E.P.; Eddy, J.A.; Papin, J.A. Dynamic analysis of integrated signaling, metabolic, and regulatory networks. PLoS Comput. Biol. 2008. [Google Scholar] [CrossRef]
- Covert, M.W.; Xiao, N.; Chen, T.J.; Karr, J.R. Integrating metabolic, transcriptional regulatory and signal transduction models in Escherichia coli. Bioinformatics 2008, 24, 2044–2050. [Google Scholar] [CrossRef] [PubMed]
- Herrgård, M.J.; Lee, B.-S.; Portnoy, V.; Palsson, B.Ø. Integrated analysis of regulatory and metabolic networks reveals novel regulatory mechanisms in saccharomyces cerevisiae. Genome Res. 2006, 16, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Covert, M.W.; Palsson, B.Ø. Transcriptional regulation in constraints-based metabolic models of Escherichia Coli. J. Biol. Chem. 2002, 277, 28058–28064. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, S.; Price, N.D. Probabilistic integrative modeling of genome-scale metabolic and regulatory networks in Escherichia coli and Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2010, 107, 17845–17850. [Google Scholar] [CrossRef] [PubMed]
- Yizhak, K.; Benyamini, T.; Liebermeister, W.; Ruppin, E.; Shlomi, T. Integrating quantitative proteomics and metabolomics with a genome-scale metabolic network model. Bioinformatics 2010, 26, i255–i260. [Google Scholar] [CrossRef] [PubMed]
- Gowen, C.M.; Fong, S.S. Genome-scale metabolic model integrated with rnaseq data to identify metabolic states of Clostridium thermocellum. Biotech. J. 2010, 5, 759–767. [Google Scholar] [CrossRef]
- May, P.; Christian, N.; Ebenhöh, O.; Weckwerth, W.; Walther, D. Integration of proteomic and metabolomic profiling as well as metabolic modeling for the functional analysis of metabolic networks. Methods Mol. Biol. 2011, 694, 341–363. [Google Scholar] [PubMed]
- Borenstein, E.; Kupiec, M.; Feldman, M.W.; Ruppin, E. Large-scale reconstruction and phylogenetic analysis of metabolic environments. Proc. Natl. Acad. Sci. USA 2008, 105, 14482–14487. [Google Scholar] [CrossRef] [PubMed]
- Zengler, K.; Palsson, B.O. A road map for the development of community systems (COSY) biology. Nat. Rev. Microbiol. 2012, 10, 366–372. [Google Scholar] [PubMed]
- Grimm, V.; Railsback, S.F. Individual-Based Modeling and Ecology; Princeton University Press: Princeton, NJ, USA, 2005. [Google Scholar]
- Hansen, S.K.; Rainey, P.B.; Haagensen, J.A.J.; Molin, S. Evolution of species interactions in a biofilm community. Nature 2007, 445, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Mitri, S.; Xavier, J.B.; Foster, K.R. Social evolution in multispecies biofilms. Proc. Natl. Acad. Sci. USA 2011, 108, 10839–10846. [Google Scholar] [CrossRef] [PubMed]
- Freilich, S.; Zarecki, R.; Eilam, O.; Segal, E.S.; Henry, C.S.; Kupiec, M.; Gophna, U.; Sharan, R.; Ruppin, E. Competitive and cooperative metabolic interactions in bacterial communities. Nat. Commun. 2011. [Google Scholar] [CrossRef]
- Costello, E.K.; Stagaman, K.; Dethlefsen, L.; Bohannan, B.J.M.; Relman, D.A. The application of ecological theory toward an understanding of the human microbiome. Science 2012, 336, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Hyduke, D.R.; Palsson, B.Ø. Towards genome-scale signalling-network reconstructions. Nat. Rev. Genet. 2010, 11, 297–307. [Google Scholar] [CrossRef] [PubMed]
- An, G. Introduction of an agent-based multi-scale modular architecture for dynamic knowledge representation of acute inflammation. Theor. Biol. Med. Model. 2008. [Google Scholar] [CrossRef]
- An, G.; Nieman, G.; Vodovotz, Y. Toward computational identification of multiscale “tipping points” in acute inflammation and multiple organ failure. Annal. Biomed. Engin. 2012, 40, 2414–2424. [Google Scholar] [CrossRef]
- Castiglione, F.; Pappalardo, F.; Bianca, C.; Russo, G.; Motta, S. Modeling biology spanning different scales: An open challenge. BioMed Res. Int. 2014, 902545. [Google Scholar]
- Alizon, S.; Magnus, C. Modelling the course of an hiv infection: Insights from ecology and evolution. Viruses 2012, 4, 1984–2013. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, R.M. Dynamics of CD4+T cells in HIV-1 infection. Immunol. Cell Biol. 2007, 85, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Abuelezam, N.N.; Rough, K.; Seage, G.R. Individual-based simulation models of HIV transmission: Reporting quality and recommendations. PLoS One 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Beerenwinkel, N.; Sing, T.; Lengauer, T.; Rahnenführer, J.; Roomp, K.; Savenkov, I.; Fischer, R.; Hoffmann, D.; Selbig, J.; Korn, K.; et al. Computational methods for the design of effective therapies against drug resistant HIV strains. Bioinformatics 2005, 21, 3943–3950. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Reiner, R.C.; Perkins, T.A.; Barker, C.M.; Niu, T.; Chaves, L.F.; Ellis, A.M.; George, D.B.; le Menach, A.; Pulliam, J.R.C.; Bisanzio, D.; et al. A systematic review of mathematical models of mosquito-borne pathogen transmission: 1970–2010. J. R. Soc. Interface 2013. [Google Scholar] [CrossRef]
- Mandal, S.; Sarkar, R.R.; Sinha, S. Mathematical models of malaria—A review. Malaria J. 2011. [Google Scholar] [CrossRef]
- Buckee, C.O.; Gupta, S. Modelling malaria population structure and its implications for control. Adv. Exp. Med. Biol. 2010, 673, 112–126. [Google Scholar] [PubMed]
- Marino, S.; Linderman, J.J.; Kirschner, D.E. A multifaceted approach to modeling the immune response in tuberculosis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 479–489. [Google Scholar] [CrossRef] [PubMed]
- White, P.; Garnett, G. Mathematical Modelling of the Epidemiology of Tuberculosis. In Modelling Parasite Transmission and Control; Michael, E., Spear, R., Eds.; Springer: New York, NY, USA, 2010; Volume 673, pp. 127–140. [Google Scholar]
- Ozcaglar, C.; Shabbeer, A.; Vandenberg, S.L.; Yener, B.; Bennett, K.P. Epidemiological models of Mycobacterium tuberculosis complex infections. Math. Biosci. 2012, 236, 77–96. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Ribeiro, R.M. Modeling the viral dynamics of influenza a virus infection. Crit. Rev. Immunol. 2010, 30, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.J.; Lye, D.C.; Wilder-Smith, A. Combination strategies for pandemic influenza response—A systematic review of mathematical modeling studies. BMC Med. 2009. [Google Scholar] [CrossRef]
- Lon, H.-K.; Liu, D.; Jusko, W.J. Pharmacokinetic/pharmacodynamic modeling in inflammation. Crit. Rev. Biomed. Engin. 2012, 40, 295–312. [Google Scholar] [CrossRef]
- Vodovotz, Y.; Constantine, G.; Rubin, J.; Csete, M.; Voit, E.O.; An, G.C. Mechanistic simulations of inflammation: Current state and future prospects. Math. Biosci. 2009, 217, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Vodovotz, Y.; An, G. Complex Systems and Computational Biology Approaches to Acute Inflammation; Springer: New York, NY, USA, 2013. [Google Scholar]
- Lo, W.-C.; Martin, E.W.; Hitchcock, C.L.; Friedman, A. Mathematical model of colitis-associated colon cancer. J. Theor. Biol. 2013, 317, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Lo, W.-C.; Arsenescu, R.I.; Friedman, A. Mathematical model of the roles of t cells in inflammatory bowel disease. Bull. Math. Biol. 2013, 75, 1417–1433. [Google Scholar] [CrossRef] [PubMed]
- Vodovotz, Y.; Clermont, G.; Chow, C.; An, G. Mathematical models of the acute inflammatory response. Curr. Opin. Crit. Care 2004, 10, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Wendelsdorf, K.V.; Alam, M.; Bassaganya-Riera, J.; Bisset, K.; Eubank, S.; Hontecillas, R.; Hoops, S.; Marathe, M. Enteric immunity simulator: A tool for in silico study of gastroenteric infections. IEEE Trans. Nanobiosci. 2012, 11, 273–288. [Google Scholar] [CrossRef]
- Pigozzo, A.B.; Macedo, G.C.; Santos, R.W.D.; Lobosco, M. On the computational modeling of the innate immune system. BMC Bioinform. 2013. [Google Scholar] [CrossRef]
- Nathan, C. Points of control in inflammation. Nature 2002, 420, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- An, G.C.; Christley, S. Addressing the translational dilemma: Dynamic knowledge representation of inflammation using agent-based modeling. Crit. Rev. Biomed. Engin. 2012, 40, 323–340. [Google Scholar] [CrossRef]
- Barber, J.; Tronzo, M.; Harold Horvat, C.; Clermont, G.; Upperman, J.; Vodovotz, Y.; Yotov, I. A three-dimensional mathematical and computational model of necrotizing enterocolitis. J. Theor. Biol. 2013, 322, 17–32. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Thakar, J.; Albert, R. Boolean models of within-host immune interactions. Curr. Opin. Microbiol. 2010, 13, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Vodovotz, Y.; Csete, M.; Bartels, J.; Chang, S.; An, G.C. Translational systems biology of inflammation. PLoS Comput. Biol. 2008. [Google Scholar] [CrossRef]
- An, G.C. Closing the scientific loop: Bridging correlation and causality in the petaflop age. Sci. Transl. Med. 2010. [Google Scholar] [CrossRef]
- Huttenhower, C.; Kostic, A.D.; Xavier, R.J. Inflammatory bowel disease as a model for translating the microbiome. Immunity 2014, 40, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, M.M.; Corrin, M.S.C.; Johnson, M.T.J. Adaptive evolution in ecological communities. PLoS Biol. 2012. [Google Scholar] [CrossRef]
- Schluter, J.; Foster, K.R. The evolution of mutualism in gut microbiota via host epithelial selection. PLoS Biol. 2012. [Google Scholar] [CrossRef]
- Delaux, P.-M.; Varala, K.; Edger, P.P.; Coruzzi, G.M.; Pires, J.C.; Ané, J.-M. Comparative phylogenomics uncovers the impact of symbiotic associations on host genome evolution. PLoS Genet. 2014. [Google Scholar] [CrossRef]
- Werner, G.D.A.; Strassmann, J.E.; Ivens, A.B.F.; Engelmoer, D.J.P.; Verbruggen, E.; Queller, D.C.; Noë, R.; Johnson, N.C.; Hammerstein, P.; Kiers, E.T. Evolution of microbial markets. Proc. Natl. Acad. Sci. USA 2014, 111, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Filotas, E.; Grant, M.; Parrott, L.; Rikvold, P.A. Positive interactions and the emergence of community structure in metacommunities. J. Theor. Biol. 2010, 266, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Estrela, S.; Brown, S.P. Metabolic and demographic feedbacks shape the emergent spatial structure and function of microbial communities. PLoS Comput. Biol. 2013. [Google Scholar] [CrossRef]
- Carbo, A.; Bassaganya-Riera, J.; Pedragosa, M.; Viladomiu, M.; Marathe, M.; Eubank, S.; Wendelsdorf, K.; Bisset, K.; Hoops, S.; Deng, X.; et al. Predictive computational modeling of the mucosal immune responses during Helicobacter pylori infection. PLoS One 2013. [Google Scholar] [CrossRef]
- Seal, J.B.; Alverdy, J.C.; Zaborina, O.; An, G.C. Agent-based dynamic knowledge representation of Pseudomonas aeruginosa virulence activation in the stressed gut: Towards characterizing host-pathogen interactions in gut-derived sepsis. Theor. Biol. Med. Model. 2011. [Google Scholar] [CrossRef]
- Kim, M.; Christley, S.; Alverdy, J.C.; Liu, D.; An, G. Immature oxidative stress management as a unifying principle in the pathogenesis of necrotizing enterocolitis: Insights from an agent-based model. Surg. Infect. 2012, 13, 18–32. [Google Scholar] [CrossRef]
- Arciero, J.; Bard Ermentrout, G.; Siggers, R.; Afrazi, A.; Hackam, D.; Vodovotz, Y.; Rubin, J. Modeling the interactions of bacteria and toll-like receptor-mediated inflammation in necrotizing enterocolitis. J. Theor. Biol. 2013, 321, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Gennari, J.H.; Neal, M.L.; Galdzicki, M.; Cook, D.L. Multiple ontologies in action: Composite annotations for biosimulation models. J. Biomed. Inform. 2011, 44, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Christley, S.; An, G.C. A proposal for augmenting biological model construction with a semi-intelligent computational modeling assistant. Comput. Math. Organ. Theory 2012, 18, 380–403. [Google Scholar] [CrossRef] [PubMed]
- Knüpfer, C.; Beckstein, C.; Dittrich, P.; Novère, N.L. Structure, function, and behaviour of computational models in systems biology. BMC Syst. Biol. 2013. [Google Scholar] [CrossRef]
- Cockrell, C.; Christley, S.; An, G.C. Investigation of inflammation and tissue patterning in the gut using a spatially explicit general-purpose model of enteric tissue (SEGMEnT). PLoS Comput. Biol. 2014. [Google Scholar] [CrossRef]
- Smolen, P.; Baxter, D.A.; Byrne, J.H. A reduced model clarifies the role of feedback loops and time delays in the Drosophila circadian oscillator. Biophys. J. 2002, 83, 2349–2359. [Google Scholar] [CrossRef] [PubMed]
- Lema, M.A.; Golombek, D.A.; Echave, J. Delay model of the circadian pacemaker. J. Theor. Biol. 2000, 204, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J. Autoinhibition with transcriptional delay: A simple mechanism for the zebrafish somitogenesis oscillator. Curr. Biol. 2003, 13, 1398–1408. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.-P. Time-delay systems: An overview of some recent advances and open problems. Automatica 2003, 39, 1667–1694. [Google Scholar] [CrossRef]
- Fenton, A.; Lello, J.; Bonsall, M.B. Pathogen responses to host immunity: The impact of time delays and memory on the evolution of virulence. Proc. Biol. Sci. 2006, 273, 2083–2090. [Google Scholar] [CrossRef] [PubMed]
- Bewick, S.; Yang, R.; Zhang, M. The danger is growing! A new paradigm for immune system activation and peripheral tolerance. PLoS One 2009. [Google Scholar] [CrossRef]
- Tan, J.; Pan, R.; Qiao, L.; Zou, X.; Pan, Z. Modeling and dynamical analysis of virus-triggered innate immune signaling pathways. PLoS One 2012. [Google Scholar] [CrossRef]
- Lagoa, C.E.; Bartels, J.; Baratt, A.; Tseng, G.; Clermont, G.; Fink, M.P.; Billiar, T.R.; Vodovotz, Y. The role of initial trauma in the host’s response to injury and hemorrhage: Insights from a correlation of mathematical simulations and hepatic transcriptomic analysis. Shock 2006, 26, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Nieman, G.; Brown, D.; Sarkar, J.; Kubiak, B.; Ziraldo, C.; Dutta-Moscato, J.; Vieau, C.; Barclay, D.; Gatto, L.; Maier, K.; et al. A two-compartment mathematical model of endotoxin-induced inflammatory and physiologic alterations in swine. Crit. Care Med. 2012, 40, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Clermont, G.; Bartels, J.; Kumar, R.; Constantine, G.; Vodovotz, Y.; Chow, C. In silico design of clinical trials: A method coming of age. Crit .Care Med. 2004, 32, 2061–2070. [Google Scholar] [CrossRef] [PubMed]
- An, G.C. In silico experiments of existing and hypothetical cytokine-diyected clinical trials using agent-based modeling. Crit. Care Med. 2004, 32, 2050–2060. [Google Scholar] [CrossRef] [PubMed]
- Mi, Q.; Li, N.Y.-K.; Ziraldo, C.; Ghuma, A.; Mikheev, M.; Squires, R.; Okonkwo, D.O.; Verdolini-Abbott, K.; Constantine, G.; An, G.C.; et al. Translational systems biology of inflammation: Potential applications to personalized medicine. Per. Med. 2010, 7, 549–559. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christley, S.; Cockrell, C.; An, G. Computational Studies of the Intestinal Host-Microbiota Interactome. Computation 2015, 3, 2-28. https://doi.org/10.3390/computation3010002
Christley S, Cockrell C, An G. Computational Studies of the Intestinal Host-Microbiota Interactome. Computation. 2015; 3(1):2-28. https://doi.org/10.3390/computation3010002
Chicago/Turabian StyleChristley, Scott, Chase Cockrell, and Gary An. 2015. "Computational Studies of the Intestinal Host-Microbiota Interactome" Computation 3, no. 1: 2-28. https://doi.org/10.3390/computation3010002
APA StyleChristley, S., Cockrell, C., & An, G. (2015). Computational Studies of the Intestinal Host-Microbiota Interactome. Computation, 3(1), 2-28. https://doi.org/10.3390/computation3010002